Abstract
The discovery of clonal hematopoiesis (CH) in older individuals has changed the way hematologists and stem cell biologists view aging. Somatic mutations accumulate in stem cells over time. While most mutations have no impact, some result in subtle functional differences that ultimately manifest in distinct stem cell behaviors. With a large pool of stem cells and many decades to compete, some of these differences confer advantages under specific contexts. Approximately 20 genes are recurrently found as mutated in CH, indicating they confer some advantage. The impact of these mutations has begun to be analyzed at a molecular level by modeling in cell lines and in mice. Mutations in epigenetic regulators such as DNMT3A and TET2 confer an advantage by enhancing self-renewal of stem and progenitor cells and inhibiting their differentiation. Mutations in other genes involved in the DNA damage response may simply enhance cell survival. Here, we review proposed mechanisms that lead to CH, specifically in the context of stem cell biology, based on our current understanding of the function of some of the CH-associated genes.
Introduction
The recent discovery of the prevalence of clonal hematopoiesis (CH) has changed the way hematologists think about hematopoietic stem cells (HSCs). While fluctuation in the activity of stem cell clones has long been appreciated, overall, the contribution of stem cells to blood production was thought to be fairly stable in the absence of overt disease such as leukemia or bone marrow failure. Fundamentally, CH is the result of competition among long-lived stem cells in the bone marrow. CH evolves over a very long period of time, analogous to a marathon race. In a marathon that would start with runners initially well matched, a small endurance advantage can pay off, while a runner that expends a large amount of energy in the early phase may not last. Similarly, runners that are injured will drop out along the way. Over a long race, many minor factors can come into play, including psychology, weather, and terrain. Chance always plays some role, and finally, the likelihood of winning also depends also on number of competing runners. When we extrapolate these concepts to CH, we ask how can a stem cell “win”?
This review will focus on the proposed mechanisms that lead to CH, specifically in the context of stem cell biology, based on our current understanding of the function of some of the CH-associated genes.
Stem cells by the numbers
For decades, it has been appreciated that we harbor an abundance of HSCs, most of which are superfluous. Generally, HSCs are quiescent, dividing infrequently, while occasionally becoming activated to differentiate. Estimates of the total number of HSCs in human bone marrow vary from ∼10 0001,2 to ∼200 000.3,4
Throughout life, all somatic cells continuously accumulate mutations, and HSCs are no exception, acquiring ∼10 mutations per year (1-2 per cell division; Figure 1).5 Thus, by adulthood (∼20 years old), most of our HSCs will harbor 2 coding mutations and ∼200 noncoding mutations (which could also impact function). If we have 100 000 HSCs, that 20 year old will have accumulated on the order of 200 000 coding mutations across the ∼20 000 genes spread among the entire stem cell pool, rendering all of the HSCs slightly different. The number of lesions per gene continues to increase in a linear fashion with age.5
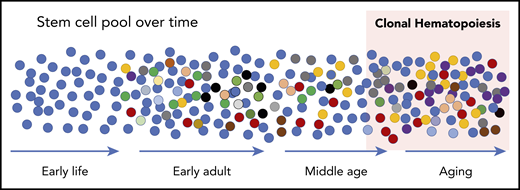
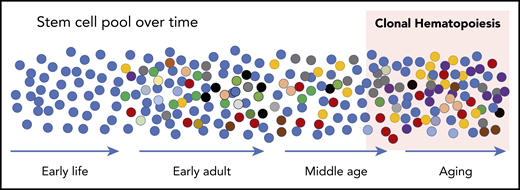
Schematic of development of CH. At birth, the HSC pool is relatively uniform. Over time, somatic mutations accumulate at a rate of ∼10 per year such that all HSCs are slightly different in early adults. These differences manifest in disadvantages and advantages for survival and contribution to peripheral blood production, resulting in some HSCs that “win” with advanced age (red cells at the far right). While expanded clones can be detected in middle age with sensitive sequencing techniques, the current accepted definition is when a clone reaches a proportion of ∼4% of cells measured in the peripheral blood. This equates to a variant allele frequency (VAF) of 2% when the variants (mutations) are heterozygous.
