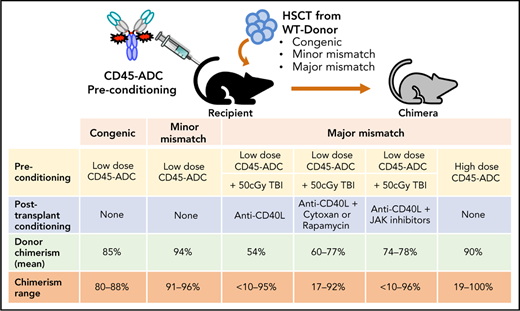
Key Points
Antibody-drug-conjugate (ADC) targeting hematopoietic cells is a promising strategy for the engraftment of donor stem cells.
Anti-mouse CD45-ADC pretreatment facilitates robust donor cells engraftment and immune reconstitution in several preclinical mouse models.

Abstract
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a potentially curative treatment of patients with nonmalignant or malignant blood disorders. Its success has been limited by graft-versus-host disease (GVHD). Current systemic nontargeted conditioning regimens mediate tissue injury and potentially incite and amplify GVHD, limiting the use of this potentially curative treatment beyond malignant disorders. Minimizing systemic nontargeted conditioning while achieving alloengraftment without global immune suppression is highly desirable. Antibody-drug-conjugates (ADCs) targeting hematopoietic cells can specifically deplete host stem and immune cells and enable alloengraftment. We report an anti-mouse CD45-targeted-ADC (CD45-ADC) that facilitates stable murine multilineage donor cell engraftment. Conditioning with CD45-ADC (3 mg/kg) was effective as a single agent in both congenic and minor-mismatch transplant models resulting in full donor chimerism comparable to lethal total body irradiation (TBI). In an MHC-disparate allo-HSCT model, pretransplant CD45-ADC (3 mg/kg) combined with low-dose TBI (150 cGy) and a short course of costimulatory blockade with anti-CD40 ligand antibody enabled 89% of recipients to achieve stable alloengraftment (mean value: 72%). When CD45-ADC was combined with pretransplant TBI (50 cGy) and posttransplant rapamycin, cyclophosphamide (Cytoxan), or a JAK inhibitor, 90% to 100% of recipients achieved stable chimerism (mean: 77%, 59%, 78%, respectively). At a higher dose (5 mg/kg), CD45-ADC as a single agent was sufficient for rapid, high-level multilineage chimerism sustained through the 22 weeks observation period. Therefore, CD45-ADC has the potential utility to confer the benefit of fully myeloablative conditioning but with substantially reduced toxicity when given as a single agent or at lower doses in conjunction with reduced-intensity conditioning.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is an effective treatment of many patients with hematological malignancies or inherited blood disorders.1 Prior to allo-HSCT, substantial cytoreduction with irradiation and/or cytotoxic drugs is required to reliably achieve durable engraftment of donor allogeneic hematopoietic stem cells (HSCs) or genetically corrected autologous HSCs.2-4 HSCT could provide benefit in the treatment of type I diabetes,5 AIDS,6 and tolerance induction in solid organ transplantation.7 However, allo-HSCT immune complications including graft-versus-host disease (GVHD) and nonspecific, multiorgan toxicity associated with conditioning that can incite and amplify GVHD have limited more widespread allogeneic HSCT use.8-11
While reduced-intensity conditioning (RIC) regimens in HLA-mismatched transplant settings can improve safety and tolerability, toxicity due to broad, nontargeted conditioning remains.12 RIC regimens that deliver an agent selectively targeting host cells that interfere with donor alloengraftment combined with minimal or no irradiation could facilitate wider allo-HSCT applications. RIC is especially beneficial for diseases associated with DNA damage and cancer predisposition, such as dyskeratosis congenita, Schwachman-Diamond syndrome, and Fanconi anemia (FA). Unconjugated monoclonal antibodies (mAbs) that specifically target host hematopoietic cells, including HSCs and hematopoietic stem and progenitor cells (HSPCs), often have low utility in immunocompetent as well as immunocompromised mouse models.13-16 In contrast, recent findings in syngeneic immunocompetent and FA mouse models using an antibody-drug conjugate (ADC) composed of a mAb conjugated to saporin, a ribosomal inactivating plant toxin, was more effective in achieving engraftment than mAb alone.17-19
CD45 is a cell-surface antigen present on all immune and hematopoietic cells, including HSCs and HSPCs. When administered as a single pretransplant dose alone or with other nontoxic reagents as a conditioning agent to mice receiving congenic, minor histocompatibility antigen-disparate or MHC-disparate donor bone marrow (BM), CD45-ADC, an anti-CD45 mAb engineered for rapid half-life clearance conjugated with a DNA crosslinker payload from the pyrrolobenzodiazepine (PBD) class, effectively promoted engraftment. Preclinical studies, including ours, have shown that blocking CD40/CD40L costimulatory pathway via anti-CD40L (anti-CD154) mAb can effectively induce tolerance in solid organ transplantation models and augment donor BM alloengraftment in MHC-disparate recipients conditioned with sublethal irradiation.20-26 Murine engraftment in the complete absence of cytoreductive conditioning has been achieved by repetitive infusions of donor BM and anti-CD40L mAb23 or by high donor BM dose(s), CD28/CTLA4:B7 blocking fusion protein, CTLA4Ig, and anti-CD40L mAb,24 the latter also effective in inhibiting donor antihost T-cell GVHD.22,27 Although anti-CD40L mAb monotherapy facilitated BM, chimerism levels were 6% to 12% despite 8 repeated BM doses totaling 160 × 106 cells (∼8 × 109/kg).23
Here, we report a nonmyeloablative regimen of single CD45-ADC (3 mg/kg) dose with low-dose TBI (50 cGy) and transient anti-CD40L mAb enabled durable alloengraftment, even in fully MHC-disparate HSCT recipients. Adding a JAK1/2 or JAK3 inhibitor posttransplant further increased alloengraftment. By increasing the CD45-ADC to 5 mg/kg as a single dose, well-tolerated monotherapy enabled complete and durable donor chimerism in fully allogeneic recipients. Thus, CD45-ADC given pretransplant proved to be a promising targeted conditioning strategy for autologous or allogeneic HSCT either as a primary agent at a lower dose or at a high dose as monotherapy.
