Key Points
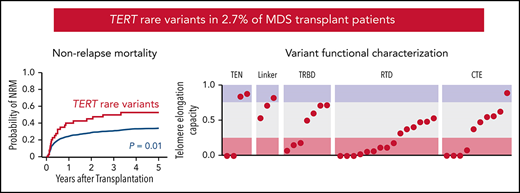
TERT rare variants are present in 2.7% of MDS patients and associated with increased nonrelapse mortality after stem cell transplant.
As a group, TERT rare variants have impaired telomere elongation capacity in cells and likely disrupt domain-specific functions.

Abstract
Germline pathogenic TERT variants are associated with short telomeres and an increased risk of developing myelodysplastic syndrome (MDS) among patients with a telomere biology disorder. We identified TERT rare variants in 41 of 1514 MDS patients (2.7%) without a clinical diagnosis of a telomere biology disorder who underwent allogeneic transplantation. Patients with a TERT rare variant had shorter telomere length (P < .001) and younger age at MDS diagnosis (52 vs 59 years, P = .03) than patients without a TERT rare variant. In multivariable models, TERT rare variants were associated with inferior overall survival (P = .034) driven by an increased incidence of nonrelapse mortality (NRM; P = .015). Death from a noninfectious pulmonary cause was more frequent among patients with a TERT rare variant. Most variants were missense substitutions and classified as variants of unknown significance. Therefore, we cloned all rare missense variants and quantified their impact on telomere elongation in a cell-based assay. We found that 90% of TERT rare variants had severe or intermediate impairment in their capacity to elongate telomeres. Using a homology model of human TERT bound to the shelterin protein TPP1, we inferred that TERT rare variants disrupt domain-specific functions, including catalysis, protein–RNA interactions, and recruitment to telomeres. Our results indicate that the contribution of TERT rare variants to MDS pathogenesis and NRM risk is underrecognized. Routine screening for TERT rare variants in MDS patients regardless of age or clinical suspicion may identify clinically inapparent telomere biology disorders and improve transplant outcomes through risk-adapted approaches.
Introduction
Impaired telomere maintenance is implicated in the pathogenesis of myelodysplastic syndrome (MDS),1-5 for which allogeneic hematopoietic stem cell transplantation (HSCT) is the only potential cure.6,7 Shorter pretransplant blood telomere length is independently associated with an increased risk of early nonrelapse mortality (NRM) in MDS patients,8 but the genetic determinants of telomere length in MDS are incompletely characterized.
Germline pathogenic variants affecting telomerase- and telomere-associated proteins cause a global impairment in telomere maintenance and short telomeres in all tissues.1,9–11 Individuals with dyskeratosis congenita, an early-onset syndromic telomere biology disorder, have characteristic mucocutaneous features, bone marrow failure, and a markedly increased risk of developing MDS and acute myeloid leukemia.1,2,4,12,13 In contrast, adult patients with a telomere biology disorder more frequently present with aplastic anemia,14-16 idiopathic pulmonary fibrosis,17-22 and liver cirrhosis.20,23,24 Affected families may show anticipation, marked by changes in the onset, phenotype, and severity of clinical disease across successive generations.20,25–27 Clinical suspicion for a telomere biology disorder is based on the presence of syndromic features and disease phenotypes in relatives.3,4,20 However, clinical manifestations are highly variable, and up to 40% of affected patients lack a family history of hematologic, pulmonary, or hepatic abnormalities.4 In contrast to dyskeratosis congenita, the risk of developing MDS in older adults with late-presenting or unrecognized telomere biology disorders is unknown.
TERT is the most frequently mutated gene among patients with a telomere biology disorder4 and can cause disease in an autosomal dominant form.1,4 TERT encodes telomerase reverse transcriptase that binds to the telomerase RNA component (TERC) and functions within the multisubunit telomerase holoenzyme complex to extend telomere ends during DNA replication.9,28 Telomerase is composed of 4 structural domains with distinct functional roles29,30 : the telomerase essential/N-terminal (TEN) domain, telomerase RNA-binding domain (TRBD), reverse transcriptase domain (RTD), and C-terminal extension (CTE) domain. Disease-associated germline TERT variants are predominantly missense substitutions and occur within all structural domains.31 Novel TERT missense variants are classified as variants of unknown significance (VUS) by consensus guidelines in the absence of additional supporting computational or functional evidence of pathogenicity.32,33 However, in silico prediction algorithms have limited utility in assessing genotype–phenotype relationships for missense substitutions.34-36 Furthermore, the cellular effects of TERT variants may not be recapitulated by in vitro functional assays.37-39
The prevalence and clinical significance of TERT variants among MDS patients unselected for suspicion of a telomere biology disorder are unknown. Here we analyzed the clinical and functional effects of TERT variants in a registry-level cohort of MDS patients who underwent allogeneic HSCT.
Methods
Patients and samples
We previously described a cohort of 1514 MDS patients who were enrolled in The Center for International Blood and Marrow Transplant Research repository and research database who had banked whole peripheral blood DNA samples.8 Sequencing and annotation of MDS somatic mutation was performed previously using a panel of 129 genes with known or suspected involvement in myeloid disease.40 The median follow-up time for censored patients was 5.0 years. A separate cohort of 401 adult patients with non-Hodgkin lymphoma (NHL) treated with high-dose chemotherapy with autologous stem cell rescue at the Dana-Farber Cancer Institute had banked mobilized whole peripheral blood DNA samples.41 This study was conducted with the approval of the institutional review board at the Dana-Farber Cancer Institute. Research was conducted in accordance with the Declaration of Helsinki.
TERT sequencing and variant annotation
In the MDS cohort, we sequenced the TERT coding region (exons 1-16) and known germline single nucleotide polymorphisms (SNPs). In the NHL cohort, we sequenced TERT in DNA extracted from mobilized whole peripheral blood samples. The genetic analysis was completed and locked before merging with clinical data. Detailed sequencing methods are in the supplemental Methods, available on the Blood Web site.