Schematic of development of CH. At birth, the HSC pool is relatively uniform. Over time, somatic mutations accumulate at a rate of ∼10 per year such that all HSCs are slightly different in early adults. These differences manifest in disadvantages and advantages for survival and contribution to peripheral blood production, resulting in some HSCs that “win” with advanced age (red cells at the far right). While expanded clones can be detected in middle age with sensitive sequencing techniques, the current accepted definition is when a clone reaches a proportion of ∼4% of cells measured in the peripheral blood. This equates to a variant allele frequency (VAF) of 2% when the variants (mutations) are heterozygous.
While the precise number of HSCs in adults is still unclear,2 each gene will acquire multiple “hits” over a life-time (dependent on gene size and codon composition4 ), and the total number of coding mutations spread across the whole stem cell pool is astonishing (potentially on the order of 1 million by the age of 70 (see also the review by Jaiswal6 ).
These mutations make each HSC a unique “runner” with slightly different features that may help or hurt its chances to win over time. The combination of the total numbers of mutations accumulated, our long lifespan, and the initial HSC pool size accounts for the inevitability of CH that can be observed by deep sequencing even at relatively young ages.7
Most of the acquired mutations are irrelevant, with many even deleterious to HSC function, leading to their demise. Rare mutations that impart any kind of advantage on the HSC will increase its likelihood of dominating its competitors over time; however, only certain kinds of advantage will manifest as a larger HSC “clone” over time (Figure 2). The most potent mutations will be those that result in increased self-renewal of HSCs, making more HSCs rather than balanced differentiation. Indeed, in a survey of 150 genes in which the impact of gene knockout (KO) in mice was examined, the majority had a deleterious effect on HSC function, while a few led to HSC expansion.8 Interestingly, 3 of those genes (Dnmt3a, Tet2, and Cbl) were later shown to be recurrently mutated in CH. Because it takes time to accumulate a sufficient number of mutations and then for those mutations to show biological expansion in humans, CH is rare before the age of 70 years using the classical definition of >2% clone VAF in the blood. These thresholds and their clinical relevance are discussed further in other reviews in this series.
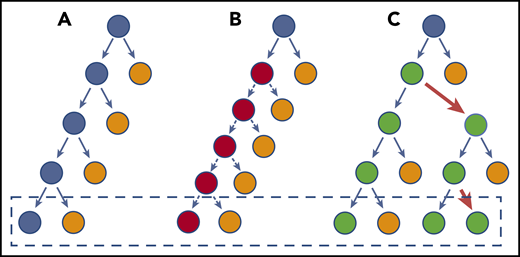
Model of HSC divisions that can result in CH. (A) There is a balance between regeneration of stem cells (blue) and differentiated cells (orange). This schematic does not necessarily imply an asymmetric division, but depicts the net result of the stem cell decisions (dashed box). (B) A stem cell divides faster, but each decision has the same net outcome as in panel A. Therefore, the net, after more cell divisions, does not increase the stem cell pool and does not outcompete normal HSCs. (C) The HSC has a slight bias toward self-renewal. Every few divisions (red arrows), it generates an imbalance such that the net, over time, is generation of more stem cells. The speed with which it results in truly biased outputs will depend on the frequency of imbalanced decisions. For most CH genes, this is probably a very subtle bias initially, explaining the very long time lag for CH to become apparent.
Model of HSC divisions that can result in CH. (A) There is a balance between regeneration of stem cells (blue) and differentiated cells (orange). This schematic does not necessarily imply an asymmetric division, but depicts the net result of the stem cell decisions (dashed box). (B) A stem cell divides faster, but each decision has the same net outcome as in panel A. Therefore, the net, after more cell divisions, does not increase the stem cell pool and does not outcompete normal HSCs. (C) The HSC has a slight bias toward self-renewal. Every few divisions (red arrows), it generates an imbalance such that the net, over time, is generation of more stem cells. The speed with which it results in truly biased outputs will depend on the frequency of imbalanced decisions. For most CH genes, this is probably a very subtle bias initially, explaining the very long time lag for CH to become apparent.
Cell-intrinsic contribution to clonal dominance: molecular mechanisms that enhance HSC fitness
There are a limited number of genes whose mutation will increase HSC self-renewal and manifest as CH (Figure 3). A sufficient number of humans with advanced age would need to be sequenced to exhaustively identify all of the potential genes,4 but the major players have certainly been identified. The types of genes recurrently mutated in CH broadly fall into 3 main classes, epigenetic regulators, splicing factors, and DNA damage response (DDR) genes, with rarer contributions from cohesin members, signaling molecules, and hematopoietic transcription factors.