Methods
ADC
The variable region (VH/VL) of anti-mouse CD45.2 antibody (clone 104) was grafted onto a human IgG1 Fc mutated in the heavy chain to abolish murine FcRn binding to enhance in vivo clearance.28 Cysteine residues29 incorporated in the Fc region for site-specific conjugation to the PBD linker payload tesirine30 yielded CD45-ADC. A hen egg lysozyme isotype-ADC with matching engineering and conjugation served as a control (Magenta Therapeutics, MA).
BM transplantation (BMT)
Conditioning: Female recipients received isotype-ADC or CD45-ADC IV on day (d)-2 or d-3 (single dose), or d-5 and d-3 (multidose), and/or TBI on d-1 followed by BM from female donors injected IV on d0 into recipients.
Congenic model: Wild-type (WT) B6 (H-2b,CD45.2) recipients received isotype-ADC (3 mg/kg) or CD45-ADC (0.3, 1, or 3 mg/kg) on d-2 or RIC by TBI (500 cGy by X-ray; d-1). On d0, 20 × 106 BM from B6.SJL-Ptprca Pepcb/BoyJ (H-2b,CD45.1) donors were transplanted.
Minor-mismatch model: DBA/2 (H-2d,CD45.2) recipients received 3 mg/kg isotype-ADC or CD45-ADC on d-3 or TBI (900 cGy; d-1) and CByJ.SJL(B6)-Ptprca/J (H-2d,CD45.1 allelic) donor BM (20 × 106) on d0.
MHC-disparate models: WT B6 (H-2b) recipients received 3 mg/kg isotype-ADC or CD45-ADC on d-2 or TBI on d-1 followed by BALB/c (H-2d) donor BM cells (40 × 106; d0). Where indicated, 200 µg anti-CD40L mAb (clone MR1; BioXcell, NH) or irrelevant hamster IgG (Rockland, PA) was administered intraperitoneally daily from d-1 through d+5, then 2 times per week until d+14. Alternatively, WT B6 (H-2b,CD45.2) recipients received isotype-ADC or CD45-ADC (5 mg/kg; d-3) or TBI (900 cGy; d-1) and CByJ.SJL(B6)-Ptprca/J (H-2d,CD45.1) donor BM cells (40 × 106) on d0. Experimental mice were observed daily following BMT and were electively sacrificed only if moribund as per Institutional Animal Care and Use Committee approved protocol.
Clinical immune-modulatory drugs
Where indicated, mice were given rapamycin (Sigma, MO) (1.5 mg/kg per day intraperitoneally in 0.5% carboxymethyl cellulose [Sigma] d-1 through d+13),31 cyclophosphamide (Cytoxan) (Sigma) (25 mg/kg per day; intraperitoneally d+3, d+4),32 ruxolitinib or tofacitinib (Selleck Chemicals, TX) (30 mg/kg twice daily by mouth d0 through d+1433 or baricitinib (Selleck Chemicals) (200 μg daily by mouth Monday through Friday for 2 weeks post-BMT).34
Supplemental methods
Descriptions of mice, statistical methods, and additional details of ADC testing, for example, in vitro assay, pharmacokinetics, peripheral blood (PB) and BM hematopoiesis, flow cytometry, and histology, are provided.
Results
CD45-ADC broadly depletes hematopoietic cells
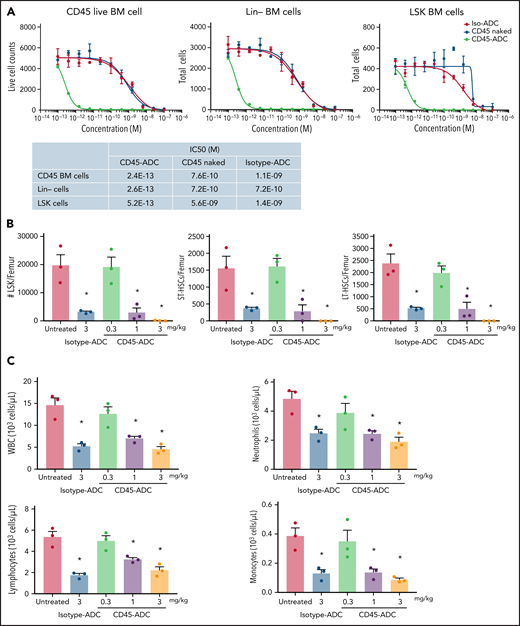
In vitro treatment of WT B6 BM, lineage-depleted (Lin−) BM, and BM progenitors and HSCs (LSK: Lin−Sca1+cKit+) showed each population highly susceptible to CD45-ADC killing compared with unconjugated CD45 mAb or isotype-ADC (CD45-ADC IC50 of 0.24 pM, 0.26 pM, and 0.52 pM, for BM, Lin−, and LSK cells, respectively) (Figure 1A). WT B6 BM harvested 2 days after in vivo CD45-ADC (1 or 3 mg/kg) or isotype-ADC (3 mg/kg) conditioning showed significant reduction in total numbers of LSK cells, short-term HSCs (ST-HSC: LSK,CD150+CD48+), and long-term HSCs (LT-HSC: LSK,CD150+CD48−) compared with untreated control (P < .05); CD45-ADC (3 mg/kg) completely depleted all populations (supplemental Figure S1A; Figure 1B). CD45-ADC (1 or 3 mg/kg) or isotype-ADC (3 mg/kg) treatment resulted in partial and equivalent depletion of BM white blood cells (WBCs), neutrophils, lymphocytes, and monocytes 2 days postdosing (Figure 1C).