Telomere length measurements
Relative telomere length of MDS patients was measured by quantitative polymerase chain reaction (qPCR) as previously reported.8 K562 cell DNA was extracted using the QIAamp Blood Mini Kit (Qiagen). Telomere length was measured by 2 orthogonal methods: (1) qPCR as previously described8 and (2) telomere restriction fragment analysis using TeloTAGGG Telomere Length Assay (Sigma-Aldrich) according to the manufacturer's protocol.
Plasmids, cloning, and site-directed mutagenesis
Human TERT cDNA38 was cloned into the Gateway pDONR221 plasmid (Invitrogen). TERT variants were generated using the Q5 Site-Directed Mutagenesis Kit (NEB) and primers designed using NEBase changer (supplemental Table 1). Transfer of each construct to the pCW57.1 destination plasmid (Addgene) was performed using LR clonase (ThermoFisher), and the complete sequence was confirmed by Sanger sequencing.
Lentivirus production and cell line generation
Lentivirus for each TERT variant was produced in HEK 293T cells. TP53-repaired K562 cells (gift from the Ebert laboratory)42 were transduced at a multiplicity of infection of 1 followed by puromycin selection (2 μg/mL) to generate bulk cell lines. K562 cells were treated with doxycycline (1 μg/mL) for 27 days. RPMI media were added every 2 days, and cells were split every 3 days.
Western blotting for hTERT expression
Protein extracts were prepared in Laemmli 2× buffer, and western blots were performed using SDS gels (BioRad) and Trans-Blot Turbo Transfer System. hTERT expression was visualized using TERT antibody (1:1,000 dilution, #600-401-252; Rockland Immunochemicals) with anti-rabbit secondary antibody. B-actin was used as loading control (1:10000 dilution, #ab20272; Abcam). Chemiluminescence images were obtained using Bio-Rad ChemiDoc and analyzed using Image Laboratory software.
Structural modeling of residues mutated in TERT rare variants
A homology model of human telomerase bound to oligonucleotide/oligosaccharide-binding (OB) domain of TPP1 (TPP1-OB) and parts of TERC was generated using the cryo-electron microscopy structure of the Tetrahymena thermophilia telomerase holoezyme,43,44 the crystal structure of human TPP1-OB45 as described previously, and additional crystal structures of TERT/TERC domains from various species43,45-47 using Phyre 2.48 TERT rare variant positions were manually annotated in the final homology model. Details of the homology model are listed in supplemental Methods.
Statistical analyses
Fisher’s exact test was used to test for association between pairs of categorical variables. The Wilcoxon rank-sum test was used to assess a location shift in the distribution of continuous variables between 2 groups. For associations with ordered categorical variables, the Cochran-Armitage trend test was used for singly ordered contingency tables. All P values were 2 sided.
Overall survival (OS) was defined as the time from transplant until death from any cause. Subjects not confirmed dead were censored at the time last known to be alive. Differences in survival curves were assessed using log-rank tests. NRM was defined as death without relapse. NRM, with relapse as a competing risk, was assessed with the use of Gray’s test. For relapse, death without relapse was considered a competing risk. Univariate and multivariable analyses of OS were performed using Cox regression. OS estimates were calculated using the method of Kaplan-Meier and reported with 95% confidence intervals (Cis) based on Greenwood’s formula. Hazard ratios (HRs) with 95% CIs and Wald P values were reported for covariates in multivariable Cox models. Multivariable models for competing risks of relapse and NRM were generated using the Fine and Gray method.
Results
TERT variants in MDS and NHL cohorts
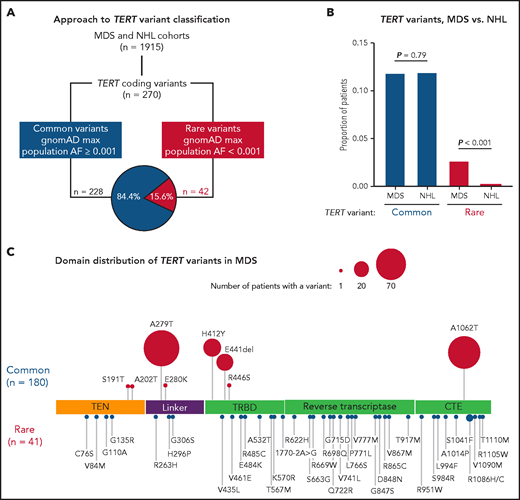
In total, we identified 270 nonsynonymous TERT coding variants among the MDS and NHL cohorts (Figure 1A; supplemental Figure 1). Pathogenic genetic variants are observed infrequently in the general population because of strong negative selection.33,49 Therefore, we grouped TERT variants based on their maximum gnomAD population allele frequency in any reference population,50 where common variants had a maximum allele frequency ≥0.001 and rare variants had a maximum allele frequency <0.001. Using this approach, 228 variants (84.4%) were classified as common and 42 variants as rare (15.6%; Figure 1A; supplemental Figure 1). The frequency of TERT common variants was similar in the MDS and NHL cohorts (11.8% vs 11.2%, P = .79) and primarily included the SNPs p.A279T (rs61748181), p.H412Y (rs34094720), p.E441del (rs377639087), and p.A1062T (rs35719940) (Figure 1C; supplemental Figure 2). In contrast, TERT rare variants were significantly more common in patients with MDS (41 of 1514, 2.7% MDS vs 1 of 401, 0.25% NHL, P < .001; Figure 1B).