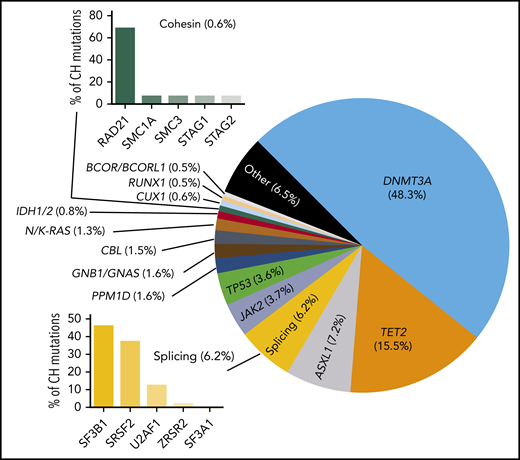
Proportion of common CH mutations in individuals in which a driver can be identified. These vary from study to study, but the general proportions are indicated.
Proportion of common CH mutations in individuals in which a driver can be identified. These vary from study to study, but the general proportions are indicated.
Mouse models have been useful to begin to dissect the role that mutations in these genes play. Genetic manipulation of many of these genes leads to increased HSC fitness and self-renewal. Aligned with the concept that small advantages are important in a phenomenon that develops over decades, the advantage afforded by these mutations is often subtle, leading to minor changes in molecular and cellular function, making mechanistic elucidation of how these mutations work challenging.
DNMT3A mutations in CH
Mutations in epigenetic regulators dominate the landscape of CH, with ∼50% of all CH mutations being variants in DNMT3A.9-11 Most DNMT3A mutations in CH are heterozygous and probably lead to loss of protein function by divergent mechanisms. DNMT3A mutations in CH are spread throughout the length of the gene, with missense mutations clustering in known structural and functional domains (Figure 4). Importantly, the variant spectrum of DNMT3A in CH is distinct from that observed in myeloid neoplasms, being notably less enriched for the R882 hotspot residue.12
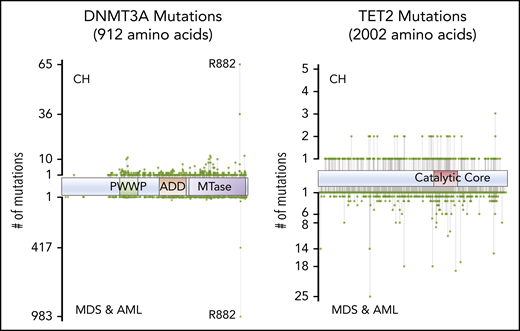
Positions of common variants in DNMT3A and TET2, comparing CH and AML/myelodysplastic syndrome (MDS).
Positions of common variants in DNMT3A and TET2, comparing CH and AML/myelodysplastic syndrome (MDS).
The DNMT3AR882 variant is a hypomorph with dominant negative activity.13,14 Thus, in patients where 1 wild-type (WT) allele is still expressed, functional DNMT3A activity may be reduced to only ∼20% of normal levels, approaching that of a complete null situation. Heterozygous DNMT3A mutations in other domains presumably reduce the effective DNMT3A function to as low as 50% of WT. We speculate that this results in a less vigorous HSC expansion, whereas CH+ individuals with DNMT3AR882 are more “at-risk” for acquiring acute myeloid leukemia (AML).4 An understanding of the relative risk of different DNMT3A mutations for subsequent disease will be important as more individuals with CH are identified as part of routine practice. Overall, we predict that the level of DNMT3A activity that remains after mutation, effectively the DNMT3A “dosage,” will correlate with the rate of expansion of mutant clones. If borne out by data, this may help stratify patients and prioritize them for closer monitoring for conversion to AML and perhaps for prophylactic intervention for CH expansion.