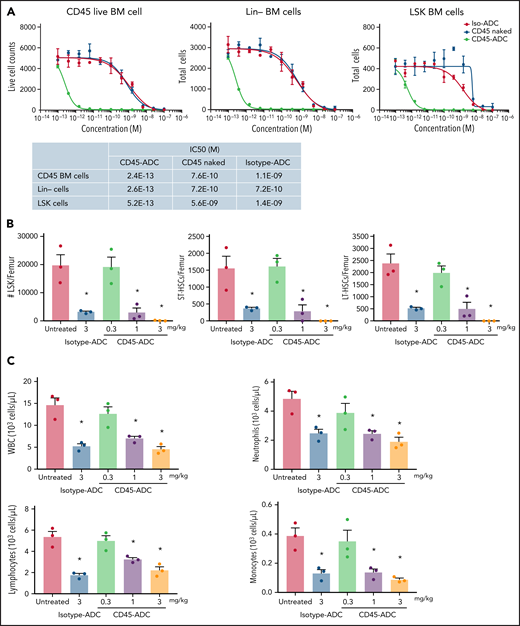
CD45-ADC broadly depletes hematopoietic cells. (A) Total bone marrow (BM) cells, lineage (Lin)-depleted BM cells, or LSK (Lin-Sca1+ cKit+) cells isolated from WT B6 mice were cultured in the media along with different concentrations of unconjugated CD45 Ab, CD45-ADC, or isotype-ADC for 5 days. Viability of cells was analyzed by flow cytometry on d5. (B) CD45-ADC-mediated depletion of hematopoietic progenitor cells (LSKs) and HSCs (ST-HSC: LSKCD150+CD48+ or LT-HSC: LSKCD150+CD48−) in BM was assessed 2 days after IV administration in WT B6 mice (n = 3 per group, 10- to 12-week-old female). The frequencies of progenitors and HSCs were analyzed by flow cytometry, and absolute cell counts/femur were calculated. Nontreated mice served as control. (C) The number of WBCs, neutrophils, lymphocytes, and monocytes in BM were assessed 2 days after IV administration of CD45-ADC or isotype-ADC in WT B6 mice (n = 3 per group). Nontreated mice served as control. Data in (B) and (C) represent mean ± SEM. Experiments were performed twice, and data shown are from 1 representative experiment. *P < .05 vs untreated mice, using ANOVA with posthoc Tukey’s multiple comparisons test.
CD45-ADC broadly depletes hematopoietic cells. (A) Total bone marrow (BM) cells, lineage (Lin)-depleted BM cells, or LSK (Lin-Sca1+ cKit+) cells isolated from WT B6 mice were cultured in the media along with different concentrations of unconjugated CD45 Ab, CD45-ADC, or isotype-ADC for 5 days. Viability of cells was analyzed by flow cytometry on d5. (B) CD45-ADC-mediated depletion of hematopoietic progenitor cells (LSKs) and HSCs (ST-HSC: LSKCD150+CD48+ or LT-HSC: LSKCD150+CD48−) in BM was assessed 2 days after IV administration in WT B6 mice (n = 3 per group, 10- to 12-week-old female). The frequencies of progenitors and HSCs were analyzed by flow cytometry, and absolute cell counts/femur were calculated. Nontreated mice served as control. (C) The number of WBCs, neutrophils, lymphocytes, and monocytes in BM were assessed 2 days after IV administration of CD45-ADC or isotype-ADC in WT B6 mice (n = 3 per group). Nontreated mice served as control. Data in (B) and (C) represent mean ± SEM. Experiments were performed twice, and data shown are from 1 representative experiment. *P < .05 vs untreated mice, using ANOVA with posthoc Tukey’s multiple comparisons test.
We next assessed CD45-ADC mediated mature effector cell depletion in WT B6 mice. Isotype-ADC- or CD45-ADC-treated mice were tracked over 3 weeks postadministration. PB analysis confirmed that CD45-ADC (≤1 mg/kg) treatment was nonmyeloablative and did not cause sustained lymphopenia or neutropenia. A dose-dependent transient decrease in PB WBC, neutrophil, lymphocyte, and monocyte counts was observed by d7, rebounding to baseline levels by d21. A transient decrease in PB red blood cell (RBC) and platelet counts also was seen by d7 (supplemental Figure S1B). Mice administered a higher CD45-ADC dose (3 mg/kg) had deep and sustained RBC and platelet reduction, neutropenia, and were killed on d11 due to hematopoietic failure with poor body condition and significant weight loss, establishing CD45-ADC (3 mg/kg) as fully myeloablative. In contrast, mice receiving isotype-ADC (3 mg/kg) did not present evidence of sustained hematopoietic failure (supplemental Figure S1B), indicating CD45-ADC-specific targeting of HSCs and immune cells.
Plasma drug concentration of IV-administered CD45-ADC (3 or 6 mg/kg single dose, or 3 mg/kg Q2D × 2) vs isotype-ADC (3 mg/kg single dose) was evaluated over a time course. At all doses, CD45-ADC was detectable immediately postinfusion, and plasma drug concentration declined rapidly, reaching lower quantification limits by 48 hours (supplemental Figure S2). Calculated CD45-ADC clearance and exposure established a substantial target-mediated drug disposition (TMDD) due to a broad CD45 expression profile coupled with a desired rapid half-life (2 to 4 hours for CD45-ADC groups) suitable for juxtaposition to hematopoietic cell infusion. Compared with 3 mg/kg CD45-ADC, isotype-ADC at 3 mg/kg demonstrated a longer half-life (15.3 hours) and higher AUC-based exposure (3.8-fold), as expected due to lack of TMDD.
CD45-ADC depletes hematopoietic cells and lymphocytes in the lymph node, thymus, and BM
To determine single isotype-ADC (3 mg/kg) or CD45-ADC (0.3 to 3 mg/kg) dose effects on adaptive immune system cells, we analyzed splenic, lymph node, and thymic CD45+ cells by flow cytometry at 48 hours and 72 hours postdosing. At 48 hours, CD45-ADC conditioning modestly depleted thymocytes, hematopoietic cells, T cells, and B cells in lymph nodes but not the spleen (supplemental Figure S3A). At 72 hours, ∼80% depletion of CD45+ hematopoietic cells was seen in the thymus and BM; the spleen was depleted by 35% (supplemental Figure S3B).
Residual hematopoietic cells in the spleen are functionally inactivated
The CD45-ADC PBD payload is a potent DNA crosslinker and is particularly cytotoxic against dividing cells. We hypothesized that residual splenic lymphocytes of CD45-ADC-treated animals were quiescent because CD45-ADC would be expected to kill dividing lymphocytes. To test the CD45-ADC activity on cycling cells, splenic T cells from naïve mice were cultured with plate-bound antimouse-CD3 in the presence of CD45-ADC or isotype-ADC for 5 days and evaluated for ex vivo expansile capacity. CD45-ADC vs isotype-ADC decreased T-cell numbers, including Ki-67+ T cells ex vivo (supplemental Figure S4A). The impact of 3 mg/kg ADC treatment on proliferating T cells (splenic, blood) in vivo was evaluated on d5 posttreatment in mice undergoing active GVHD (DBA/2 CD45.2−>BALB/c CD45.1). We observed reductions in the number and proliferation status of allogeneic T cells with CD45-ADC treatment (supplemental Figure S4B).