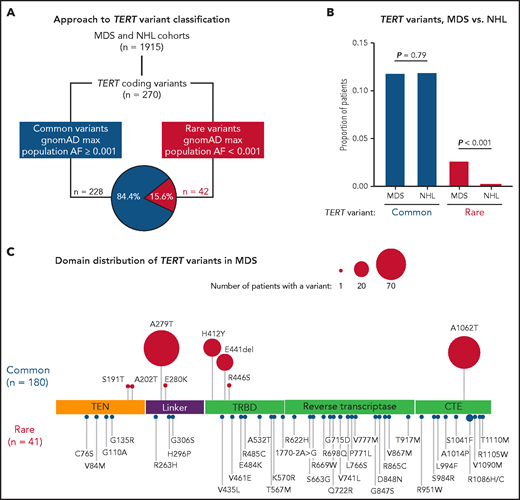
TERT variants in MDS and NHL. (A) Classification approach of nonsynonymous TERT coding variants identified in the MDS and NHL cohorts. (B) Frequency of TERT common and TERT rare variants within the MDS and NHL cohorts. (C) Domain distribution of TERT variants within the MDS cohort. TERT common variants (n = 180) and rare variants (n = 41, R1086H in 2 patients) are located above and below the coding region, respectively. The size of each ball is proportional to the number of patients with that variant. TERT rare variants are colored in red and TERT common variants in blue.
TERT variants in MDS and NHL. (A) Classification approach of nonsynonymous TERT coding variants identified in the MDS and NHL cohorts. (B) Frequency of TERT common and TERT rare variants within the MDS and NHL cohorts. (C) Domain distribution of TERT variants within the MDS cohort. TERT common variants (n = 180) and rare variants (n = 41, R1086H in 2 patients) are located above and below the coding region, respectively. The size of each ball is proportional to the number of patients with that variant. TERT rare variants are colored in red and TERT common variants in blue.
TERT rare variants in MDS occurred within all structural domains: RTD (n = 15), CTE domain (n = 11), TRBD (n = 8), TEN domain (n = 4), and linker region between TEN and TRBD (n = 3; Figure 1C). Most variants were missense substitutions (40 of 41) with 1 splice site variant (c.1770-2A>G). According toAmerican College of Medical Genetics and Genomics (ACMG) and Association of Molecular Pathology (AMP) guidelines32 and Sherloc criteria,33 40 variants were classified as VUS and only 1 variant (p.R865C) as likely pathogenic (Table 1). One TERT rare variant (R1086H) was identified in 2 patients. Twenty-three variants (56.1%) were absent from gnomAD, 17 variants were listed in ClinVar (41.5%), and 11 variants (26.8%) have been published in patients with telomere biology disorders. In silico combined annotation-dependent depletion49 Phred-like scores >20 and ClinPred51 scores >0.5 were observed in 52.5% and 47.5% of variants, respectively (Table 1; supplemental Table 2). In contrast, TERT common variants were classified as benign or likely benign (supplemental Table 3), and most have been experimentally determined to have comparable activity to wild-type TERT.39
TERT rare variants and clinical characteristics
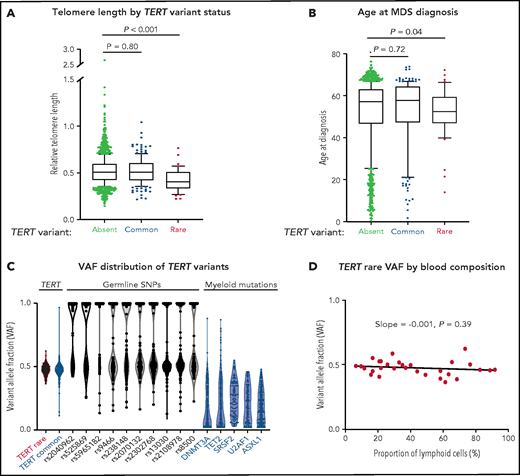
Patients with a TERT rare variant had shorter telomere length (Δ cycle threshold [ddCT] = 0.405 vs 0.507, P < .001) and younger age at MDS diagnosis (median age, 52 vs 59 years, P = .03) than those without a TERT variant (Figure 2A-B; supplemental Table 4). The frequency of TERT rare variants was similar between patients younger or older than age 40 (1.6% vs 2.9%, P = .38; supplemental Figure 3). All other clinical characteristics, including Karnofsky performance status, hematopoietic stem cell transplant comorbidity index (HCT-CI) score, peripheral blood counts at transplant, frequency of somatic mutations, frequency of therapy-related MDS, and Revised International Prognostic Scoring System (IPSS-R) risk group were similar in patients with and without a TERT rare variant (Table 2; supplemental Tables 5 and 6). Specifically, there was no significant difference in the prevalence of pretransplant pulmonary or hepatic dysfunction based on TERT rare variant status (supplemental Table 7). Abnormalities of chromosome 7 (−7 or del7p), which have been reported to occur frequently in patients with telomere biology disorders,4 were observed in a similar proportion of patients with and without a TERT rare variant (20% vs 19%, P > .99; supplemental Table 8). Myeloid somatic mutations were detected in 71% of patients with a TERT rare variant compared with 79% in patients without a TERT rare variant (P = .24; supplemental Figure 4; Table 2; supplemental Tables 6 and 9). Among the 12 patients without detected somatic mutations, 6 patients had either excess blasts or MDS-defining cytogenetic abnormalities, 2 had other cytogenetic clones, and 4 did not have available cytogenetic data (supplemental Figure 5).
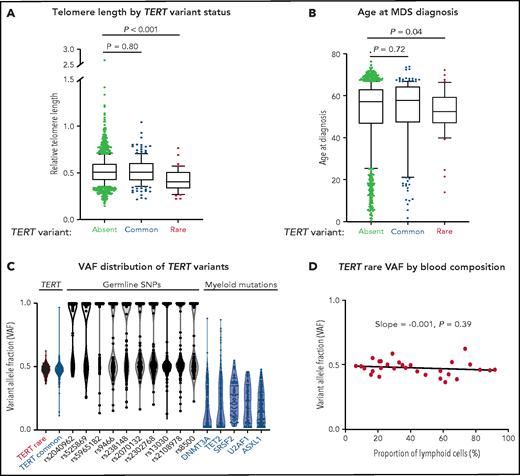
Association of TERT variants with telomere length and age at MDS diagnosis. (A) Pretransplant whole blood relative telomere length by TERT variant status. (B) Age at MDS diagnosis by TERT variant status. TERT variant groups are labeled: no TERT variant (black), TERT common variant (gray), and TERT rare variant (red). (C) Variant allele fraction distribution of TERT variants, germline SNPs (black), and myeloid mutations (blue). (D) Variant allele fraction distribution of TERT rare variants as a function of the proportion of blood lymphocytes. Linear regression slope with P value is shown.