DNMT3A function
Given the prevalence of DNMT3A mutations in CH and malignancies, there has been tremendous interest in defining the function of this protein in normal and malignant hematopoiesis. DNMT3A encodes a de novo DNA methyltransferase enzyme responsible for establishing new DNA methylation patterns during development and stem cell fate decisions.15-17
Mouse models have shed light on how mutations in DNMT3A promote CH. Work with a conditional KO mouse model showed that Dnmt3a−/− HSCs show enhanced self-renewal and reduced differentiation efficiency, resulting in the accumulation of phenotypically normal HSCs that can engraft and expand but are impaired in blood production.18,19 The phenotype was exacerbated by stress such as repeated rounds of bone marrow transplantation that further depleted DNA methylation focally. Importantly, the expansion of Dnmt3a−/− HSCs in mouse bone marrow was truly a result of biased HSC fate decisions (more cell divisions resulting in stem cells) and not increased proliferation.20 The increased self-renewal potential of mutant HSCs was remarkable; while HSCs from WT mice will cease to contribute to engraftment after ∼3 rounds of transplantation, Dnmt3a−/− HSCs could continue to give rise to phenotypically normal HSCs after 12 rounds of transplantation, showing they were effectively immortalized, but not transformed.19 Importantly, loss of DNMT3A appears to have its biggest effect on stem cells. Any effects in downstream cells are a result of its loss of function in the HSC. Initially, HSCs with a DNMT3A mutation appear to have a slight bias toward self-renewal at the expense of differentiation; this bias becomes more pronounced with more cell divisions, leading to HSCs with greatly impeded differentiation.19 The concept that loss of Dnmt3a results in a bias toward self-renewal is consistent with the prevalence of CH in humans and the apparent requirement for additional proproliferative hits for transformation. These data also predict that individuals with significant DNMT3A mutant clones in the peripheral blood may exhibit even larger clones in the bone marrow or HSC pool.
Why does DNMT3A loss of function have such an enormous effect on HSCs? While the detailed mechanisms remain to be elucidated, there are several observations that at least partially explain its role. While genome-wide DNA methylation analysis has shown that DNA methylation is largely maintained throughout the genome after Dnmt3a KO, it becomes significantly reduced at enhancer regions, and also at the boundaries of large undermethylated domains termed “canyons.” These canyons are enriched for genes associated with HSC self-renewal such as HoxA9, Meis1, and Evi1.21 Concordant with this reduced methylation, higher expression of these self-renewal–associated genes is observed in Dnmt3a−/− cells.18 Concomitantly, the expression of genes important for differentiation is slightly reduced, consistent with the less efficient production of differentiated cells. Moreover, the cells that do differentiate from the KO HSCs continue to partially express self-renewal genes in some downstream lineages18 consistent with the inability to fully repress the self-renewal program. Human cells in which DNMT3A is mutated show a similar reduction of DNA methylation at enhancers and at the edges of DNA methylation canyons, as well as upregulation of genes involved in self-renewal.22,23
These findings lead to a model in which, under normal circumstances, when the HSC receives a signal to differentiate, DNMT3A acts to silence the self-renewal program. DNMT3A is targeted (through poorly understood mechanisms) to the promoters and enhancers associated with HSC-specific genes to epigenetically repress their expression by DNA methylation (likely recruiting other epigenetic repressors). The resultant downregulation of these genes allows the HSC to engage differentiation programs. In the absence of DNMT3A function (conditional KO in mice, somatic loss-of-function mutations in human), this process becomes inefficient, leading to ineffective differentiation. Cumulatively, this biases HSC fate decisions toward self-renewal at the expense of differentiation.
Transformation to malignancy requires additional mutations. Mice transplanted with Dnmt3a-KO cells will develop a variety of malignancies over a long period of time and acquire a variety of cooperating mutations.24,25 Enforced expression of activated signaling molecules drives both myeloid and lymphoid malignancies in a dose-related manner.26,27 Murine models with the mouse homolog of DNMT3AR882H (Dnmt3aR878H) knocked into the endogenous locus show a similar pathological phenotype, with the addition of cooperating mutations like Npm1c and Flt3ITD required to drive AML.28,29 Given the precise order and combination of genetic events required, combined with the right developmental age and selection of external stressors, it is unsurprising that the rate of progression from CH to disease is relatively low, even given the high incidence of DNMT3A-mutant CH in the general population.
