Key Points
RNF138 catalyzes ubiquitination in MYD88L265P but not in wild-type MYD88.
A20 suppresses RNF138 expression to inhibit the oncogenic activity of MYD88L265P.

Abstract
Myeloid differentiation primary response protein 88 (MYD88) is a critical universal adapter that transduces signaling from Toll-like and interleukin receptors to downstream nuclear factor-κB (NF-κB). MYD88L265P (leucine changed to proline at position 265) is a gain-of-function mutation that occurs frequently in B-cell malignancies such as Waldenstrom macroglobulinemia. In this study, E3 ligase RING finger protein family 138 (RNF138) catalyzed K63-linked nonproteolytic polyubiquitination of MYD88L265P, resulting in enhanced recruitment of interleukin-1 receptor–associated kinases and elevated NF-κB activation. However, RNF138 had little effect on wild-type MYD88 (MYD88WT). With either RNF138 knockdown or mutation on MYD88 ubiquitination sites, MYD88L265P did not constitutively activate NF-κB. A20, a negative regulator of NF-κB signaling, mediated K48-linked polyubiquitination of RNF138 for proteasomal degradation. Depletion of A20 further augmented MYD88L265P-mediated NF-κB activation and lymphoma growth. Furthermore, A20 expression correlated negatively with RNF138 expression and NF-κB activation in lymphomas with MYD88L265P and in those without. Strikingly, RNF138 expression correlated positively with NF-κB activation in lymphomas with MYD88L265P, but not in those without it. Our study revealed a novel mutation-specific biochemical reaction that drives B-cell oncogenesis, providing a therapeutic opportunity for targeting oncogenic MYD88L265P, while sparing MYD88WT, which is critical to innate immunity.
Introduction
Myeloid differentiation primary response protein 88 (MYD88) is a general adaptor protein that acts downstream of the Toll-like receptor (TLR) and interleukin (IL)-1 or -18 receptors to mediate the activation of nuclear factor-κB (NF-κB).1 MYD88 gain-of-function mutations are prevalent in hematologic B-cell malignancies. The most common MYD88 mutation is a single amino acid change of leucine to proline at position 265 (MYD88L265P).2-4 MYD88L265P is detected at high frequencies in Waldenstrom macroglobulinemia (WM; ∼90%) and in the activated B-cell subtype of diffuse large B-cell lymphoma (ABC-DLBCL, ∼30%), but is rarely found in solid tumors.5 MYD88L265P drives constitutive activation of NF-κB and signal transducer and activator of transcription 3 (STAT3) in lymphoma cells.2
Many components of the NF-κB signaling pathway undergo posttranslational modifications (PTMs) in both lymphoid and myeloid malignancies.6-11 PTMs, including phosphorylation and ubiquitination, control the function of wild-type (WT) MYD88 (MYD88WT). SYK kinase phosphorylates MYD88, promoting MYD88-dependent IL-1 signaling.12 For ubiquitination, the type of linkage chains generally determines the fate of targeted proteins: lysine 48 (K48)–linked ubiquitin chains for protein degradation by the proteasome and K63-linked ubiquitin chains for regulating proteasome-independent pathways, such as signal transduction and endocytosis.13 Three E3 ubiquitin ligases (NRDP1 [RNF41],14 SMURF1, and SMURF215 ) ubiquitinate MYD88WT through K48-linked ubiquitin chains and promote its degradation. In mice, speckle-type poz protein (SPOP), a substrate-binding adaptor protein of the CUL3-RBX1 E3 ubiquitin ligase complex, restrains the inflammatory activation of hematopoietic stem cells by promoting the ubiquitination and degradation of Myd88WT.16 In human lymphoma cells, SPOP triggers the nondegradative mixed-linkage ubiquitination of both MYD88WT and MYD88L265P, to inhibit NF-κB signaling.17 Yet PTMs specific to MYD88L265P have never been reported.
Mice with B-cell–specific Myd88L252P (the orthologous position of the human MYD88L265P mutation) develop a lymphoproliferative disease, which occasionally transforms into clonal lymphoma.18 Additional genetic hits, including loss of function of TNF-α–induced protein-3 (TNFAIP3; encodes for the A20 protein) or upregulation of BCL2 significantly shortens time to lymphoma onset.18,19 In this work, we explored the role of the ubiquitination of MYD88L265P in driving prosurvival oncogenic signaling in lymphomagenesis. We found that MYD88L265P-specific ubiquitination by the E3 ligase RNF138 drove constitutive NF-κB activation and promoted lymphoma growth. We also elucidated a mechanism by which A20 negatively regulated the oncogenic signaling of MYD88L265P by downregulating the RNF138 protein level through K48-linked polyubiquitination. Our data describe a novel enzymatic reaction that can be exploited in therapeutic development to treat lymphomas with an MYD88L265P mutation.
Methods
Immunofluorescence
Cells were seeded into a chamber slide for 24 hours, fixed in 4% paraformaldehyde (cat. no. sc-281692; Santa Cruz Biotechnology) for 10 minutes, permeabilized in 0.2% Triton X-100 (cat. no. 13444259; Thermo Fisher) for 20 minutes, and treated in 2% bovine serum albumin (cat. no. B2518; Sigma-Aldrich) for 1 hour. Slides were incubated with specific primary antibody at 4°C in a humidified chamber overnight. After they were washed 3 times with phosphate-buffered saline, the slides were incubated with a fluorescence-labeled secondary antibody. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (cat. no. P36935; Thermo Fisher). The following antibodies were used: goat anti-rabbit IgG (Alexa Fluor 488; cat. no. ab15007; 1:500), goat anti-mouse IgG (Alexa Fluor 488; cat. no. ab150117; 1:500), donkey anti-rabbit IgG (Alexa Fluor 647; cat. no. ab150075; 1:500; all from Abcam). Images were obtained with an inverted SP8 microscope at ×100 magnification (Leica Biosystems, Wetzlar, Germany).
Viral shRNA and single guide RNA
Short hairpin RNAs (shRNAs) in SMARTvector targeting 3′ untranslated region of genes were obtained from Dharmacon (Lafayette, CO). Lentiviruses were made according to the manufacturer’s protocol. In brief, shRNA constructs and packaging plasmids (pVSVg and psPAX2) were transfected into HEK293T cells by using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) in serum-free Opti-MEM. The cells were incubated at 37°C, and the medium was replaced with fresh medium after 6 hours. Viral supernatant was collected 48 hours after the cotransfection. Targeted cells were infected with viral supernatant with the addition of 8 μg/mL polybrene (Sigma, St. Louis, MO), and the cells were selected with medium containing 0.5 μg/mL puromycin (Sigma) 24 hours after infection and maintained for 5 days. MYD88L265P cell lines with shRNF138 grow slowly but can be cultured over 2 months. The RNF138 gene were also genetically knocked out by using the CRISPR/Cas9 system (the lentiCRISPR v2-based single guide RNA [sgRNA] vector; cat. no. 52961; Addgene). The sgRNAs were cloned into the vector with the following primers (sgRNA against EGFP as a control): EGFP-sgRNA-forward (F)-CACCGGAAGTTCGAGGGCGACACCC; EGFP-sgRNA-reverse (R)-AAACGGGTGTCGCCCTCGAACTTCC; RNF138-sgRNA 1F-CACCGGTGATCCAGTAA ACGCTGTC; RNF138-sgRNA 1R-AAACGACAGCGTTTACTGGATCACC; RNF138-sgRNA 2F-CACCGTAGTCACATTTCCACGACAT; and RNF138-sgRNA 2R-AAACATGTCGTGGAAATGTGACTAC. Lentiviruses were produced as just described. Infected cells were selected with 0.5 μg/mL puromycin 2 days after infection and maintained for 7 days. After sgRNA viral infection and puromycin selection, the number of cells was noted for 8 days before they were counted or for 3 days for MTS. MYD88L265P cell lines with sgRNF138 cannot be cultured long term and were used within 2 weeks of puromycin selection.