Association of TERT variants with telomere length and age at MDS diagnosis. (A) Pretransplant whole blood relative telomere length by TERT variant status. (B) Age at MDS diagnosis by TERT variant status. TERT variant groups are labeled: no TERT variant (black), TERT common variant (gray), and TERT rare variant (red). (C) Variant allele fraction distribution of TERT variants, germline SNPs (black), and myeloid mutations (blue). (D) Variant allele fraction distribution of TERT rare variants as a function of the proportion of blood lymphocytes. Linear regression slope with P value is shown.
Patients with a TERT common variant were similar to those without any TERT variant with respect to telomere length (ddCT = 0.509 vs 0.507, P = .80), age at MDS diagnosis (median age, 59 vs 59 years, P = .72), and other clinical characteristics (Figure 2A-B; supplemental Table 5). Clinical characteristics among patients with different TERT common variants were also similar. Specifically, there were no differences between patients with the TERT A1062T variant, which has previously been associated with acute myeloid leukemia (AML),52 compared with those with other TERT common variants (supplemental Figure 6).
In the absence of available constitutional reference tissue, we used genetic characteristics to determine whether TERT rare variants were likely present in the germline or acquired somatically within the malignant clone. Germline variants are present in all cells and have a variant allele fraction (VAF) around 0.5 (heterozygous) or 1 (homozygous), whereas somatic mutations are present only in a subset of clonal cells with a wider range of VAF that falls below 0.5 in diploid cells. TERT rare variants and control germline SNPs had a median VAF of 0.48 (95% CI, 0.457-0.502) and 0.56 (95% CI, 0.557-0.564), respectively (Figure 2C), whereas MDS somatic mutations typically present in the founding clone displayed lower median VAFs (DNMT3A: 0.10, TET2: 0.16, SRSF2: 0.25, U2AF1: 0.20, ASXL1: 0.18). Additionally, the VAF of TERT rare variants did not vary with the proportion of blood lymphocytes (Figure 2D), indicating that the variants were present in all nucleated cells, including both the clonal myeloid compartment and the putatively nonclonal lymphoid compartment.
TERT rare variants and clinical outcomes
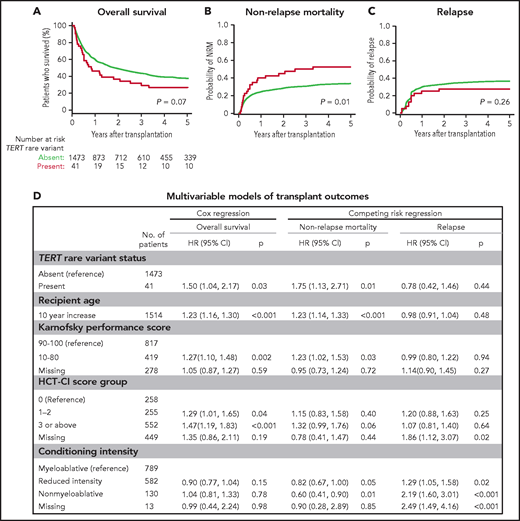
To determine whether TERT variant status was associated with clinical outcomes after transplantation, we evaluated OS and the cumulative incidences of relapse and NRM. Among 41 MDS patients with a TERT rare variant, OS was 24% at 5 years (Figure 3A), and the cumulative incidences of NRM and relapse at 5 years were 52.5% and 27.5%, respectively (Figure 3B-C). In multivariable analysis, the presence of a TERT rare variant compared with the absence of a TERT rare variant was associated with inferior OS (HR for death, 1.50; 95% CI: 1.04-2.20; P = .03) and an increased rate of NRM (HR, 1.75; 95% CI, 1.13-2.72; P = .01) but not a higher rate of relapse (HR, 0.78; 95% CI: 0.42-1.16; P = .44; Figure 3D). The overall presence or absence of somatic genetic evolution, indicated by the acquisition of somatic MDS mutations (supplemental Figure 4), had no effect on outcomes in patients with a TERT rare variant (supplemental Figure 7). Nongenetic factors also impacted the rate of NRM in this model, including recipient age (per 10-year increase: HR, 1.23; 95% CI: 1.14-1.33; P < .01), Karnofsky performance score <90 (HR, 1.23; 95% CI: 1.02-1.53; P = .03), and reduced-intensity conditioning (HR, 0.82; 95% CI: 0.67-1.00; P = .05). The effect of TERT rare variant status on NRM in patients receiving reduced-intensity conditioning and myeloablative conditioning is shown in supplemental Figure 8. The complete results of the multivariable Cox model for OS and the Fine–Gray model for the rates of NRM and relapse, along with adjusted covariates for each model, are provided in supplemental Table 10. Transplant outcomes were similar among patients without a TERT variant and those with a TERT common variant, including patients with the TERT A1062T variant (supplemental Figures 6 and 9).