Despite the knowledge gained from analysis of murine systems, the precise molecular mechanisms through which DNMT3A controls HSC function remain to be fully elucidated and are likely much more complex than current models. Although DNA hypomethylation in the absence of DNMT3A can lead to upregulation of genes important for HSC identity, the impacted genes reflect the minority of DNA methylation changes in these cells. In both mouse and human cells with DNMT3A variants, there is relatively poor overall correlation between alterations in DNA methylation and concomitant changes in expression levels of downstream genes.18,22 DNMT3A has complex interactions with other epigenetic regulators. DNA methylation canyons are decorated with active or repressive histone marks,21 which leads to different behaviors after methylation loss.19,21,30 The Ten-eleven translocation (TET) proteins have also been shown to be active at canyons, and the combination of histone marks and DNMT3A activity influences the outcome in terms of gene expression changes.30-32 DNA methylation alterations may also affect 3D genome interactions,33 which may in turn affect stem cell function. Finally, DNMT3A may play some roles independent of DNA methylation.28 More work is required to fully understand the mechanisms through which mutation of DNMT3A leads to CH.
TET2 mutations in CH
The second most commonly mutated gene in CH encodes the epigenetic regulator TET2. The TET family of dioxygenases promote DNA demethylation via the conversion of 5-methylcytosine to 5-hydroxymethylcytosine and other oxidized derivatives.34,35 The 5-hydroxymethylcytosine mark generated by TET2 is not maintained by DNMT1, and therefore, DNA methylation is lost in the next cell division if not actively replaced by a de novo DNA methyltransferase such as DNMT3A.
TET2 mutations in CH are also loss of function and distributed throughout the coding region. Mouse models of Tet2 loss have also been instructive. Overall, Tet2−/− HSCs have some similarities to Dnmt3a−/− HSCs, including increased self-renewal.36-38 However, while Dnmt3a loss of function more specifically impacts HSCs, conditional deletion of Tet2 in the hematopoietic system leads to a broader expansion of progenitors as well as HSCs.39 Indeed, the major manifestation of Tet2 loss of function appears to be driving myeloproliferation of downstream progenitor cells rather than direct effects on HSCs.39 TET2 also impacts DNA methylation in a broad variety of sites in the genome, including gene bodies,40 enhancers,41 and canyons.21,30,42 Some sites of DNA methylation alteration overlap in Tet2−/− and Dnmt3a−/− cells (ie, CpGs or differentially methylated regions that are hypomethylated in Dnmt3a−/− cells, but not Tet2−/− cells), suggesting these regions are acted on competitively by both proteins. But there are many clear differences, with some CpGs seemingly targeted uniquely by the individual proteins.42
A major conundrum in the field is that DNMT3A and TET2 antagonize each other at the biochemical level, with one normally adding DNA methylation (DNMT3A) and the other promoting its erasure (TET2), yet, at the genetic level, mutations in either lead to stem and progenitor cell expansion, impeded differentiation, CH, and ultimately malignancy development. This suggests a common end result of their DNA methylation regulatory activity, such as impeded differentiation, but the mechanistic details are yet unclear. Alternatively, the proteins could share roles beyond from regulation of DNA methylation. Underscoring their parallel action is the observation that some blood cancers harbor mutations in both DNMT3A and TET2, indicating that their loss can sometimes be complementary or additive.43,44 As with DNMT3A, we broadly understand that TET2 normally plays a role in changing gene expression at least in part through impacting DNA methylation levels to promote differentiation and suppress self-renewal, but much more remains to be elucidated.
DDR regulators in CH
A number of genes involved in the DDR are recurrently mutated in CH, notably TP53 and PPM1D.10,11 Mutations in PPM1D (protein phosphatase Mg2+/Mn2+–dependent 1D), a negative regulator of the DDR, are seen in 3% to 4% of cases of CH.11
PPM1D was a particular surprise, as previously, it had only been shown to be associated with nonhematopoietic malignancies. Upon further study, they were found particularly associated with therapy-related AML,45-48 particularly in the context of specific exposures. This has led to the concept that PPM1D mutations are selected for due to their enhanced resistance to cytotoxic exposures, much like TP53 mutations.49
PPM1D functions as a negative regulator of the DDR. PPM1D becomes upregulated by p53 after DNA damage and then serves to dephosphorylate numerous proteins to turn down the DDR after it peaks (Figure 5); one of its major targets is p53. Mutations of PPM1D truncate the protein, leading to a highly stabilized form. This in turn has the effect of attenuating the entire DDR.45,50

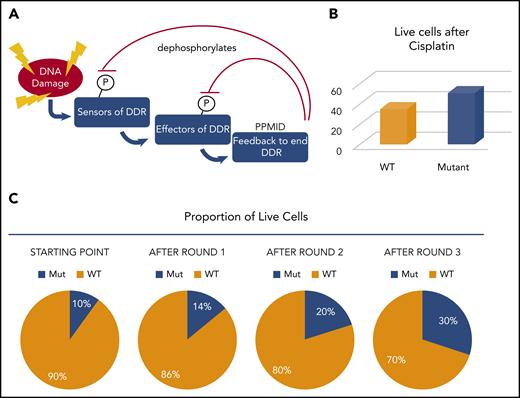
PPM1D mutations contribute to CH by suppressing the DDR and enhancing cell survival. (A) Schematic of the DDR and the role of PPM1D in dephosphorylating the positive regulators of DDR. (B) After 1 round of cisplatin treatment, 16% more PPM1D-mutant cells were alive. (C) If mutant cells start as 10% of the population, after each round of cisplatin, 16% more cells are surviving. This compounds in each round such that the proportion climbs to ∼30% after only 3 rounds. In patients, the mutant cells would be represented as 1 or a few HSCs but would still increase dramatically over time.