Mouse models
All procedures for the mouse model were conducted according to our protocol, which was approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic. Xenograft tumors were established by subcutaneous injection of lymphoma cells (1 × 106) into the right flank of 6-week-old NOD-SCID IL2rgnull (NSG) mice (5 mice per group; cat. no. 005557; The Jackson Laboratory, Bar Harbor, ME). Tumors were measured by caliper every 3 days. Tumor volume was calculated according to the formula: tumor volume (mm3) = (length × width × width/2), where length is the longest diameter and width is the shortest. Mice were killed when the tumor volume reached 1200 mm3, and xenografted tumors were dissected and analyzed.
Cellular and biochemical analyses
These procedures are described in detail in supplemental Methods, available on the Blood Web site.
Statistical analysis
All quantitative results were expressed as the mean ± standard error of the mean of 3 independent experiments. Differences between the means were evaluated by Student t test or analysis of variance when data were normally distributed. When data were not normally distributed, we used the nonparametric Wilcoxon exact matched-pair signed-rank test. For multiple comparisons, a 1- or 2-way analysis of variance followed by Tukey’s multiple-comparisons test was applied. The clinicopathologic significance of clinical samples was evaluated by the χ2 test or Fisher’s exact test for categorical data. The Mann-Whitney U test was used when the data did not fit a normal distribution. Kaplan-Meier analysis and the log-rank test (Mantel-Cox) were used for survival analysis. The Pearson rank correlation was used for correlation tests. P ≤ .05 indicated statistical significance.
Results
Identification and characterization of E3 ligase RNF138 for MYD88L265P K63-linked polyubiquitination
We sought to identify MYD88L265P-interacting proteins in MYD88-null 293T cells by using mass spectrometry (MS) analysis (supplemental Figure 1A) and found that RNF138 and 5 other E3 ligases bound MYD88L265P (supplemental Figure 1B). Coimmunoprecipitation (co-IP) was performed to confirm the interaction between MYD88L265P and RNF138 in 293T cells ectopically overexpressing these 2 proteins (Figure 1A; supplemental Figure 1C). This interaction was also observed in vitro with purified proteins (Figure 1B). Interestingly, RNF138 interacted with both MYD88L265P and MYD88WT, but its binding to MYD88L265P was stronger (supplemental Figure 1D). The endogenous co-IP assay (Figure 1C) and in situ proximity-ligation assay (PLA; Figure 1D; supplemental Figure 1E) revealed that RNF138 preferred MYD88L265P over MYD88WT and MYD88S222R in lymphoma cells. Deleting the death domain or Toll/interleukin-1 receptor (TIR) domains of MYD88L265P markedly impaired the RNF138-MYD88 interaction (supplemental Figure 1F). The zinc finger (ZNF) domain and ubiquitin-interacting motif domains of RNF138 were essential for its binding with MYD88L265P (supplemental Figure 1G).
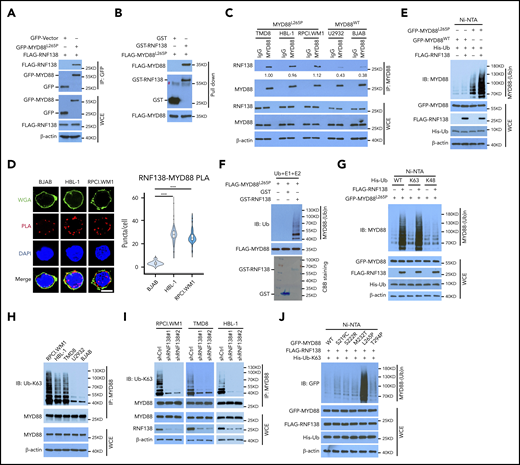
RNF138 interacts with MYD88L265P and promotes MYD88L265P ubiquitination. (A) Co-IP analysis of MYD88L265P and RNF138 interaction in 293T cells transfected with the indicated constructs. (B) Direct binding between RNF138 and MYD88L265P. FLAG-MYD88L265P immunoprecipitated from 293T cells bound to GST-RNF138 in vitro. #, non-specific signal. (C) Co-IP analysis of the endogenous MYD88 and RNF138 interaction in lymphoma cells. (D) PLA of MYD88 and RNF138 interaction in lymphoma cells. ***P < .001. The bar represents 5 μm. Wheat germ agglutinin (WGA), Alexa Fluor 488 (green); PLA, Alexa Fluor 568 (red). (E) In vivo MYD88L265P and MYD88WT ubiquitination by RNF138 in 293T cells. His-Ub–linked proteins were purified using Ni-NTA beads from cell lysates before they were subjected to IB analysis with MYD88 antibody. (F) MYD88L265P ubiquitination by RNF138 in vitro. FLAG-MYD88L265P was incubated with E1, E2, and ubiquitin+GST or GST-RNF138 in a cell-free system. CBB Coomassie brilliant blue. (G) RNF138-mediated MYD88L265P ubiquitination with ubiquitin mutants in 293T cells. Cells were transfected with RNF138 and MYD88L265P, along with WT or mutant ubiquitin and subjected to Ni-NTA bead purification and IB analysis as in panel E. (H) Endogenous MYD88 ubiquitination in MYD88L265P (RPCI.WM1, HBL-1, and TMD8 cells) and MYD88WT (U2932 and BJAB) cell lines. Lysates from cells were immunoprecipitated using the antibody against MYD88 before they were subjected to IB with Ub-K63 antibody. (I) Endogenous MYD88L265P ubiquitination in RPCI.WM1, TMD8, and HBL-1 cells with RNF138 knockdown. Lysates from control and RNF138 stable knockdown cells were immunoprecipitated using the antibody against MYD88 before they were subjected to IB. (J) RNF138-mediated ubiquitination on MYD88 mutants in 293T cells. His-Ub–linked proteins were purified using Ni-NTA beads from cell lysates before IB analysis with GFP antibody. WCE, whole-cell extract.
RNF138 interacts with MYD88L265P and promotes MYD88L265P ubiquitination. (A) Co-IP analysis of MYD88L265P and RNF138 interaction in 293T cells transfected with the indicated constructs. (B) Direct binding between RNF138 and MYD88L265P. FLAG-MYD88L265P immunoprecipitated from 293T cells bound to GST-RNF138 in vitro. #, non-specific signal. (C) Co-IP analysis of the endogenous MYD88 and RNF138 interaction in lymphoma cells. (D) PLA of MYD88 and RNF138 interaction in lymphoma cells. ***P < .001. The bar represents 5 μm. Wheat germ agglutinin (WGA), Alexa Fluor 488 (green); PLA, Alexa Fluor 568 (red). (E) In vivo MYD88L265P and MYD88WT ubiquitination by RNF138 in 293T cells. His-Ub–linked proteins were purified using Ni-NTA beads from cell lysates before they were subjected to IB analysis with MYD88 antibody. (F) MYD88L265P ubiquitination by RNF138 in vitro. FLAG-MYD88L265P was incubated with E1, E2, and ubiquitin+GST or GST-RNF138 in a cell-free system. CBB Coomassie brilliant blue. (G) RNF138-mediated MYD88L265P ubiquitination with ubiquitin mutants in 293T cells. Cells were transfected with RNF138 and MYD88L265P, along with WT or mutant ubiquitin and subjected to Ni-NTA bead purification and IB analysis as in panel E. (H) Endogenous MYD88 ubiquitination in MYD88L265P (RPCI.WM1, HBL-1, and TMD8 cells) and MYD88WT (U2932 and BJAB) cell lines. Lysates from cells were immunoprecipitated using the antibody against MYD88 before they were subjected to IB with Ub-K63 antibody. (I) Endogenous MYD88L265P ubiquitination in RPCI.WM1, TMD8, and HBL-1 cells with RNF138 knockdown. Lysates from control and RNF138 stable knockdown cells were immunoprecipitated using the antibody against MYD88 before they were subjected to IB. (J) RNF138-mediated ubiquitination on MYD88 mutants in 293T cells. His-Ub–linked proteins were purified using Ni-NTA beads from cell lysates before IB analysis with GFP antibody. WCE, whole-cell extract.