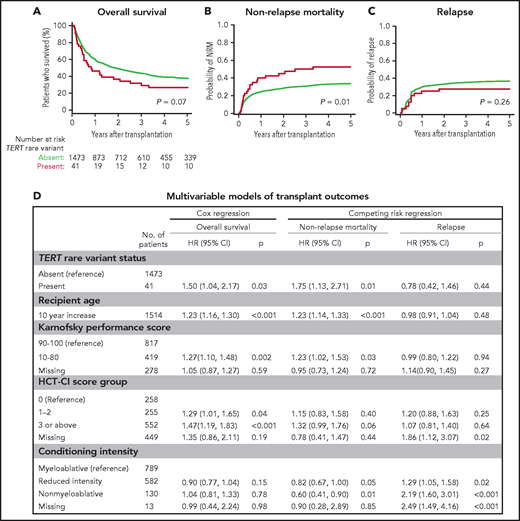
Transplant outcomes by TERT rare variant status. (A) Kaplan-Meier curve for overall survival. (B) Cumulative incidence curves for nonrelapse mortality. (C) Cumulative incidence curves for relapse. Patients with a TERT rare variant are colored in red and patients without a TERT rare variant are colored in green. (D) Multivariable models of overall survival, NRM, and relapse.
Transplant outcomes by TERT rare variant status. (A) Kaplan-Meier curve for overall survival. (B) Cumulative incidence curves for nonrelapse mortality. (C) Cumulative incidence curves for relapse. Patients with a TERT rare variant are colored in red and patients without a TERT rare variant are colored in green. (D) Multivariable models of overall survival, NRM, and relapse.
Primary disease (32%), noninfectious pulmonary causes (21%), and infections (18%) were the most common causes of death in patients with a TERT rare variant (supplemental Table 11). Among these, noninfectious pulmonary causes of death occurred more frequently in patients with a TERT rare variant compared with those without a TERT rare variant. Five of 6 patients with a TERT rare variant and noninfectious pulmonary cause of death received myeloablative conditioning.
Functional effects of TERT rare variants
We next determined the impact of TERT rare variants on telomere elongation in human cells (Figure 4A). Doxycycline-inducible expression of wild-type TERT in K562 AML cells resulted in progressive increase in telomere length over 27 days, whereas expression of luciferase or a known catalytically impaired TERT variant (TERTV694M)14,39 resulted in minimal change in telomere length (Figure 4B). Bulk cell lines were generated for all 39 TERT missense rare variants and 1 TERT common variant (p.A279T). TERT expression was consistent throughout the experiment (Figure 4A; supplemental Figure 10). Telomere elongation capacity was calculated as the change in qPCR telomere length from day 0 to day 27 normalized to that of wild-type TERT (Figure 4C; supplemental Figure 11).
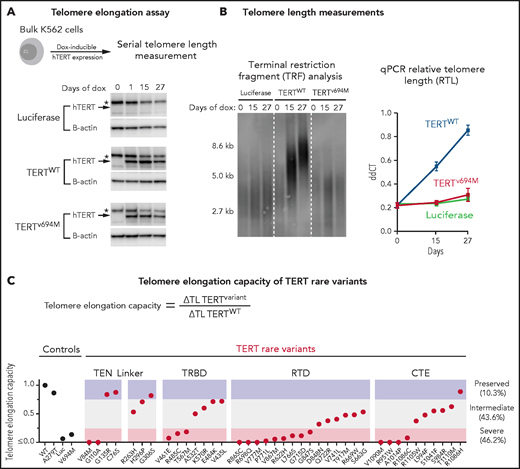
Functional characterization of TERT rare variants. (A) Cell-based telomere elongation assay in isogenic bulk K562 cell lines with doxycycline-inducible TERT expression. TERT expression throughout the experiment is shown for control conditions: luciferase, TERTWT, TERTV694M. hTERT band (∼127 kDa) is labeled with an arrow, and the asterisk corresponds to a nonspecific band seen in all conditions. (B) Telomere length measurements by terminal restriction fragment analysis and qPCR (ddCT represents RTL) for luciferase (green), TERTWT (blue), and TERTV694M (red). (C) Telomere elongation capacity of TERT rare variants normalized to TERTWT are shown in ranked order grouped by structural domain. Control conditions are colored in black and TERT rare variants in red.
Functional characterization of TERT rare variants. (A) Cell-based telomere elongation assay in isogenic bulk K562 cell lines with doxycycline-inducible TERT expression. TERT expression throughout the experiment is shown for control conditions: luciferase, TERTWT, TERTV694M. hTERT band (∼127 kDa) is labeled with an arrow, and the asterisk corresponds to a nonspecific band seen in all conditions. (B) Telomere length measurements by terminal restriction fragment analysis and qPCR (ddCT represents RTL) for luciferase (green), TERTWT (blue), and TERTV694M (red). (C) Telomere elongation capacity of TERT rare variants normalized to TERTWT are shown in ranked order grouped by structural domain. Control conditions are colored in black and TERT rare variants in red.
Most TERT rare variants exhibited impaired capacity to elongate telomeres compared with wild-type TERT (Figure 4C; supplemental Figures 11 and 12). Eighteen variants (46.2%) displayed severely impaired telomere elongation capacity (<25% of wild type), including 10 of 11 previously reported variants associated with telomere biology disorders (supplemental Tables 4 and 12). Intermediate telomere elongation capacity (25% to 75% of wild type) was observed for 17 variants (43.6%). C76S, G135R, G306S, and R1086H exhibited preserved telomere elongation capacity (>75% of wild type). Severely impaired variants occurred within all major structural domains of TERT: TEN (2 of 4), TRBD (3 of 8), RTD (9 of 15), CTE (4 of 11). As a group, linker variants had a modest impact on telomere elongation capacity (H263H: 53%, H296P: 71%, G306S: 82%).