To assess whether residual cells were unable to mount an allogeneic response following CD45-ADC exposure, splenocytes isolated from ADC-treated mice were labeled with CellTrace Violet and transferred into immunodeficient allogeneic hosts; in vivo proliferation was quantified. Splenocytes from untreated or isotype-ADC but not CD45-ADC-treated mice proliferated robustly and accumulated in PB and spleens (supplemental Figure S5), demonstrating that residual lymphocytes in CD45-ADC-treated mice were rendered functionally inactive.
CD45-ADC conditioning enables efficient multilineage donor cells engraftment in congenic recipients
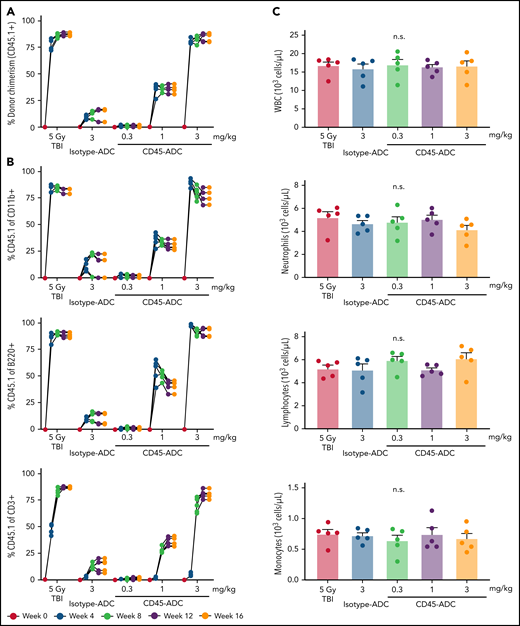
To determine whether HSC depletion induced by CD45-ADC was sufficient to permit durable multilineage engraftment, B6 (CD45.2) recipients were given isotype-ADC or CD45-ADC on d-2 or TBI (500 cGy), a RIC dose on d-1 and transplanted with B6 congenic (CD45.1) donor BM cells (20 × 106; d0). Four weeks post-BMT, CD45-ADC-conditioned but not isotype-ADC-conditioned mice had significant dose-dependent increases in PB donor chimerism, highest at 3 mg/kg (supplemental Figure S6A; Figure 2A). Maximum engraftment (>80%) achieved by 12 weeks was equivalent to a RIC dose of TBI (500 cGy). Donor chimerism remained stable through 16 weeks at ≥80% and ≥30% for all mice treated with CD45-ADC at 3 mg/kg or 1 mg/kg, respectively. At 3 mg/kg CD45-ADC dose, individual myeloid or B-cell chimerism levels exceeded 80% by 4 weeks. T-cell chimerism continued to improve until 12 weeks and remained stable at >77% thereafter (Figure 2B).

CD45-ADC conditioning enables efficient multilineage donor engraftment in congenic mice. WT B6 (CD45.2) recipients (n = 5 mice/group, 10- to 12-week-old female) were conditioned with isotype-ADC (3 mg/kg) or CD45-ADC (0.3 mg/kg, 1 mg/kg, or 3 mg/kg) on d-2, or 500 cGy (5 Gy) TBI on d-1, and transplanted with 20 × 106 B6 (CD45.1) donor BM on d0. (A) Overall donor (CD45.1) engraftment in the PB of transplanted mice was assessed by flow cytometry. Each individual line represents a single mouse tracked over 16 weeks, as indicated. (B) Donor-derived (CD45.1) lineage subsets in the PB of transplanted mice were assessed by flow cytometry. (C) Immune cell subsets in BM were analyzed at 16 weeks post-BMT in transplanted recipients. (D) Frequencies of donor-derived (CD45.1) hematopoietic progenitor cells (LSKs) and HSCs in the BM of transplanted recipients were analyzed at 16 weeks post-BMT by flow cytometry. *P < .05 vs isotype-ADC (3 mg/kg), using 1-way ANOVA with posthoc Dunnett’s multiple comparisons test. Data in (C) and (D) represent mean ± SEM. Experiments were performed twice, and data shown are from 1 representative experiment.
CD45-ADC conditioning enables efficient multilineage donor engraftment in congenic mice. WT B6 (CD45.2) recipients (n = 5 mice/group, 10- to 12-week-old female) were conditioned with isotype-ADC (3 mg/kg) or CD45-ADC (0.3 mg/kg, 1 mg/kg, or 3 mg/kg) on d-2, or 500 cGy (5 Gy) TBI on d-1, and transplanted with 20 × 106 B6 (CD45.1) donor BM on d0. (A) Overall donor (CD45.1) engraftment in the PB of transplanted mice was assessed by flow cytometry. Each individual line represents a single mouse tracked over 16 weeks, as indicated. (B) Donor-derived (CD45.1) lineage subsets in the PB of transplanted mice were assessed by flow cytometry. (C) Immune cell subsets in BM were analyzed at 16 weeks post-BMT in transplanted recipients. (D) Frequencies of donor-derived (CD45.1) hematopoietic progenitor cells (LSKs) and HSCs in the BM of transplanted recipients were analyzed at 16 weeks post-BMT by flow cytometry. *P < .05 vs isotype-ADC (3 mg/kg), using 1-way ANOVA with posthoc Dunnett’s multiple comparisons test. Data in (C) and (D) represent mean ± SEM. Experiments were performed twice, and data shown are from 1 representative experiment.
At 16 weeks, absolute numbers of WBCs, neutrophils, lymphocytes, and monocytes in BM were comparable for RIC (TBI), CD45-ADC, and isotype-ADC conditioning (Figure 2C). Although PB analysis at 16 weeks showed reduced WBC and lymphocyte but not neutrophil or monocyte numbers in CD45-ADC-treated compared with TBI-conditioned recipients (supplemental Figure S6B), PB neutrophil, lymphocyte, RBC, and platelet counts were within the normal range compared with untreated BMT mice (supplemental Figure S6C). At 16 weeks post-BMT, CD45.1+ donor cells in BM populations were comparable in RIC by TBI and 3 mg/kg CD45-ADC-conditioned recipients, whereas <10% donor chimerism was observed in the isotype-ADC-treated group (Figure 2D).