We next investigated whether MYD88L265P was directly ubiquitinated by RNF138. Cotransfection with RNF138 promoted the ubiquitination of MYD88L265P, but not MYD88WT, in 293T cells (Figure 1E). Among the E3 ligases, RNF138 had the strongest effect in promoting the polyubiquitination of MYD88L265P (supplemental Figure 1H). The catalytically inactive RNF138 mutant Cys18/21→Ala (C18/21A) failed to ubiquitinate MYD88L265P (supplemental Figure 2A), although it interacted with MYD88L265P (supplemental Figure 2B). Recombinant RNF138 ubiquitinated purified MYD88L265P in a cell-free in vitro ubiquitination assay, supporting that RNF138 is a bona fide E3 ligase for MYD88L265P (Figure 1F). However, RNF138 did not affect MYD88L265P protein levels (supplemental Figure 2C), and treatment with the proteasome inhibitor MG132 had no significant impact on RNF138-mediated MYD88L265P ubiquitination in 293T cells (supplemental Figure 2D). Next, we identified the type of ubiquitin chains that formed on MYD88L265P. RNF138 promoted MYD88L265P polyubiquitination in the presence of ubiquitin K63, but not K48 (K63/48 indicates that all K residues in ubiquitin were mutated to arginine [R] except K63/48; Figure 1G). When individual K residues in ubiquitin mutated, only the K63R (K63 mutated to R) mutation impaired RNF138-induced MYD88L265P ubiquitination (supplemental Figure 2E). Previous studies have reported that RNF41,14 SMURF1, and SMURF215 mediate MYD88WT ubiquitination; however, we found that they had no effect on K63-linked ubiquitination of MYD88L265P (supplemental Figure 2F). Furthermore, MYD88 K63-linked ubiquitination was stronger in MYD88L265P than in MYD88WT lymphoma cell lines (Figure 1H). RNF138 knockdown by shRNAs diminished endogenous K63-linked ubiquitination significantly in MYD88L265P, but not in MYD88WT, lymphoma cells (Figure 1I; supplemental Figure 2G). Downregulation of RNF138 did not affect MYD88 protein levels in these cells. In addition, RNF138 elicited the strongest K63-linked polyubiquitination of MYD88L265P among recurrent MYD88 mutants found in lymphoma2 (Figure 1J). Altogether, our data indicate that RNF138 catalyzes K63-linked polyubiquitination of MYD88L265P.
RNF138 promotes NF-κB activation and tumorigenicity of MYD88L265P lymphoma cells
Hyperactivation of NF-κB is a cardinal feature in lymphoid malignancies with MYD88L265P, so we investigated whether RNF138 is essential for NF-κB activation in lymphoma cells. The MYD88L265P mutant nucleates a signaling complex containing IL-1 receptor–associated kinase -1 (IRAK-1) and -4, to activate NF-κB.2 We first characterized the interaction between MYD88L265P and IRAK-1 or IRAK-4 in MYD88L265P+ TMD8 cells by using a PLA. RNF138 knockdown significantly decreased the interaction between MYD88L265P and IRAK-1/4 (Figure 2A-B). Knockdown of RNF138 also drastically decreased the phosphorylation of p65 and STAT3 in MYD88L265P+ RPCI.WM1, TMD8, and HBL-1 cells (Figure 2C), but not in MYD88WT-U2932 and BJAB cells (supplemental Figure 3A). Similarly, an immunoblot (IB) analysis showed that RNF138 knockdown decreased the nuclear fraction of p65 in RPCI.WM1, TMD8, and HBL-1 cells (Figure 2D; supplemental Figure 3B). The immunofluorescence (IF) (supplemental Figure 3C) results consistently revealed that depletion of RNF138 by shRNA reduced the positive staining of p65 in the nucleus of TMD8 cells. In addition, RNF138 knockdown reduced the mRNA levels of NF-κB-transactivated genes in RPCI.WM1, TMD8, and HBL-1 cells, but not in U2932 cells (Figure 2E). Furthermore, secretion levels of IL-6 and -10 were significantly lower in MYD88L265P+HBL-1 and TMD8 cells after RNF138 knockdown (supplemental Figure 3D).
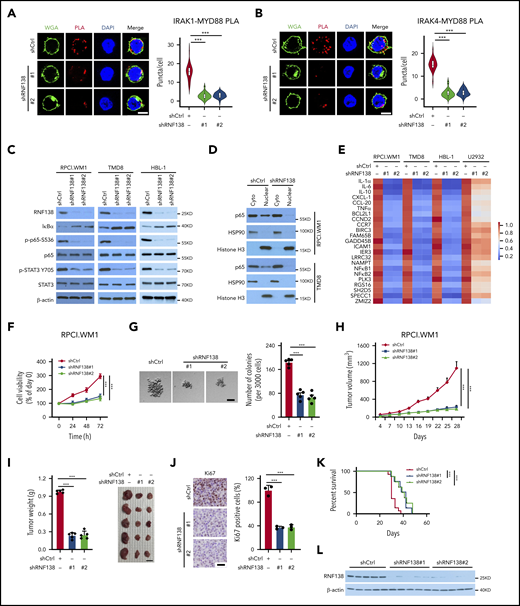
RNF138 is required for NF-κB activation and tumor growth in MYD88L265P lymphoma cells. (A-B) PLA to analyze the interaction between MYD88L265P and IRAK-1 (A) or IRAK-4 (B) in TMD8 cells with RNF138 knockdown. ***P < .001. The bars represent 5 μm. WGA, Alexa Fluor 488 (green); PLA, Alexa Fluor 568 (red). (C) IB analysis of NF-κB and STAT3 signaling in RPCI.WM1, TMD8, and HBL-1 cells with RNF138 knockdown. (D) IB analysis of p65 in cytosolic (cyto) and nuclear fractions from RPCI.WM1 and TMD8 cells with RNF138 knockdown. (E) Real-time polymerase chain reaction analysis of NF-κB downstream target genes in RPCI.WM1, TMD8, HBL-1, and U2932 cells after RNF138 knockdown. (F-G) MTS (F) and colony-forming unit (G) assays to examine cell viability and colony formation of RPCI.WM1 cell. ***P < .001. The bar represents 50 μm. (H-J) Effect of RNF138 knockdown on tumor growth of RPCI.WM1 cells in NSG mice. Tumor volumes were monitored (H), and tumors were dissected and weighed (I). The bar represents 1 cm. (J) Ki67 was examined using IHC analysis. Ki67 was stained with DAB as the chromogen (brown). The bar represents 25 μm. ***P < .001. (K) Kaplan-Meier curve analysis of the survival of mice bearing RPCI.WM1 xenograft tumors. ***P < .001. (L) IB analysis of RNF138 protein levels in individual RPCI.WM1-derived xenograft tumors. WCE, whole-cell extract.