Structural analysis of TERT rare variants
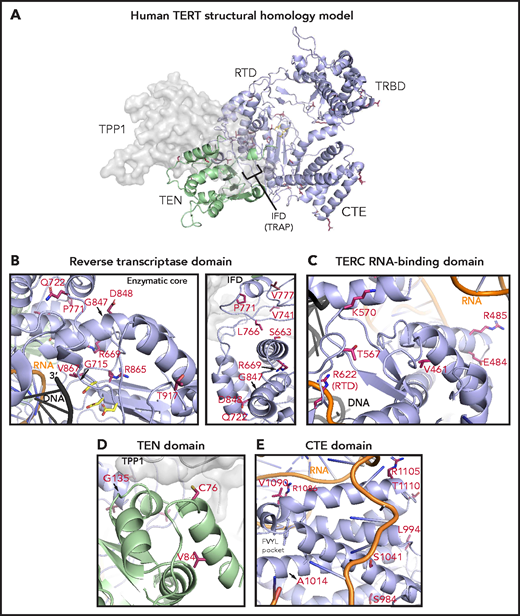
To study the potential structural effects of TERT rare variants, we constructed a homology model of human telomerase bound to the OB domain of the shelterin protein TPP1. In this model, the TRBD, RTD, and CTE domains form a closed ring structure that is consistent with the cryo-electron microscopy structure of human telomerase (Figure 5A).53 The TEN domain straddles the insertion in fingers domain (IFD) portion of the RTD and contacts the CTE domain, thereby trapping the TERC template and DNA substrate within the active site (TERC and DNA omitted in Figure 5A for simplicity). The TPP1 OB domain docks at the TEN-IFD interface of TERT, as described previously.53
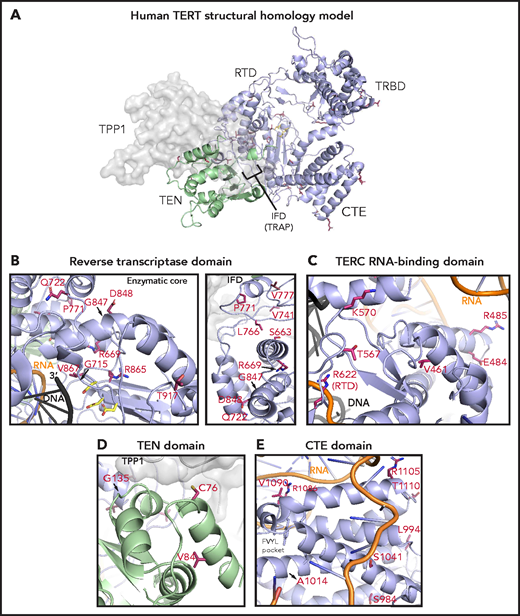
Structural analysis of TERT rare variants. (A) Human TERT homology model. The ring, formed by the TRBD, RTD, and CTE domain, is colored in purple and the TEN domain in green. Panels B, C, D, and E show the TERT rare variants within the RTD (enzymatic core and IFD regions), TRBD, TEN, and CTE domains (including the FVYL pocket), respectively. R622 belongs to the RTD but is displayed in panel C because of its proximity to the TRBD. The side-chains for the residues mutated in the MDS rare variants (crimson carbon atoms) and the catalytic residues in the active site (yellow carbon atoms) are shown in stick representation. The modeled regions of TERC are in orange, whereas the DNA substrate is depicted in black.
Structural analysis of TERT rare variants. (A) Human TERT homology model. The ring, formed by the TRBD, RTD, and CTE domain, is colored in purple and the TEN domain in green. Panels B, C, D, and E show the TERT rare variants within the RTD (enzymatic core and IFD regions), TRBD, TEN, and CTE domains (including the FVYL pocket), respectively. R622 belongs to the RTD but is displayed in panel C because of its proximity to the TRBD. The side-chains for the residues mutated in the MDS rare variants (crimson carbon atoms) and the catalytic residues in the active site (yellow carbon atoms) are shown in stick representation. The modeled regions of TERC are in orange, whereas the DNA substrate is depicted in black.
Of the 15 TERT rare variants that map to the RTD, 10 localize within the catalytic core region (Figure 5B) that is structurally similar to other known reverse transcriptases.30 Four of these 10 variants (R622H, G715D, R865C, and V867M) are proximal to the active site pocket and the RNA-DNA duplex (Figure 5B; see Figure 5C for R662). In contrast, variants S663G, R669W, R698Q (data not shown), G847S, D848N, and T917M map distal to the active site (Figure 5B). We also identified 5 RTD variants that lie within the IFD.53 The IFD consists of 2 bracing helices with an intervening TERT-specific TRAP subdomain that entraps the template-primer duplex within the active site (Figure 5B). Q722R resides at the base of the N-terminal bracing helix, and 4 variants (V777M, P771L, V741L, and L766S) lie within the TRAP region. Notably, V777 resides within an α helix that contacts the TEN domain, whereas P771 lies immediately adjacent to it.
The TEN domain facilitates telomere repeat addition processivity of telomerase and mediates telomerase recruitment to the ends of chromosomes through specific interactions with the TERT IFD-TRAP and the N-terminal OB domain of TPP1.53 Four variants (C76S, V84M, G110A [not modeled], and G135R) localize to the TEN domain and are proximal to a region implicated in recognizing TPP1 (Figure 5D).
The CTE domain, known as the thumb domain in other polymerases, is composed of 4 highly conserved motifs (E-I, E-II, E-III, E-IV) essential for biological activity through RNA-DNA duplex binding, as well as repeat addition processivity.54 Nine CTE variants localize to E-I (R951W, S984R, L994F, A1014P), E-II (S1041F), and E-III (R1086H in 2 patients, R1086C, V1090M; Figure 5C). At opposite ends of a loop connecting E-III and E-IV, R1105W sits in close proximity to TERC, whereas T1110M resides on a solvent-exposed face.
The TRBD interacts extensively with TERC via the CR4/5 domain and pseudoknot/template region.46 Among the 8 TRBD variants, R485C, E484K, and A532T (data not shown) localize to α-helices in contact with the CR4/5 domain of TERC in the homology model, whereas T567M and K570R reside on a hydrophilic loop in close proximity to the RNA–DNA duplex (Figure 5C). V461E alters a residue buried within the protein hydrophobic core. In contrast, variant V435L (data not shown) occurs at a poorly conserved residue within an unstructured region. The remaining 3 variants (R263H, H296P, and G306S) localize to a weakly conserved linker region that connects the TEN domain to the TRBD (not modeled).