PPM1D mutations contribute to CH by suppressing the DDR and enhancing cell survival. (A) Schematic of the DDR and the role of PPM1D in dephosphorylating the positive regulators of DDR. (B) After 1 round of cisplatin treatment, 16% more PPM1D-mutant cells were alive. (C) If mutant cells start as 10% of the population, after each round of cisplatin, 16% more cells are surviving. This compounds in each round such that the proportion climbs to ∼30% after only 3 rounds. In patients, the mutant cells would be represented as 1 or a few HSCs but would still increase dramatically over time.
Experiments have shown that PPM1D-mutant cells outcompete WT cells through a mechanism distinct from that conferred by DNMT3A and TET2 mutations. PPM1D mutant cells have enhanced survival after chemotherapeutic exposure. In vitro, a modestly higher proportion (16% in some experiments) of PPM1D mutant cells than WT cells survived after a single round of cisplatin treatment. Even a small survival difference, after multiple rounds of treatment, compounds (Figure 5), resulting in a significant difference in mutant vs WT cells after several rounds of treatment such that a small starting population can end up dominating the pool of cells after several rounds of treatment.45 Once the proportion of mutant to WT cells changed, this remained steady in murine experiments for >1 year. If we extrapolate this to imagine the situation in humans undergoing cisplatin (or other) treatments, they would start with very small numbers of PPM1D-mutant cells initially. If the selective advantage for the human HSCs was similar to that measured in cell lines, even if only 0.01% of HSCs were PPM1D mutant, they would reach nearly 0.5% of the HSC pool over 10 rounds of treatment. Clearly, the starting fraction, the total number of HSCs in the patient, and the number of rounds of treatment will impact whether or not mutant cells will appear in the blood with a detectable VAF. While most of the impact of PPM1D mutation can be accounted for by a slightly lower death rate, this does not preclude other mechanisms working in parallel, such as a small difference in proliferation over time.50
While these findings broadly explain the selective advantage of PPM1D mutant cells there are major questions remaining. First, how does hyperactive PPM1D suppress apoptosis, and what affect does this have on the accumulation of mutations in HSC? Similarly, whether PPM1D mutations act largely via dephosphorylation of p53 is unclear. PPM1D has many DDR targets, so there could be multiple contributors to its outcome. Importantly, the phenotype of PPM1D mutants is distinct from those of TP53. While TP53 mutations are associated with permissiveness to chromosomal copy-number changes and a particularly dismal prognosis, PPM1D mutations do not appear to be associated with genomic instability to the same degree. Furthermore, while PPM1D mutations are prevalent in therapy-related AML patients, their low variant allele frequency is not consistent with a major role in driving AML. Therefore, their precise role in CH and subsequent cancer development remains to be determined. Finally, PPM1D mutations were reported in the original CH cohorts of “normal” individuals.11 It is not known if these were individuals who had unreported chemotoxic exposures or if there are additional contexts in which mutant cells could gain an advantage.