RNF138 is required for NF-κB activation and tumor growth in MYD88L265P lymphoma cells. (A-B) PLA to analyze the interaction between MYD88L265P and IRAK-1 (A) or IRAK-4 (B) in TMD8 cells with RNF138 knockdown. ***P < .001. The bars represent 5 μm. WGA, Alexa Fluor 488 (green); PLA, Alexa Fluor 568 (red). (C) IB analysis of NF-κB and STAT3 signaling in RPCI.WM1, TMD8, and HBL-1 cells with RNF138 knockdown. (D) IB analysis of p65 in cytosolic (cyto) and nuclear fractions from RPCI.WM1 and TMD8 cells with RNF138 knockdown. (E) Real-time polymerase chain reaction analysis of NF-κB downstream target genes in RPCI.WM1, TMD8, HBL-1, and U2932 cells after RNF138 knockdown. (F-G) MTS (F) and colony-forming unit (G) assays to examine cell viability and colony formation of RPCI.WM1 cell. ***P < .001. The bar represents 50 μm. (H-J) Effect of RNF138 knockdown on tumor growth of RPCI.WM1 cells in NSG mice. Tumor volumes were monitored (H), and tumors were dissected and weighed (I). The bar represents 1 cm. (J) Ki67 was examined using IHC analysis. Ki67 was stained with DAB as the chromogen (brown). The bar represents 25 μm. ***P < .001. (K) Kaplan-Meier curve analysis of the survival of mice bearing RPCI.WM1 xenograft tumors. ***P < .001. (L) IB analysis of RNF138 protein levels in individual RPCI.WM1-derived xenograft tumors. WCE, whole-cell extract.
We next asked whether RNF138 regulates lymphoma cell proliferation and tumor growth. Downregulation of RNF138 attenuated cell viability in 3 MYD88L265P-lymphoma cell lines (Figure 2F; supplemental Figure 3E,I), but not that of MYD88WT-cells (supplemental Figure 3M). Upon RNF138 knockdown, colony formation of RPCI.WM1 cells decreased (Figure 2G). Xenograft tumors derived from RPCI.WM1 cells with RNF138 knockdown showed a significant decrease in tumor growth, tumor mass, and the population of Ki67+ cells, and extended animal survival compared with the control (Figure 2H-K). Similar in vivo tumor growth results were observed for HBL-1 (supplemental Figure 3F-H) and TMD8 cells (supplemental Figure 3J-L). RNF138 remained repressed in the RPCI.WM1-derived xenograft tumors (Figure 2L). However, RNF138 knockdown in U2932 cells had little impact on tumorigenesis and animal survival (supplemental Figure 3M-Q). Reconstitution of RNF138 promoted MYD88 K63-linked ubiquitination, activated NF-κB phosphorylation, and increased cell viability in RPCI.WM1 and TMD8 cells, whereas these effects were not seen with the catalytically inactive RNF138 mutant (C18/21A; supplemental Figure 3R-S). In addition, we used the CRISPR/Cas9 technology to knock out RNF138 in 4 lymphoma cell lines. RNF138 deletion attenuated MYD88 K63-linked ubiquitination, NF-κB phosphorylation (supplemental Figure 4A), and cell viability (supplemental Figure 4B-C) in MYD88L265P cell lines, but not in the MYD88WT U2932 line. A TLR7 agonist induced p65 phosphorylation and K63-linked ubiquitination of MYD88WT in U2932 cells, which were not affected by sgRNA-mediated RNF138 knockout. In RPCI.WM1 cells with a high basal level of p65 phosphorylation and K63-linked ubiquitination of MYD88L265P, treatment with the ligand increased these PTMs. However, they were significantly reduced by knockout of RNF138, even in the presence of TLR stimulation (supplemental Figure 4D). Taken together, these results support that RNF138 is essential for constitutive NF-κB activation in MYD88L265P lymphoma cells and sustains K63 ubiquitination and tumorigenesis.
Mutation of MYD88L265P ubiquitination sites abolishes its oncogenic activity
We wanted to identify the sites on MYD88L265P where RNF138-mediated polyubiquitination takes place. Human MYD88 contains 15 K residues, yet no single K-to-R substitution significantly affected MYD88L265P polyubiquitination (supplemental Figure 5A) or NF-κB–dependent luciferase activity (supplemental Figure 5B). We next performed MS analysis of purified MYD88L265P from 293T cells, which ectopically overexpress MYD88L265P and RNF138, and identified 5 residues on MYD88L265P (K108, K140, K227, K263, and K275) that were linked to ubiquitin (supplemental Figure 6). When these 5 Ks were mutated to Rs (termed 5KR), MYD88L265P-5KR displayed significantly less ubiquitination than MYD88L265P by RNF138 (Figure 3A), yet its binding to RNF138 appeared to be unaffected (Figure 3B).
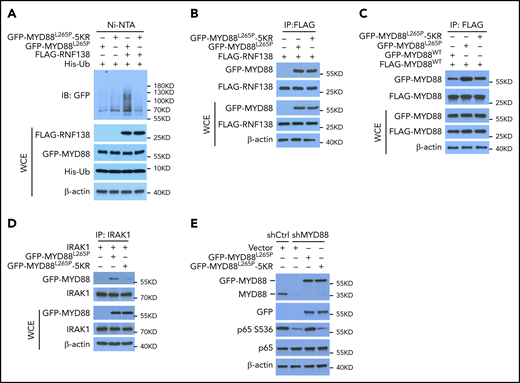
K63-linked polyubiquitination of MYD88L265P is critical for NF-κB activation. (A) MYD88L265P and MYD88L265P-5KR ubiquitination by RNF138 in 293T cells. Ubiquitination was analyzed as in Figure 1E. (B) Co-IP analysis of the interaction between RNF138 and MYD88L265P or MYD88L265P-5KR in 293T cells. (C) Co-IP analysis of the interaction between FLAG-MYD88WT and GFP-tagged MYD88WT, MYD88L265P, or MYD88L265P-5KR in 293T cells. Equimolar amounts of MYD88 constructs were introduced into the cells. (D) Co-IP analysis of the interaction between IRAK-1 and MYD88L265P or MYD88L265P-5KR in 293T cells. (E) IB analysis of p65 phosphorylation in RPCI.WM1 cells with MYD88 knockdown and reintroduction of MYD88L265P or MYD88L265P-5KR. WCE, whole-cell extract.
K63-linked polyubiquitination of MYD88L265P is critical for NF-κB activation. (A) MYD88L265P and MYD88L265P-5KR ubiquitination by RNF138 in 293T cells. Ubiquitination was analyzed as in Figure 1E. (B) Co-IP analysis of the interaction between RNF138 and MYD88L265P or MYD88L265P-5KR in 293T cells. (C) Co-IP analysis of the interaction between FLAG-MYD88WT and GFP-tagged MYD88WT, MYD88L265P, or MYD88L265P-5KR in 293T cells. Equimolar amounts of MYD88 constructs were introduced into the cells. (D) Co-IP analysis of the interaction between IRAK-1 and MYD88L265P or MYD88L265P-5KR in 293T cells. (E) IB analysis of p65 phosphorylation in RPCI.WM1 cells with MYD88 knockdown and reintroduction of MYD88L265P or MYD88L265P-5KR. WCE, whole-cell extract.