TERT rare variant in NHL
In the NHL cohort, we identified a single patient with a TERT rare variant (p.V826F) that was classified as a VUS according to AMP/ACMG criteria. V826F is localized within the enzymatic core of the RTD and demonstrated intermediate telomere elongation capacity (43% of wild type) in our functional assay (supplemental Figure 12). Clinically, this patient had delayed engraftment and persistent cytopenias after autologous stem cell transplant for follicular lymphoma. Eight years later, the patient developed therapy-related AML with prolonged aplasia after intensive induction. At the time of AML diagnosis, the patient's lymphocyte telomere length was 4.6 kb, which was low (<10th percentile) for age.
Discussion
The prevalence, prognostic significance, and functional effects of TERT variants in MDS patients have not been systematically evaluated. In this study, we identified all TERT variants in a registry-level cohort of 1514 MDS patients unselected for suspicion of telomere biology disorder and studied their clinical and functional consequences. TERT variants that are frequent in population databases, such as A279T, H412Y, E441del, and A1062T, had no apparent phenotypic consequences, consistent with studies showing that common polymorphisms do not contribute measurably to telomere-related diseases.39 In contrast, TERT rare variants (<0.1% in all reference populations) were present in 2.7% of MDS patients and were associated with characteristics of disease-causing germline mutations, including shorter telomere length, younger age at MDS diagnosis, and impaired telomere elongation capacity in human cells.
As a group, patients with a TERT rare variant had poor survival after allogeneic HSCT owing to an increased risk of NRM. The prognostic impact of TERT rare variants was independent of established clinical predictors of NRM, such as recipient age, Karnofsky performance status, HCT-CI score, donor-recipient HLA matching, and conditioning intensity. In particular, patients with a TERT rare variant were more likely to die of a noninfectious pulmonary cause than those without a TERT rare variant. This increased risk of NRM and post-transplantation pulmonary complications evokes studies that have reported high rates of NRM and fatal posttransplant pulmonary complications in patients with dyskeratosis congenita.2,55-58 In this context, a global defect in telomere maintenance and constitutionally short telomeres may render patients susceptible to nonhematopoietic end-organ toxicity after conditioning with radiation or DNA alkylating agents.
Most germline TERT variants observed in telomere biology disorder patients are missense substitutions.31 In the absence of compelling family history or functional data, novel variants thus present a clinical dilemma, where accurate variant classification relies on multitiered evidence to support interpretation.32,33,59 Most of the TERT rare variants we identified in this MDS cohort were classified as VUS and would not be definitively actionable in clinical practice. In silico modeling approaches have shown limited utility in predicting the effects of missense variants in patients.34 Moreover, in vitro functional assays may fail to reveal nonenzymatic defects that are essential for in vivo activity, including nuclear localization,60 holoenzyme assembly,61 and telomerase recruitment to telomeres.53,62 We therefore determined the functional effects of all candidate TERT rare variants in a cell-based assay with in vivo telomere extension as the readout. Using this approach, we showed that 90% of rare variants caused a quantifiable defect in telomere elongation compared with wild-type TERT, whereas the SNP A279T had preserved function. The degree of impairment among rare missense substitutions was variable, with 18 having severe functional effect (<25% of wild-type telomere elongation capacity) and 17 having intermediate function (25% to 75% of wild type). Variants that displayed preserved telomere elongation capacity in our assay (>75% of wild type) may represent private genetic variants with no significant biological impact. Conversely, a mild functional impairment of these variants may not be evident in our assay but nevertheless contribute to clinical disease due to genetic anticipation or in combination with other factors increasing hematopoietic cell turnover.
TERT rare variants were distributed across multiple domains, suggesting that there are multiple mechanisms by which TERT variants can impair telomere extension. By correlating the functional data with the human telomerase homology model, we inferred the mechanistic basis of each variant’s effect. For example, although all 15 TERT rare variants within the RTD demonstrated reduced telomere extension, 10 were in proximity to the active site and potentially impair catalysis directly,14 whereas the 5 IFD variants likely alter TPP1-dependent telomere association.63 In this regard, the 2 variants within the TEN domain that showed severely reduced telomere elongation capacity (V84M and G110A) are also positioned to likely disrupt TPP1-mediated telomerase recruitment.53 In contrast, variants within the TRBD and CTE likely impair interactions with regions of TERC, including its CR4/5 and template/pseudoknot domains, that are important for both ribonucleoprotein complex stability and catalysis.29 Our complementary functional and structural analysis provides a powerful framework for evaluation of novel TERT variants and may enhance establishment of TERT-specific variant classification guidelines with broad clinical applicability.