In contrast to PPM1D, TP53 has been studied in greater depth in hematopoiesis. Despite this, the precise impact of TP53 mutations in HSCs and how this confers an advantage is currently unclear. Certainly, cells with heterozygous mutation in TP53 will have a selective advantage after cancer therapy, as has been widely reported.49 However, TP53 mutations appear in CH in the absence of known selection. p53 has myriad activities in the genome and has been implicated in stem cell differentiation in pluripotent stem cells. Furthermore, loss of TP53 confers an advantage on aged51 and young HSCs in a dosage-dependent manner.52 Therefore, TP53 mutations may confer an advantage in CH through multiple mechanisms not all related to increased genomic instability (which likely plays a larger role once both alleles are mutated and cytotoxic exposures are present, ultimately contributing more to the onset of transformation53 ). Furthermore, different classes of TP53 mutation may also have different effects on CH vs leukemia development, conferring different levels of advantage in the cell.54
Together, genes involved in regulating the DDR are found mutated at significant frequencies in CH. In addition to PPM1D and TP53, mutations in BRCC3, ATM, and SRCAP probably fall in this group.47 There is still significant work to be done to understand the mechanisms through which they impact HSC competitiveness. This class of genes may offer unique opportunities to impede the development of CH. For example, because PPM1D acts dominantly, there have been efforts to develop inhibitors55 that may offer an opportunity to slow clonal expansion. An improved understanding of the mechanisms and opportunities may drive different choices of chemotherapeutic agents in contexts where CH or secondary malignancies may be deemed a particular risk.
It is now well appreciated that cytotoxic therapies will promote dominance of HSC clones with specific mutations such as PPM1D and TP53 (see more below and the article by Warren and Link56 ). Whether there are other environmental exposures that mimic chemotherapeutic treatments in otherwise-normal individuals remains unclear, but expansion of PPM1D or TP53 mutant cells could be the proverbial “canary in the coal mine” for cytotoxic exposures.
Additional CH mutation classes
Two other classes of mutations that, as groups of mutations in the same functional pathway, are found recurrently mutated are those involved in the cohesin complex and splicing-associated factors. The mechanisms through which mutations in these contribute to CH are still poorly understood and will not be reviewed in depth here. Mutations in cohesin associated genes (RAD21, SMC3, STAG2, and others10,11 ) may contribute through a similar overall effect as that of DNMT3A and TET2, as Stag2 loss of function in mice has been shown to lead to increased expression of self-renewal genes and decreased expression of differentiation genes57 ; nevertheless, much more work needs to be done on this important class. ASXL1 mutations also likely increase self-renewal capacity of HSCs, but the mechanisms are obscure.58 Similarly, the role of mutations in splicing regulators is enigmatic. While key genes involved in differentiation and self-renewal have been shown to be mis-spliced in the context of some of these mutations,59,60 the molecular outcomes of splicing factors mutations are inconsistent, and there is no unified view on their mechanism of action. Finally, a curious CH mutation is the JAK2V617F variant, which can be found in ∼3% of individuals with CH.61 This variant is virtually the only mutation in an activating signaling molecule in CH. The JAK2V617F mutation is common in myeloproliferative neoplasms, where it is often the only driver mutation that can be identified in these patients.62-64 This fact alone points to something special about this signaling mutation in hematopoiesis. Initially, mouse models suggested that JAK2V617F-mutant HSCs have a modest competitive advantage.65,66 However, more quantitative single-cell analyses indicate JAK2V617F reduces self-renewal of individual HSCs but leads to expansion of progenitor cells, which provides a window in which collaborating mutations can accumulate to drive disease progression.67
Cell-extrinsic contribution to clonal dominance: environmental influences on clone selection
As discussed above, everyone will harbor HSCs with coding mutations in CH-related genes by age 50 years.7 However, only in a fraction of individuals will mutant HSCs expand sufficiently by age 70 years to be detectable at the currently accepted 2% VAF threshold. Therefore, it is either chance and time4 or additional extrinsic factors that drive mutant clones to expand to various degrees in different individuals, as well as progress to leukemia, which happens in an even smaller fraction of people. Consequently, there is intense interest in identifying factors that promote CH.68
The role of extrinsic influences on clonal expansion is suggested from some human bone marrow transplantation data.69,70 In one study of sibling transplants, CH originating from the donor was able to be identified. In 4 of 5 cases, the CH clone was larger in the recipient than in the donor, suggesting that either some aspect of the transplantation process promoted clonal expansion or perhaps the milieu of the recipient bone marrow contributed.71 While the number of examined cases is limited, these data show that well-designed studies of allogeneic transplantation may be able to lend additional future insights into factors that promote CH.