Because MYD88 recruits IRAK-1 for activation of downstream signaling in its dimer form,20 we characterized the binding between MYD88WT, MYD88L265P, and MYD88L265P-5KR and between MYD88 and IRAK-1 in 293T cells by using co-IP. MYD88WT/MYD88L265P binding was stronger than that of MYD88WT/MYD88WT, whereas 5KR substantially reduced the binding of MYD88WT/MYD88L265P (Figure 3C). Also, 5KR reduced binding between MYD88L265P and IRAK-1 (Figure 3D). In RPCI.WM1 cells, MYD88 knockdown decreased p65 phosphorylation, which was restored by exogenous MYD88L265P, but not by MYD88L265P-5KR (Figure 3E). In addition, MYD88L265P, but not MYD88L265P-5KR, increased NF-κB–dependent luciferase activity >20-fold (supplemental Figure 5C) and resulted in predominant p65 nuclear localization in 293T cells (supplemental Figure 5D). Exogenous MYD88L265P enhanced IRAK-1 phosphorylation in U2932 DLBCL cells, as previously reported,2 whereas 5KR did not (supplemental Figure 5E). These results suggest that K63-linked polyubiquitination of MYD88L265P is critical to NF-κB activation. It is notable that K263 is also ubiquitinated by the presumed CUL3-RBX1 E3 ubiquitin ligase complex with assistance from SPOP.17 Thus, we cannot exclude the possibility that other E3 ligases ubiquitinate MYD88L265P at these 5 sites and that 5KR alter the normal functions of MYD88.
A20 inhibits the oncogenic activity of MYD88L265P indirectly by promoting RNF138 degradation
Because NF-κB signaling is interwoven by both E3 ligases and deubiquitinases (DUBs),10,21 we performed a DUB screening by overexpressing 55 individual human DUBs in 293T-NF-κB-Luc cells with exogenous MYD88L265P or MYD88WT and RNF138 (Figure 4A). A20, an ovarian tumor (OTU) superfamily member, caused the greatest decrease in MYD88L265P-mediated NF-κB–dependent luciferase activity (Figure 4B) and significantly diminished MYD88L265P-induced p65 nuclear localization (Figure 4C). When 9 OTU family DUBs and cylindromatosis were tested in 293T-NF-κB-Luc cells, A20 downregulated the expression of RNF138, but not MYD88L265P; A20 was the only one that reduced (by ∼70%); NF-κB luciferase activity mediated by RNF138 and MYD88L265P (supplemental Figure 7A-B). Knockdown of A20 in RPCI.WM1 or TMD8 cells increased RNF138 expression and further enhanced MYD88L265P-mediated NF-κB activation, as determined by an increase in p65 phosphorylation (Figure 4D) and increased expression of NF-κB downstream target genes (supplemental Figure 7C). Cell viability and colony formation of RPCI.WM1 cells were also augmented (supplemental Figure 7D-E).
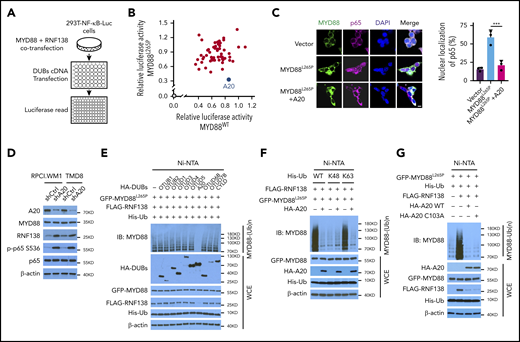
A20 inhibits MYD88L265P K63-linked polyubiquitination and abrogates NF-κB activation. (A) Schematic workflow of DUB screening assay using 293T-NF-κB-Luc cells. (B) Effect of DUB on NF-κB luciferase activity in 293T-NF-κB-Luc cells. Cells were transfected with vector control or cotransfected MYD88L265P /MYD88WT, RNF138, and DUBs and examined for luciferase activity. Blue spot, A20; gray spot, vector control. (C) Effect of A20 on MYD88L265P-mediated p65 nuclear localization in 293T cells. Cells were transfected with MYD88L265P and A20 and analyzed by IF. ***P < .001. The bar represents 10 μm. MYD88, Alexa Fluor 488 (green); p65, Alexa Fluor 647 (red). (D) The effect of A20 knockdown on p65 phosphorylation and RNF138 level in RPCI.WM1 and TMD8 cells. (E) Effect of DUBs on RNF138-mediated MYD88L265P ubiquitination in 293T cells. (F) Effect of A20 and Ub mutants on MYD88L265P ubiquitination in 293T cells. (G) Effect of A20 deubiquitination activity on MYD88L265P ubiquitination in 293T cells. Cells were transfected with GFP-MYD88L265P, FLAG-RNF138, and His-Ub, along with HA-A20 or HA-A20 C103A. The ubiquitination in panels A-G was analyzed as in Figure 1E. WCE, whole-cell extract.
A20 inhibits MYD88L265P K63-linked polyubiquitination and abrogates NF-κB activation. (A) Schematic workflow of DUB screening assay using 293T-NF-κB-Luc cells. (B) Effect of DUB on NF-κB luciferase activity in 293T-NF-κB-Luc cells. Cells were transfected with vector control or cotransfected MYD88L265P /MYD88WT, RNF138, and DUBs and examined for luciferase activity. Blue spot, A20; gray spot, vector control. (C) Effect of A20 on MYD88L265P-mediated p65 nuclear localization in 293T cells. Cells were transfected with MYD88L265P and A20 and analyzed by IF. ***P < .001. The bar represents 10 μm. MYD88, Alexa Fluor 488 (green); p65, Alexa Fluor 647 (red). (D) The effect of A20 knockdown on p65 phosphorylation and RNF138 level in RPCI.WM1 and TMD8 cells. (E) Effect of DUBs on RNF138-mediated MYD88L265P ubiquitination in 293T cells. (F) Effect of A20 and Ub mutants on MYD88L265P ubiquitination in 293T cells. (G) Effect of A20 deubiquitination activity on MYD88L265P ubiquitination in 293T cells. Cells were transfected with GFP-MYD88L265P, FLAG-RNF138, and His-Ub, along with HA-A20 or HA-A20 C103A. The ubiquitination in panels A-G was analyzed as in Figure 1E. WCE, whole-cell extract.
Ectopic expression of A20, but not other OTU family members, strongly inhibited MYD88L265P ubiquitination in 293T cells (Figure 4E). Cylindromatosis22 and OTUD423 are reported to remove K63-linked polyubiquitin chains from MYD88WT, yet they displayed no effect on MYD88L265P (Figure 4E). That A20 reduced K63-linked polyubiquitination of MYD88L265P was further confirmed with ubiquitin mutants (Figure 4F). Consistently, neither the protein level of MYD88L265P nor the ubiquitination of MYD88WT was affected by A20 (supplemental Figure 7F-G). The A20 OTU domain is essential for its DUB function, whereas its ZNFs confer E3 ligase activity.24 We next investigated whether the DUB activity of A20 is necessary for inhibition of MYD88L265P polyubiquitination. The Cys103→Ala (C103A) mutant of A20, which abolished its DUB activity, did not affect MYD88L265P ubiquitination (Figure 4G). Overexpression of A20 ZNFs, but not of A20 OTU, markedly reduced MYD88L265P ubiquitination to an extent similar to that of A20 (supplemental Figure 7H-I).