Single-agent CD45-ADC results in complete donor chimerism in a minor mismatch transplant model
We next extended our findings to a minor histocompatibility antigen mismatch engraftment model. DBA/2 mice (H-2d,CD45.2) were conditioned with a lethal dose of TBI (900 cGy, d-1) or treated with 3 mg/kg isotype-ADC or CD45-ADC (d-3) and given CByJ.SJL(B6)-Ptprca/J (H-2d,CD45.1) donor BM cells (20 × 106; d0). Three mice per group were killed on d0 to assess HSC and mature effector cell depletion prior to BM infusion. Complete BM progenitor (LSK) and HSC depletion were observed in ADC (CD45, isotype)-conditioned recipients and were comparable with lethal-TBI conditioning (supplemental Figure S7A). PB analysis on d0 showed B-cell and T-cell but not myeloid-cell depletion with lethal-TBI conditioning that was not observed with CD45-ADC or isotype-ADC treatment (supplemental Figure S7B).
PB analysis as early as 4 weeks post-BMT revealed ∼75% donor engraftment in CD45-ADC-conditioned recipients, increasing to >85% by week 12 and remaining stable through week 16, at which time donor chimerism was equivalent to mice conditioned with lethal-TBI (Figure 3A). Donor myeloid- and B-cell chimerism were achieved with equal kinetics and remained stable over 20 weeks in recipients conditioned with CD45-ADC or lethal-TBI. Isotype-ADC treatment did not result in successful engraftment as assessed in PB. Despite faster donor T-cell chimerism kinetics in TBI-conditioned mice, overall engraftment in the CD45-ADC group was comparable at 20 weeks post-BMT (Figure 3A). Donor chimerism in BM was comparable for CD45-ADC and lethal-TBI conditioning at week 20 (Figure 3B). Isotype-ADC-treated recipients showed the presence of donor LSK and ST-HSCs but not LT-HSCs in BM at week 20.
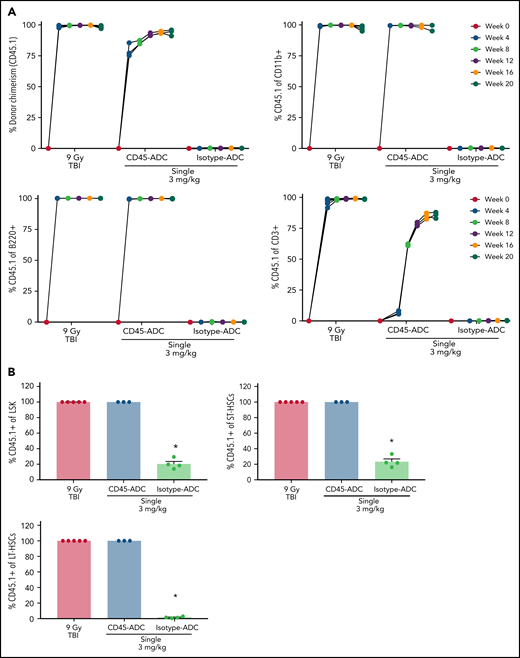
Single-agent CD45-ADC results in complete donor chimerism in a minor mismatch transplant model. DBA/2 (H-2d, CD45.2) recipients (n = 5 mice/group, 20- to 22-week-old females) were conditioned with 3 mg/kg isotype-ADC or CD45-ADC on d-3, or 900 cGy (9 Gy) TBI on d-1, and transplanted with 20 × 106 CByJ.SJL(B6)-Ptprca/J (H-2d, CD45.1) donor BM on d0. Two mice from the CD45-ADC group died 3 weeks posttransplant, and 1 mouse from the isotype-ADC group died at 14 weeks due to unknown causes. (A) Engraftment of donor cells (CD45.1+): overall chimerism and chimerism in lineage subsets in the PB of transplanted mice were assessed by flow cytometry. Each individual line represents a single mouse tracked over 20 weeks posttransplant as indicated. (B) Donor (CD45.1+)-derived hematopoietic progenitor cells (LSKs) and HSCs in BM were analyzed at 20 weeks posttransplant by flow cytometry. Data represent mean ± SEM. (A,B) Experiments were performed twice, and data shown are from 1 representative experiment. (B) *P < .05 vs 9 Gy TBI-treated mice using 1-way ANOVA with posthoc Dunnett’s multiple comparisons test.
Single-agent CD45-ADC results in complete donor chimerism in a minor mismatch transplant model. DBA/2 (H-2d, CD45.2) recipients (n = 5 mice/group, 20- to 22-week-old females) were conditioned with 3 mg/kg isotype-ADC or CD45-ADC on d-3, or 900 cGy (9 Gy) TBI on d-1, and transplanted with 20 × 106 CByJ.SJL(B6)-Ptprca/J (H-2d, CD45.1) donor BM on d0. Two mice from the CD45-ADC group died 3 weeks posttransplant, and 1 mouse from the isotype-ADC group died at 14 weeks due to unknown causes. (A) Engraftment of donor cells (CD45.1+): overall chimerism and chimerism in lineage subsets in the PB of transplanted mice were assessed by flow cytometry. Each individual line represents a single mouse tracked over 20 weeks posttransplant as indicated. (B) Donor (CD45.1+)-derived hematopoietic progenitor cells (LSKs) and HSCs in BM were analyzed at 20 weeks posttransplant by flow cytometry. Data represent mean ± SEM. (A,B) Experiments were performed twice, and data shown are from 1 representative experiment. (B) *P < .05 vs 9 Gy TBI-treated mice using 1-way ANOVA with posthoc Dunnett’s multiple comparisons test.