Much recent interest has focused on the potential role of inflammation, a pervasive feature of aging tissues. The term “inflammaging” has been coined to describe the chronic low-grade systemic inflammation in aging, and it is a significant risk factor for morbidity and mortality in elderly people.72 In CH, TET2 mutant progenitor cells in particular are particularly responsive to changes in levels of proinflammatory cytokines. While consistent exposure to proinflammatory cytokines such as tumor necrosis factor α and interleukin-6 (IL-6) are detrimental to the long-term function of normal HSCs, Tet2 mutant HSCs are refractory to these signals and maintain functional integrity, providing them with a competitive advantage in an inflammatory environment.73 TET2 is also a negative regulator of the inflammatory response in myeloid cells,74 in part by recruiting HDAC2 to suppress IL-6.75 Thus, Tet2 mutations potentially establish a positive feedback loop whereby the mutant myeloid cells secrete more IL-6, which then further augments the competitive advantage of mutant progenitor cells in the bone marrow by having a negative impact on the WT HSCs. These features of HSCs with CH-associated mutations may contribute to the role of CH in non-hematologic diseases (see also the review by Jaiswal6 in this issue).
A second source of inflammaging includes harmful products produced by the microbial constituents of the human body, such as oral or gut microbiota, which can leak into surrounding tissues and the circulation.76 A recent study showed that dysfunction of the small intestinal barrier leading to bacterial translocation and increased IL-6 production is important for disease progression of Tet2-mutant hematopoietic progenitor cells, can be reversed by antibiotic treatment, and fails to develop in germ-free mice.77 Moreover, germline inactivating IL-6R variants attenuate the risk of cardiovascular disease in people with CH, further supporting a role for inflammation in disease progression.78 Concurrent evidence is emerging that different proinflammatory cytokines may induce expansion of CH clones with specific mutations, such as positive selection of DNMT3A mutant clones by type II interferons.79 Moreover, HSCs are known to be stimulated by interferons and other inflammatory molecules.80,81 While this area of research is in its infancy, we envision that enhanced understanding of these issues could guide potential interventions. Inhibition of different nodes of the inflammatory response could be used to suppress certain CH clones and mitigate their potential for leukemic transformation, a potential mechanism of disease prevention in “at-risk” individuals. But much more work is required to identify how different types of inflammatory stress select for clones with specific mutations.
Emerging concepts
The discovery of CH was followed very quickly by the observation that similar changes in tissue mosaicism driven by somatic mutations with age occurs in other tissues such as the skin,82 esophagus,83 and brain.84 Computational modeling suggests that clonal expansion is present in almost every human tissue85 ; therefore, CH is likely a natural consequence of aging not unique to the blood system. Germline mutations, in the same and other genes, also give rise to CH, albeit with different kinetics and clinical implications (see the article by Tsai and Lindsley86 ).
Going forward, it will be important to discriminate which variants are potentially pathogenic vs passengers, as well as the relationship to cancer predisposition. One view is that the CH mutations simply increase the pool of susceptible HSCs that then serve as targets for second hits that drive malignancy development. Alternatively, the specific cellular changes that the mutations cause may promote malignancy in downstream cells by activating programs such as self-renewal. In this case, we might expect that all individuals with CH would develop malignancies if they lived long enough (see Warren and Link,56 this issue). An alternative view is that CH clones might actually be beneficial in some ways, potentially by suppressing other potentially pathogenic clones. Perhaps CH has become a mechanism by which HSC clones can extend their lifespan to sustain blood production in individuals that survive with current lifespans.
Over the past 5 years, the power of sequencing technologies has transformed the concept of CH from a curious observation, to a potential mechanism of early blood cancer identification and prevention. Given that overall survival rates for adult AML and myelodysplastic syndrome patients have not improved dramatically in the last 30 years, a compelling way to treat these diseases may be to prevent them from actually evolving in at-risk individuals (see the review by Steensma and Bolton87 ). Before this can be realized, much more research is required to determine the mechanisms by which certain clones can “win” in competition in the bone marrow over age, the interplay between somatic mutations and different environmental pressures, and the correlation of specific variants with propensity for disease progression.
Authorship
Contribution: M.A.G. and G.A.C. wrote and edited the manuscript.
Conflicts-of-interest disclosure: M.A.G. and G.A.C. declare no competing financial interests.
Correspondence: Margaret A. Goodell, Department of Molecular and Cellular Biology, Baylor College of Medicine, One Baylor Plaza, N1030, Houston, TX 77030; e-mail: goodell@bcm.edu.