A20 decreased RNF138 protein levels in 293T cells in a dose-dependent manner (Figure 5A), most likely because of a reduced RNF138 half-life (Figure 5B) and increased RNF138 polyubiquitination (Figure 5C). In the presence of A20, MG132 treatment increased the RNF138 protein level, presumably because of reduced proteasome degradation (Figure 5D). Notably, A20 mediated RNF138 ubiquitination in the presence of WT ubiquitin or ubiquitin K48, but not ubiquitin K63 (Figure 5E). A20 knockdown in MYD88L265P-lymphoma cells prolonged the half-life of endogenous RNF138 (Figure 5F). The ubiquitination data revealed that depletion of A20 reduced the K48-linked ubiquitination of RNF138 and subsequently increased the K63-linked polyubiquitination of MYD88L265P in MYD88L265P+ lymphoma cells (Figure 5G; supplemental Figure 8A). A20 or A20-ZNFs, but not A20-OTU, was essential for RNF138 K48-linked polyubiquitination and subsequent degradation in 293T cells (supplemental Figure 8B).
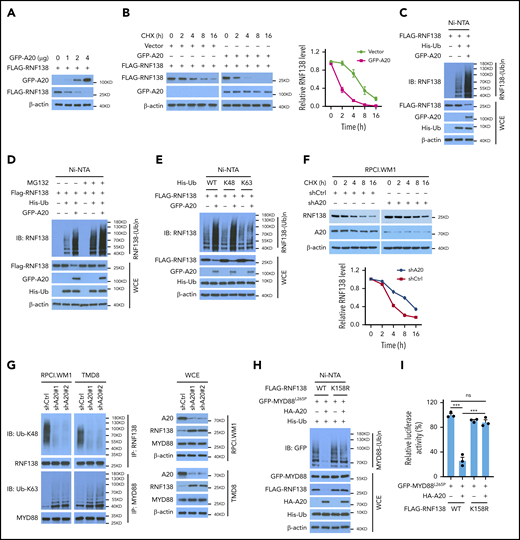
A20 mediates RNF138 degradation through K48-linked ubiquitination. (A) Effect of A20 on RNF138 protein levels in 293T cells. (B) The half-life of RNF138 in the presence of A20. FLAG-RNF138 and GFP-A20 were cotransfected into 293T cells and treated with cycloheximide (CHX). FLAG-RNF138 and GFP-A20 protein levels were analyzed by IB, quantified by densitometry, and plotted against treatment time. (C) Effect of A20 on RNF138 polyubiquitination in 293T cells. (D) A20-mediated RNF138 polyubiquitination in the presence of MG132. 293T cells were transfected with various constructs and treated with MG132 (20 μM) for 6 hours. (E) The types of polyubiquitination chains on RNF138 in 293T cells. (F) The half-life of RNF138 with A20 knockdown in RPCI.WM1 cells. Cells were transfected with shCtrl or shA20 and treated with CHX. Endogenous RNF138 protein levels were analyzed by IB, quantified by densitometry, and plotted against treatment time. (G) Endogenous RNF138 and MYD88L265P ubiquitination in RPCI.WM1 and TMD8 cells after A20 knockdown. (H) Effect of A20 and RNF138 K158R on MYD88L265P ubiquitination in 293T cells. In panels C, D, E, and H, His-Ub-linked proteins were purified by using Ni-NTA beads from cell lysates before they were subjected to IB with RNF138 antibody. (I) Effect of RNF138 K158R and A20 on NF-κB reporter activity mediated by MYD88L265P in 293T-NF-κB-Luc cells. ***P < .001. WCE, whole-cell extract.
A20 mediates RNF138 degradation through K48-linked ubiquitination. (A) Effect of A20 on RNF138 protein levels in 293T cells. (B) The half-life of RNF138 in the presence of A20. FLAG-RNF138 and GFP-A20 were cotransfected into 293T cells and treated with cycloheximide (CHX). FLAG-RNF138 and GFP-A20 protein levels were analyzed by IB, quantified by densitometry, and plotted against treatment time. (C) Effect of A20 on RNF138 polyubiquitination in 293T cells. (D) A20-mediated RNF138 polyubiquitination in the presence of MG132. 293T cells were transfected with various constructs and treated with MG132 (20 μM) for 6 hours. (E) The types of polyubiquitination chains on RNF138 in 293T cells. (F) The half-life of RNF138 with A20 knockdown in RPCI.WM1 cells. Cells were transfected with shCtrl or shA20 and treated with CHX. Endogenous RNF138 protein levels were analyzed by IB, quantified by densitometry, and plotted against treatment time. (G) Endogenous RNF138 and MYD88L265P ubiquitination in RPCI.WM1 and TMD8 cells after A20 knockdown. (H) Effect of A20 and RNF138 K158R on MYD88L265P ubiquitination in 293T cells. In panels C, D, E, and H, His-Ub-linked proteins were purified by using Ni-NTA beads from cell lysates before they were subjected to IB with RNF138 antibody. (I) Effect of RNF138 K158R and A20 on NF-κB reporter activity mediated by MYD88L265P in 293T-NF-κB-Luc cells. ***P < .001. WCE, whole-cell extract.
Among all K residues in RNF138, only K158 is predicted to be a ubiquitination site by UbPred25 (supplemental Figure 8C). K158R mutation suppressed RNF138 ubiquitination and restored its protein level in the presence of A20 in 293T cells (supplemental Figure 8D). K158R also compromised the A20 activity in negatively regulating MYD88L265P ubiquitination (Figure 5H) and NF-κB–dependent luciferase activity (Figure 5I) in 293T cells. Therefore, our findings indicate that A20 suppresses RNF138 expression and thereby reduces MYD88L265P ubiquitination and NF-κB activation.
A20-mediated degradation of RNF138 restricts the oncogenic activity of MYD88L265P
In RPCI.WM1 cells, A20 depletion significantly increased MYD88L265P polyubiquitination, which was reversed by concurrent RNF138 depletion (Figure 6A). Similar changes in NF-κB and STAT3 activation were observed (Figure 6B). Moreover, augmented oncogenesis by A20 depletion was abrogated by concurrent RNF138 depletion, as shown by cell proliferation (Figure 6C), colony formation (Figure 6D), and xenograft tumorigenesis (Figure 6E–G). In 293T cells, deletion of ZNF domains 2 to 7 (ZNF2-7) marked attenuated A20-induced RNF138 downregulation and MYD88L265P ubiquitination inhibition, whereas deletion of ZNF5-7 did not (supplemental Figure 9A-B). Deletion of ZNF2-7, but not ZNF5-7, consistently compromised the negative regulation of NF-κB activity by A20 (supplemental Figure 9C). Next, we deleted ZNF3-5, individually or together. Deletion of ZNF3-5, ZNF4, or ZNF5 in A20 partially restored RNF138 expression, MYD88L265P ubiquitination, and NF-κB activity (supplemental Figure 9D-F). In RPCI.WM1 cells, knockdown of endogenous A20 in combination with reintroduction of A20, but not the ZNF3-5 or ZNF4 deletion mutants, decreased RNF138 expression, MYD88L265P ubiquitination, p65 phosphorylation, and colony formation (supplemental Figure 9G-H). Thus, these data indicate that the ZNF domains, particularly ZNF4 of A20, are vital for A20-mediated RNF138 degradation and subsequent negative regulation of the oncogenic activity of MYD88L265P.