Together, our results indicate that TERT rare variants identify a group of MDS patients who may have an unrecognized telomere biology disorder. This conclusion is based on functional characterization of all candidate variants, telomere length measurements in primary patient samples, and annotation of clinical characteristics including age, comorbidities, and toxicity outcomes in a registry-level cohort. Our analysis is limited by the unavailability of germline reference tissue and absence of detailed family history and clinical examination. Importantly, no patient with a TERT rare variant had a clinical diagnosis of dyskeratosis congenita, and pretransplant clinical characteristics such as pulmonary and hepatic function, peripheral blood counts, and history of aplastic anemia were similar among patients with or without a TERT rare variant. This observation is consistent with previous reports that adults with telomere biology disorder rarely exhibit syndromic features and affected patients often lack a family history of hematologic, pulmonary, or hepatic abnormalities.4,20 Indeed, MDS has been reported to be a late presenting disease manifestation in patients with telomere biology disorders.2,4,20
Our results are consistent with the varied clinical presentation of patients with a telomere biology disorder and indicate that clinical criteria alone may be inadequate to identify all patients with germline TERT rare variants. Notably, 90% of MDS patients with a TERT rare variant were adults older than 40 years of age and there appeared to be no upper age limit. Furthermore, the predictive value of telomere length thresholds in identifying patients with a TERT rare variant has been shown to be poor in older patients, where the telomere length of affected patients overlaps with the lower range of the normal aging control population.20 Selection bias may have influenced the observed age distribution of TERT rare variants in the MDS cohort, as patients with nonmalignant bone marrow failure from dyskeratosis congenita and aplastic anemia, which present at a younger age than MDS,1,2,64 were not included. There can be substantial interobserver variability in reporting of morphologic dysplasia, and many patients lack the most distinctive pathologic features of MDS, such as ring sideroblasts or an excess of myeloid blasts.65 It is thus conceivable that older patients with pathogenic TERT variants who manifest clinically with cytopenias and marginal dysplasia may have had nonclonal bone marrow failure rather than a clonal neoplasm. Based on clinicopathologic and molecular data, however, we documented that nearly all patents with a TERT rare variant had characteristics of bona fide MDS. The unexpectedly high prevalence of unrecognized and clinically significant TERT variants among adult MDS transplant patients thus raises the possibility that routine TERT sequencing should be incorporated into standard transplant evaluation irrespective of age, clinical presentation, or family history. The results of screening could directly inform transplant donor selection by enabling exclusion of candidate related donors who share the germline allele. Furthermore, pretransplantation referral for evaluation of comorbid pulmonary or hepatic disease and consideration of less intensive conditioning regimens could mitigate the elevated risk of NRM. Such a strategy would require multidisciplinary assessment and gene-specific guidelines for variant classification to guide clinical decision making.66
The frequency of TERT rare variants does not fully account for the adverse effect of short telomere length on NRM in adult MDS patients.8 Genetic alterations affecting other components of the telomerase and shelterin complexes are also associated with short telomeres and clinical disease.1,2,31 Unbiased sequencing of these genes, paired with telomere length measurement and clinical outcomes, may reveal additional gene variants associated with MDS predisposition and similarly inferior transplant outcomes.
In summary, we show that TERT rare variants impair telomere elongation in cells and are associated with shorter telomeres, younger age at diagnosis, and an increased risk of NRM in MDS patients undergoing allogeneic transplantation. These results suggest that unrecognized telomere biology disorders contribute to the pathogenesis of MDS. Identifying TERT variants via systematic genetic screening in MDS transplant patients of all ages and regardless of clinical suspicion could impact clinical care by informing donor selection, family counseling, and mitigation of NRM risk.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grants K08CA204734 (National Cancer Institute [NCI]; R.C.L.), RC2DK122533 (National Institute of Diabetes and Digestive and Kidney Diseases [NIDDK]; R.C.L.), R01AG050509 (National Institute on Aging; J.N.), and R01GM120094 (National Institute of General Medical Sciences; J.N.); the Aplastic Anemia & MDS International Foundation (R.C.L.); the Jim and Lois Champy Fund (R.C.L.); the Anna Fuller Fund (R.C.L.); American Cancer Society Research Scholar grant RSF-17-037-01-DMC (J.N.); NIH/National Heart, Lung, and Blood Institute (NHLBI) fellowship training grant 2T32HL116324-06 (C.R.R.); the Sigrid Juselius Foundation (M.M.); the Maud Kuistila Memorial Foundation (M.M.); the Väre Foundation for Pediatric Cancer Research (M.M.); the Orion Research Foundation (M.M.); NIH/NCI Graduate Training in Cancer Research grant T32-CA009172 (F.D.T.); the Jock and Bunny Adams Education and Research Fund (J.H.A.); NIH/NIDDK grant 1R01DK107716 (S.A.); and the Dana-Farber/Harvard Cancer Center core grant from the NIH/NCI 5P30 CA006516 (R.R. and D.N.). The Center for International Blood and Marrow Transplant Research is supported primarily by Public Health Service grant/cooperative agreement 5U24CA076518 from the NCI, the NHLBI, and the National Institute of Allergy and Infectious Diseases (W.S., S.R.S., and Z.-H.H.), grant/cooperative agreement 4U10HL069294 from the NIH/NHLBI and NCI, Health Resources and Services Administration contract HSH250201200016C, and grants N00014-18-1-2850 and N0014-19-1-2888 from the Office of Naval Research.
The views expressed in this article do not reflect the official policy or position of the NIH, the Department of the Navy, the Department of Defense, Health Resources and Services Administration or any other agency of the US Government.
Authorship
Contribution: M.M., C.R.R., and R.C.L. designed the study, analyzed data, and wrote the manuscript; C.J.G., M.M.C., E.H.O., and I.D.V. performed sequencing; D.N. and R.R. curated clinical data, conceived the statistical plan, and performed statistical analysis; C.R.R., H.Q.R., and J.E.G. performed consensus variant classification; V.T., S.P., and J.N. performed structural modeling, analyzed data, and contributed to research discussion; F.D.T. and D.K. performed cloning and generated cell lines; W.S., S.R.S., and Z.-H.H. curated clinical data, clinical data collection and quality assurance, and contributed to research discussion; S.A., C.C., J.H.A., and D.J.D. interpreted data and contributed to research discussion; and all authors reviewed the manuscript during its preparation and approved the submission.
Conflict-of-interest disclosure: D.N. has received research support from Pharmacyclics and owns stock in Madrigral Pharmaceuticals. M.M. has received honoraria from Celgene and Sanofi. R.C.L. has received research support from Jazz Pharmaceuticals and consulting fees from Takeda Pharmaceuticals and bluebird bio. The remaining authors declare no competing financial interests.
Correspondence: R. Coleman Lindsley, Dana-Farber Cancer Institute, 450 Brookline Ave, DA-530C, Boston, MA 02215; e-mail: coleman_lindsley@dfci.harvard.edu.
Deidentified individual participant data are available indefinitely. Proposals for access should be sent to inforequest@mcw.edu.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.