Anti-CD40L mAb facilitates alloengraftment in an MHC-mismatched transplant model that is augmented by CD45-ADC
Previously we demonstrated that anti-CD40L mAb given for 2 weeks beginning d-1 to 200 cGy TBI-conditioned recipients resulted in 48% donor engraftment.26 Our initial goal was to use a single CD45-ADC injection with a low TBI dose to achieve donor chimerism in most recipients. B6 (H-2b) recipients conditioned with 350 cGy, 450 cGy, or 550 cGy TBI were transplanted with BALB/c (H-2d) BM (40 × 106).26 At 12 weeks posttransplant, donor cells were undetectable in PB of TBI-only-conditioned (350 to 550 cGy) recipients (supplemental Figure S8A). Increasing the TBI dose to 650 cGy or 750 cGy resulted in PB donor alloengraftment of 91% or 92%, respectively, in 100% of recipients (data not shown).
Adding anti-CD40L mAb to 350 cGy TBI permitted >10% PB donor chimerism in 6 of 11 (55%) recipients from 2 pooled experiments (supplemental Figure S8A,B). Pretransplant CD45-ADC (3 mg/kg) added to anti-CD40L mAb and 350 cGy TBI resulted in >62% PB mean donor chimerism in 6 of 7 (86%) recipients, significantly higher than in the cohort not given CD45-ADC. Isotype-ADC (3 mg/kg), noted to have a longer half-life and higher AUC-based exposure, also resulted in 45% mean PB donor chimerism in 86% of recipients when coupled with nonmyeloablative TBI (350 cGy) (supplemental Figure S8B). Multilineage donor PB chimerism was comprised mostly of myeloid cells along with a lower frequency of B and T cells. Recipients conditioned with TBI (350 cGy), CD45-ADC, and anti-CD40L mAb resulted in increased PB donor T-cell engraftment, albeit not statistically significant compared with conditioning with TBI and anti-CD40L mAb ± isotype-ADC (supplemental Figure S8C). The remaining host cells at 12 weeks post-BMT were mostly composed of B cells in all conditions tested (data not shown).
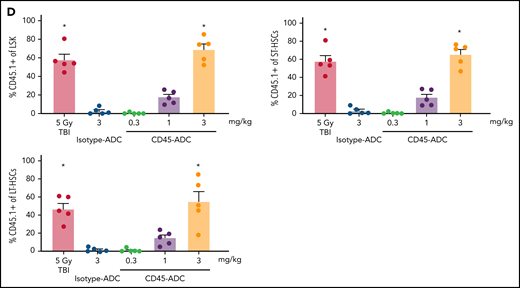
To further reduce TBI conditioning, B6 recipients were conditioned with CD45-ADC or isotype-ADC (3 mg/kg), anti-CD40L mAb, TBI (0 to 350 cGy), and transplanted with BALB/c BM cells (40 × 106). PB analysis at 4 weeks could not detect donor engraftment in recipients conditioned with CD45-ADC plus anti-CD40L mAb without TBI, whereas inclusion of minimal TBI conditioning (50 cGy) significantly improved (P = .0016) overall engraftment (Figure 4A). Engraftment was CD45-ADC specific, as conditioning with isotype-ADC plus anti-CD40L mAb and TBI (50 cGy) resulted in <5% mean PB donor engraftment. Donor engraftment in CD45-ADC-conditioned recipients remained stable and improved further at 12 weeks, albeit not statistically significant when compared with mice also receiving 150 cGy or 350 cGy to 50 cGy TBI and anti-CD40L mAb (P = .0512, P = .062, respectively) (Figure 4A). At 50 cGy TBI, multilineage PB donor chimerism comprised of T, B, and myeloid cells improved significantly in recipients conditioned with anti-CD40L mAb and CD45-ADC vs isotype-ADC (Figure 4B). Taken together, our data suggest that a nonmyeloablative conditioning regimen consisting of 50 cGy TBI, CD45-ADC (3 mg/kg), plus anti-CD40L mAb can be an effective strategy for enabling stable allogeneic donor engraftment.
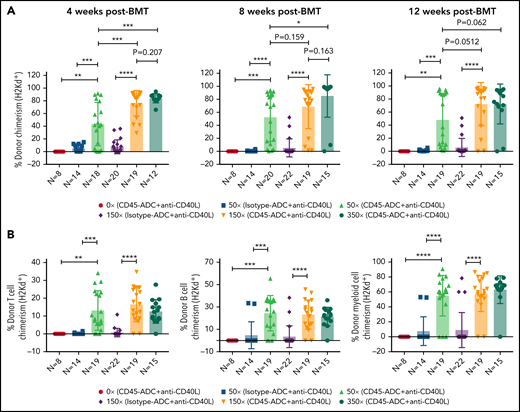
Anti-CD40L mAb treatment facilitates alloengraftment in a major mismatch transplant model that is augmented by CD45-ADC. B6 (H-2b) recipients (10- to 12-week-old female) were conditioned with 3 mg/kg isotype-ADC or CD45-ADC on d-2 and 0 to 350 cGy (x = cGy in figure) TBI on d-1. All groups of mice were transplanted with 40 × 106 BALB/c (H-2d) BM cells on d0. Recipients were treated with anti-CD40L mAb (200 μg) from d-1 to d+14 post-BMT. (A) Engraftment of donor cells (H-2d) in the PB of transplanted mice were analyzed at 4 weeks, 8 weeks, and 12 weeks post-BMT by flow cytometry (n = 8 to 22 mice per group). (B) Multilineage peripheral donor chimerism was analyzed at 12 weeks post-BMT by flow cytometry (n = 8 to 22 mice per group). (A,B) Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, 2-tailed Student t test. Pooled data from 2 to 3 experiments are shown.
Anti-CD40L mAb treatment facilitates alloengraftment in a major mismatch transplant model that is augmented by CD45-ADC. B6 (H-2b) recipients (10- to 12-week-old female) were conditioned with 3 mg/kg isotype-ADC or CD45-ADC on d-2 and 0 to 350 cGy (x = cGy in figure) TBI on d-1. All groups of mice were transplanted with 40 × 106 BALB/c (H-2d) BM cells on d0. Recipients were treated with anti-CD40L mAb (200 μg) from d-1 to d+14 post-BMT. (A) Engraftment of donor cells (H-2d) in the PB of transplanted mice were analyzed at 4 weeks, 8 weeks, and 12 weeks post-BMT by flow cytometry (n = 8 to 22 mice per group). (B) Multilineage peripheral donor chimerism was analyzed at 12 weeks post-BMT by flow cytometry (n = 8 to 22 mice per group). (A,B) Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, 2-tailed Student t test. Pooled data from 2 to 3 experiments are shown.