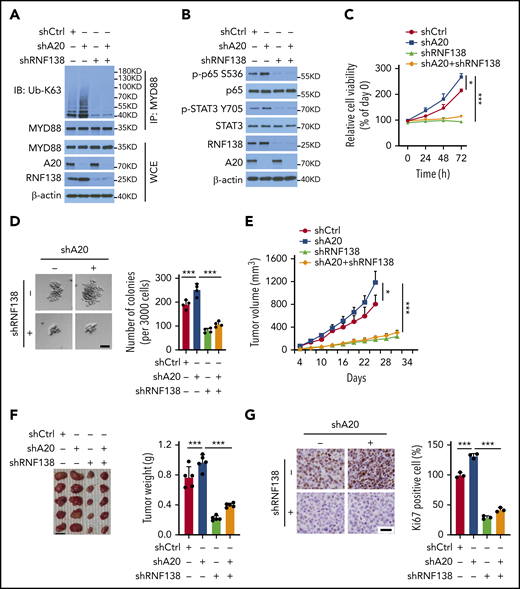
A20-mediated degradation of RNF138 restricts MYD88L265P oncogenic activity. (A-B) Effects of A20 depletion on RNF138 expression and MYD88L265P ubiquitination (A) and downstream signaling (B) in RPCI.WM1 cells. (C-D) Effects of A20 and/or RNF138 depletion in cell viability and colony formation. RPCI.WM1 cells were transfected with shRNAs targeting A20, RNF138, or both, before subjected to MTS (C) and CFU (D) assays. The bar represents 50 μm. ***P < .001. (E-G) Tumorigenesis of RPCI.WM1 cells with A20 and/or RNF138 knockdown. Cells were transfected with shRNAs targeting A20, RNF138, or both, and injected into NSG mice. The tumors were analyzed as described in panels H-J of Figure 2. Ki67 was stained with DAB as the chromogen (brown) (G). The bar in panel F represents 1 cm; panel G, 25 μm. *P < .05, ***P < .001. WCE, whole-cell extract.
A20-mediated degradation of RNF138 restricts MYD88L265P oncogenic activity. (A-B) Effects of A20 depletion on RNF138 expression and MYD88L265P ubiquitination (A) and downstream signaling (B) in RPCI.WM1 cells. (C-D) Effects of A20 and/or RNF138 depletion in cell viability and colony formation. RPCI.WM1 cells were transfected with shRNAs targeting A20, RNF138, or both, before subjected to MTS (C) and CFU (D) assays. The bar represents 50 μm. ***P < .001. (E-G) Tumorigenesis of RPCI.WM1 cells with A20 and/or RNF138 knockdown. Cells were transfected with shRNAs targeting A20, RNF138, or both, and injected into NSG mice. The tumors were analyzed as described in panels H-J of Figure 2. Ki67 was stained with DAB as the chromogen (brown) (G). The bar in panel F represents 1 cm; panel G, 25 μm. *P < .05, ***P < .001. WCE, whole-cell extract.
Aberrancies of A20-RNF138-NF-κB signaling in human lymphomas with MYD88L265P
We collected 42 cases of DLBCL specimens with MYD88L265P and 102 cases of DLBCL specimens without MYD88L265P and performed immunohistochemical (IHC) analyses of A20, RNF138, and p-p65. RNF138 and p65 phosphorylation were higher in lymphomas with MYD88L265P than in those without it (Figure 7A-B). There was a negative correlation between A20 and RNF138 protein levels in both groups (Figure 7C). Moreover, A20 expression correlated negatively with p65 phosphorylation in both groups (Figure 7D). However, RNF138 expression correlated positively with p65 phosphorylation in lymphomas with MYD88L265P, but not in those without (Figure 7E). This finding was consistent with our experimental data that showed that knockdown of RNF138 had no effect on NF-κB activation in MYD88WT lymphoma cell lines (supplemental Figure 3A).
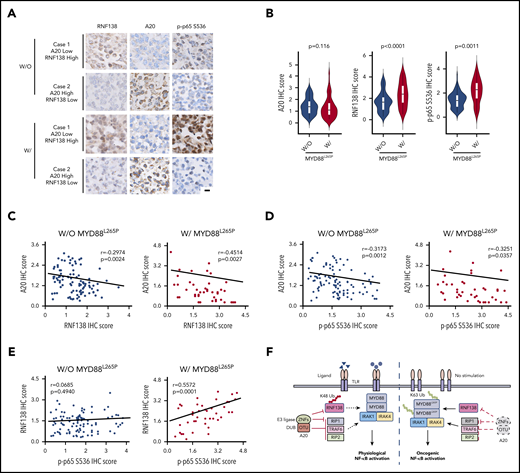
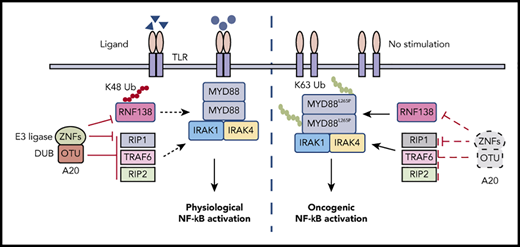
Expression of A20 and RNF138 in relationship to NF-κB activation in human lymphomas. (A) Representative images of IHC staining for RNF138, A20, and p-p65 Ser536 in lymphomas with and without MYD88L265P. The images were taken from the same area for each target in individual lymphomas. Targets were stained with DAB as the chromogen (brown). The bar represents 10 μm. (B) Comparison of A20, RNF138, and p-p65 Ser536 expression between lymphomas with and without MYD88L265P. (C) Correlation between the expression of A20 and RNF138 in lymphomas with and without MYD88L265P. (D) Correlation between the expression of A20 and p-p65 Ser536 in lymphomas with and without MYD88L265P. (E) Correlation between the expression of RNF138 and p-p65 Ser536 in lymphomas with and without MYD88L265P. Pearson’s correlation coefficient tests provided the data in panels C-E. (F) The working model of MYD88, RNF138, and A20 in regulating physiological and oncogenic NF-κB activation. W, with; W/O, without.
Expression of A20 and RNF138 in relationship to NF-κB activation in human lymphomas. (A) Representative images of IHC staining for RNF138, A20, and p-p65 Ser536 in lymphomas with and without MYD88L265P. The images were taken from the same area for each target in individual lymphomas. Targets were stained with DAB as the chromogen (brown). The bar represents 10 μm. (B) Comparison of A20, RNF138, and p-p65 Ser536 expression between lymphomas with and without MYD88L265P. (C) Correlation between the expression of A20 and RNF138 in lymphomas with and without MYD88L265P. (D) Correlation between the expression of A20 and p-p65 Ser536 in lymphomas with and without MYD88L265P. (E) Correlation between the expression of RNF138 and p-p65 Ser536 in lymphomas with and without MYD88L265P. Pearson’s correlation coefficient tests provided the data in panels C-E. (F) The working model of MYD88, RNF138, and A20 in regulating physiological and oncogenic NF-κB activation. W, with; W/O, without.
Discussion
MYD88L265P is frequently present in WM, primary testicular lymphoma, primary central nervous system lymphoma, cutaneous DLBCL leg type, IgM monoclonal gammopathy of undetermined significance, and ABC-DLBCL, but less frequently in germinal center B-cell DLBCL and other non-Hodgkin lymphomas (NHLs).5 MYD88 mutation, with nearly all being L265P, is among the most frequent of the recurrent driver mutations in NHLs.5,26 However, unlike other NHL driver mutations in TP5327 and MLL2,28 MYD88L265P has hardly ever been reported in solid tumors. The nucleotide change causing L265P (T→C) is not in a hotspot for physiological somatic hypermutation. In this work, we demonstrated that K63-linked polyubiquitination of MYD88L265P by RNF138 and subsequent enhanced recruitment of IRAK-1/4 are the underlying causes of constitutive NF-κB activation in lymphoma cells. In the absence of MYD88 polyubiquitination (by depleting RNF138 or abolishing MYD88 ubiquitination sites), the ability of MYD88L265P to drive NF-κB activation and lymphomagenesis is largely lost. RNF138 is highly expressed in the testis and the immune system, as reported in the human Protein Atlas.29 It is plausible that tissue-specific expression of RNF138 enables clonal selection of MYD88L265P+ B cells. With additional genetic hits such as A20 loss, these B cells are transformed. Tissue-specific expression of RNF138 may contribute, at least in part, to the predominance of MYD88L265P in NHLs, but not in solid tumors. It remains unclear whether the high RNF138 level in testis contributes to the pathogenesis of primary testicular lymphoma.
The consequences of MYD88L265P have led to several strategies to halt its oncogenic activation by targeting the MYD88-associated kinases (IRAK-1/4) or disrupting the “myddosome” assembly.30,31 However, these strategies would also affect the innate immunological signaling mediated by MYD88WT. MYD88L265P-derived peptides elicit leukocyte antigen class I–restricted cytotoxic T-cell responses,32,33 indicating a potential for neoantigen-based immunotherapy. Patients with WM, in whom the disease progresses after a major response to ibrutinib, acquire mutations in other genes, yet MYD88L265P remains intact.34 This observation underscores the unmet clinical need for directly targeting MYD88L265P. As an adaptor, MYD88 is considered “undruggable,” like many other nonenzyme oncoproteins that are chemically intractable, such as MYC and the androgen receptor variant 7.35 By revealing a mutant-only neoenzymatic reaction, our work opens the door to targeting this adaptor protein by exploiting its interaction with its E3 ligase. RNF138 is a member of the RING finger protein family.36 RNF138 localizes both in the cytoplasm and nuclei,37,38 which is consistent with our PLA and IHC analyses. As a nuclear E3 ligase, RNF138 targets the Wnt-activating cofactor TCF/LEF for degradation, to inhibit Wnt-β-catenin signaling.36 In addition, nuclear RNF138 ubiquitinates CtIP, Ku80, and Rad51 after a DNA double-strand break, facilitating CtIP and Rad51 access for homologous recombination.39,40 The interaction between RNF138 and MYD88L265P mostly takes place in the cytoplasm. Thus, our work describes a novel pathogenic role for cytoplasmic RNF138 in B-cell malignancies. Rnf138-knockout mice appear anatomically normal and have dysregulated spermatogonia,41 suggesting that targeting either RNF138-MYD88L265P interaction or RNF138 has limited side effects.
A20 restricts stimulated TLR-NF-κB activation because of its dichromatic properties; specifically, its OTU domain acts as a DUB and ZNF domains as an E3 ligase. First, A20 uses its OTU domain to remove K63-linked ubiquitin chains from receptor-interacting serine/threonine-protein kinase-1 (RIP-1) and its ZNF domains to add K48-linked ubiquitin chains to RIP-1 for proteasome degradation.24 Second, A20 uses its OTU domain to deubiquitinate both TRAF642 and RIP-2.43 In mouse primary B cells, Myd88L252P induces NF-κB activation, B-cell proliferation, and transformation; however, these processes are rapidly countered by A20 induction and extinguished by apoptosis.19 A recent study reported that 55% of ABC-DLBCL and 28% of WM cases that have a MYD88L265P mutation also harbor a TNFAIP3 genetic alteration.44 In another report, 24% of MYD88L265P+ABC-DLBCL cases were shown to have homozygous deletion or epigenetic silencing at the TNFAIP3 locus.2 However, the frequencies of co-occurrence in DLBCL were <5% in other studies.45-48 Such discrepancy is most likely related to differences between the cohorts (ABC-DLBCL and WM vs all DLBCLs) and the testing methods for A20 inactivation. A20 loss enhances MYD88L265P-driven signaling in DLBCL and WM cells,44,49-52 yet the biochemical connection between MYD88L265P and A20 has not been examined. We reveal a novel function for A20 during constitutive NF-κB activation caused by MYD88L265P. A20 ZNF domains target RNF138 for proteasomal degradation via K48-linked ubiquitination, thereby truncating the oncogenic function of MYD88L265P. Removing the restriction from A20, by mutating the ubiquitination site on RNF138, knocking down A20, deleting the A20 ZNFs, or deleting just the ZNF4 domain of A20, further increases NF-κB activation, lymphoma cell proliferation, and tumor development mediated by MYD88L265P. Consistent with the observation that ZNF domains of A20 are tumor suppressive, many TNFAIP3 mutations in lymphoma lead to truncated A20 proteins, which often lose 1 or more ZNF domains but retain the OTU domain.50,51,53,54 Our studies shed more light on the mechanistically distinct, but biologically interdependent, activities of the A20 domains and how these domains regulate physiological and oncogenic NF-κB signaling via ubiquitination and deubiquitination (Figure 7F). In our human lymphoma cohort, regardless of the MYD88 status, A20 expression correlates negatively with RNF138 expression and with NF-κB activation. However, RNF138 expression correlates positively with NF-κB activation, only in lymphomas with MYD88L265P, not in those without it. These pathologic data implicate that A20 suppresses RNF138 and other proteins to restrict NF-κB signaling, whereas RNF138 targets MYD88L265P, but not MYD88WT.
We speculate that the structural change in the MYD88 protein caused by the L265P mutation is responsible for the RNF138-mediated ubiquitination of MYD88L265P. L265P occurs at a residue that is invariant in evolution and contributes to a β-sheet at the hydrophobic core of the TIR domain, different from other mutants.2 In computational stimulation, the MYD88L265P mutation allosterically quenches the global conformational dynamics of its TIR domain and readjusts multiple salt bridges, resulting in stronger homodimerization that triggers the recruitment of downstream IRAK-1 and -4.20 Other MYD88 mutants induce stronger NF-κB activation than does MYD88WT, yet only L265P nucleates a signaling complex that includes phosphorylated IRAK-1.2 We showed that RNF138 exhibits much stronger ubiquitination over MYD88L265P than other mutants and MYD88WT and that loss of ubiquitination compromises the binding of MYD88L265P to phosphorylated IRAK-1. Thus, it is possible that L265P alters the conformation of MYD88 to the maximum extent that allows for full ubiquitination by RNF138, and the ubiquitinated MYD88L265P recruits phosphorylated IRAK-1 to enhance oncogenic NF-κB signaling. MYD88 mutants other than L265P may induce NF-κB activation through unknown alternative mechanisms beyond ubiquitination. These conjectures can only be experimentally tested in future studies.
In summary, this study supports the notion that RNF138 mediates a novel biochemical reaction that is specific to MYD88L265P. A20 ubiquitinates RNF138 for proteasome degradation to suppress MYD88L265P-mediated NF-κB activation and lymphomagenesis. These results reveal a novel molecular mechanism of MYD88L265P oncogenic action and offer new therapeutic opportunities for lymphomas harboring this driver mutation.
The authors will make renewable materials, data sets, and protocols available to other investigators without unreasonable restrictions in response to e-mail requests and by material transfer agreement.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Belinda Willard at the Proteomics and Metabolomics Core, Lerner Research Institute for technical support and mass spectrum analysis.
The LC-MS instrument was purchased via a National Institutes of Health (NIH) shared instrument grant (1S10RR031537-01). This work was supported by the International Waldenstrom’s Macroglobulinemia Foundation, the Cancer Prevention and Research Institute of Texas (RR190043, Y.L. is a CPRIT Scholar in Cancer Research), and NIH, National Institute of Environmental Health Sciences grant (P42ES027725).
Authorship
Contribution: X.Y., W.L., Q.D., and Y.L. designed the research; all authors performed the experiments and/or contributed to the data analyses; X.Y., W.L., Q.D., and Y.L. wrote the manuscript; and all authors provided critical review and revisions and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Youg Li, Department of Medicine, Baylor College of Medicine, 1 Baylor Plaza, Houston, TX 77030; e-mail: yong.li@bcm.edu.