Adding clinical immunomodulatory drugs to CD45-ADC and anti-CD40L mAb conditioning further increases donor alloengraftment
Immunosuppressive drugs can prevent donor T-cell activation, survival, or inhibit effector T-cell function for prevention and treatment of GVHD. For example, posttransplant cyclophosphamide combined with calcineurin inhibitors and mycophenolate mofetil is a promising allo-HSCT GVHD preventive strategy.35,36 In some clinical series, sirolimus (rapamycin) also has been shown to be effective for prevention or treatment of acute GVHD after BMT.37,38 Immunosuppressive agents can enhance murine donor alloengraftment and induce tolerance when given with nonmyeloablative conditioning and anti-CD40L mAb.39
Therefore, we investigated the possible efficacy of adding rapamycin or cyclophosphamide to B6 recipients given anti-CD40L mAb, CD45-ADC, a minimal TBI (50 cGy) dose, and BALB/c BM cells (40 × 106). PB analysis at 12 weeks showed a modest increase in the frequency of mice with ≥10% alloengraftment (100% vs 71%) with comparable mean donor chimerism levels (59% vs 47%, P = .605) when cyclophosphamide was included in the conditioning regimen. The cohort receiving rapamycin compared with no rapamycin showed that 100% vs 71% of mice had ≥10% alloengraftment with mean donor chimerism levels increasing to 77% from 47%, respectively (P = .195) (Figure 5A). Donor T-cell chimerism was significantly higher in recipients also given rapamycin or cyclophosphamide, which occurred at the expense of B-cell alloengrafment (Figure 5B). Donor and host T-cell ratios further confirmed improved T-cell engraftment in recipients treated with rapamycin vs no rapamycin but not cyclophosphamide (Figure 5C).
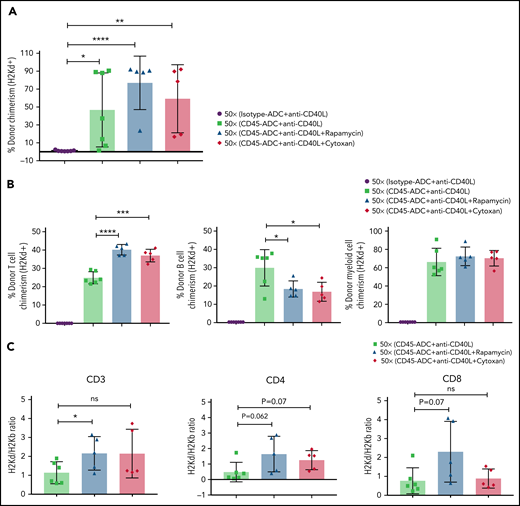
Conditioning of recipients with CD45-ADC along with anti-CD40L mAb and immunosuppressive agents can further improve allogeneic engraftment. B6 (H-2b) recipients (n = 7 mice per group, 10- to 12-week-old female) were conditioned with 3 mg/kg isotype-ADC or CD45-ADC on d-2 and 50 cGy (x = cGy in figure) TBI on d-1. All groups of mice were transplanted with 40 × 106 BALB/c (H-2d) BM cells on d0. Recipients were treated with anti-CD40L mAb (200 μg) from d-1 to d+14 post-BMT. Rapamycin (1.5 mg/kg per day) was administered from d-1 through d+13 post-BMT. Cyclophosphamide (25 mg/kg per day) was administered on d+3 and d+4 post-BMT. Two mice, each from the rapamycin group and cyclophosphamide group, died by 2 weeks of posttransplant due to unknown causes. (A) Donor cells (H-2d) engraftment in the PB of transplanted mice was analyzed at 12 weeks post-BMT by flow cytometry. (B) Multilineage peripheral donor chimerism was analyzed at 12 weeks post-BMT by flow cytometry. (C) The ratio of donor (H-2d) and host (H-2b) peripheral T-cell populations was analyzed at 12 weeks post-BMT. (A-C) Data represent mean ± SEM. Experiments were performed twice, and data shown are from 1 representative experiment. *P < .05, **P < .01, ***P < .001, ****P < .0001, 2-tailed Student t test.
Conditioning of recipients with CD45-ADC along with anti-CD40L mAb and immunosuppressive agents can further improve allogeneic engraftment. B6 (H-2b) recipients (n = 7 mice per group, 10- to 12-week-old female) were conditioned with 3 mg/kg isotype-ADC or CD45-ADC on d-2 and 50 cGy (x = cGy in figure) TBI on d-1. All groups of mice were transplanted with 40 × 106 BALB/c (H-2d) BM cells on d0. Recipients were treated with anti-CD40L mAb (200 μg) from d-1 to d+14 post-BMT. Rapamycin (1.5 mg/kg per day) was administered from d-1 through d+13 post-BMT. Cyclophosphamide (25 mg/kg per day) was administered on d+3 and d+4 post-BMT. Two mice, each from the rapamycin group and cyclophosphamide group, died by 2 weeks of posttransplant due to unknown causes. (A) Donor cells (H-2d) engraftment in the PB of transplanted mice was analyzed at 12 weeks post-BMT by flow cytometry. (B) Multilineage peripheral donor chimerism was analyzed at 12 weeks post-BMT by flow cytometry. (C) The ratio of donor (H-2d) and host (H-2b) peripheral T-cell populations was analyzed at 12 weeks post-BMT. (A-C) Data represent mean ± SEM. Experiments were performed twice, and data shown are from 1 representative experiment. *P < .05, **P < .01, ***P < .001, ****P < .0001, 2-tailed Student t test.
GVHD pathogenic cytokines can signal via the Janus-associated kinase (JAK) pathways in neutrophils and immune cell subsets, including dendritic cells, macrophages, T cells, and B cells. JAK inhibitors represent a class of immunomodulatory drugs that inhibit signaling by multiple cytokines. Ruxolitinib and baricitinib each block JAK1/2 signaling, whereas tofacitinib blocks JAK3 signaling; each is efficacious in reducing GVHD in preclinical models; ruxolitinib is FDA-approved to treat steroid-refractory acute or chronic GVHD.40-48
We investigated whether adding a JAK inhibitor to CD45-ADC, anti-CD40L mAb, and 50 cGy TBI regimen would increase the frequency of mice with ≥10% PB alloengraftment or mean chimerism levels. Cohorts of B6 recipients conditioned with CD45-ADC or isotype-ADC, anti-CD40L mAb, and 50 cGy TBI also received ruxolitinib, tofacitinib, or baricitinib (doses and schedules effective in preventing murine acute GVHD) and were transplanted with BALB/c BM (40 × 106). PB analysis at 4 to 18 weeks showed that recipients conditioned with CD45-ADC, anti-CD40L, and 50 cGy TBI had a total peak donor mean chimerism of 32% at 8 weeks that remained stable through 18 weeks (supplemental Figure S9A). Adding any of the 3 JAK inhibitors improved alloengraftment at each time point analyzed. JAK inhibitor treatment compared with no treatment showed 83% to 100% vs 67% of mice had ≥10% PB alloengraftment at 12 weeks post-BMT (supplemental Figure S9A). Pooled 12-week data also revealed improved alloengraftment in recipients treated with a JAK inhibitor that was comparable among the 3 inhibitors (Figure 6A). PB multilineage donor chimerism at 18 weeks showed significant increases in donor T- and myeloid-cell alloengraftment in recipients also given a JAK inhibitor (Figure 6B).
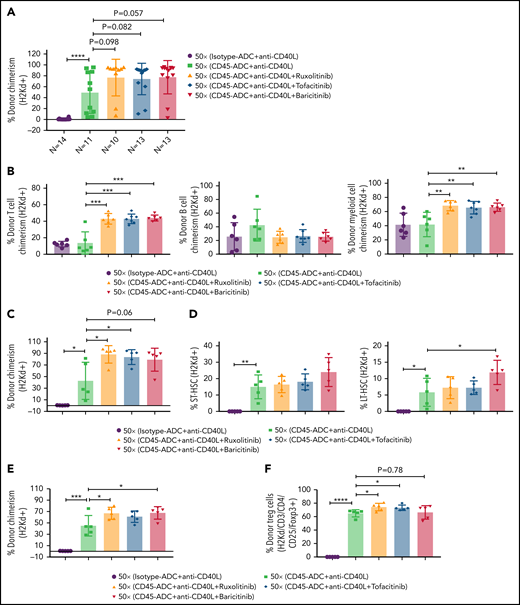

Conditioning of recipients with CD45-ADC along with anti-CD40L mAb and JAK inhibitor can improve overall allogeneic engraftment. B6 (H-2b) recipients (10- to 12-week-old female) were conditioned with 3 mg/kg isotype-ADC or CD45-ADC on d-2 and 50 cGy (x = cGy in figure) TBI on d-1. All groups of mice were transplanted with 40 × 106 BALB/c (H-2d) BM on d0. Recipients were treated with anti-CD40L mAb (200 μg) from d-1 to d+14 post-BMT. Ruxolitinib (30 mg/kg) or tofacitinib (30 mg/kg) was administered from d0 through d+14 post-BMT. Baricitinib (200 μg) was administered once daily for 5 days per week for 2 weeks post-BMT. (A) Donor cells (H-2d) engraftment in the PB of transplanted mice was assessed at 12 weeks post-BMT by flow cytometry. Pooled data from 2 experiments are shown (n = 10 to 14 mice per group). (B) Multilineage peripheral donor chimerism was analyzed at 18 weeks post-BMT by flow cytometry (n = 6 to 7 mice per group). (C-J) Recipients (n = 5 mice per group) were killed at 25 weeks post-BMT, and cells isolated from BM (C,D), thymus (E,F), and spleen (G-J) were analyzed by flow cytometry. (A-J) Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, 2-tailed Student t test.
Conditioning of recipients with CD45-ADC along with anti-CD40L mAb and JAK inhibitor can improve overall allogeneic engraftment. B6 (H-2b) recipients (10- to 12-week-old female) were conditioned with 3 mg/kg isotype-ADC or CD45-ADC on d-2 and 50 cGy (x = cGy in figure) TBI on d-1. All groups of mice were transplanted with 40 × 106 BALB/c (H-2d) BM on d0. Recipients were treated with anti-CD40L mAb (200 μg) from d-1 to d+14 post-BMT. Ruxolitinib (30 mg/kg) or tofacitinib (30 mg/kg) was administered from d0 through d+14 post-BMT. Baricitinib (200 μg) was administered once daily for 5 days per week for 2 weeks post-BMT. (A) Donor cells (H-2d) engraftment in the PB of transplanted mice was assessed at 12 weeks post-BMT by flow cytometry. Pooled data from 2 experiments are shown (n = 10 to 14 mice per group). (B) Multilineage peripheral donor chimerism was analyzed at 18 weeks post-BMT by flow cytometry (n = 6 to 7 mice per group). (C-J) Recipients (n = 5 mice per group) were killed at 25 weeks post-BMT, and cells isolated from BM (C,D), thymus (E,F), and spleen (G-J) were analyzed by flow cytometry. (A-J) Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, 2-tailed Student t test.
At 25 weeks, donor BM alloengraftment was significantly higher in recipients conditioned with CD45-ADC plus anti-CD40L mAb compared with isotype-ADC plus anti-CD40L mAb; adding a JAK inhibitor further improved overall engraftment (Figure 6C). In the baricitinib compared with no JAK inhibitor cohort, the percentage of donor LT-HSC was significantly higher (Figure 6D). Thymus (Figure 6E) and spleen (Figure 6G; supplemental Figure S9C) analyses revealed significantly higher alloengraftment in those cohorts receiving a JAK inhibitor. Although no difference in overall donor CD4 T-cell engraftment was seen in the thymus (supplemental Figure S9B), the frequency of donor regulatory T cells (Tregs) in the thymus and spleen was significantly higher, and effector/memory (CD44hiCD62Llo) T cells significantly lower with ruxolitinib or tofacitinib (Figure 6F-J). Notably, recipients in all groups remained healthy with long-term survival after allogeneic BMT and did not develop signs of GVHD (supplemental Figure S10A,B). Although we experienced minor death cases in certain experiments following BMT (supplemental Table 1), most of the BMT recipients remained healthy until the end of experiments.
Higher single dose CD45-ADC (5 mg/kg) enables full allogeneic donor engraftment
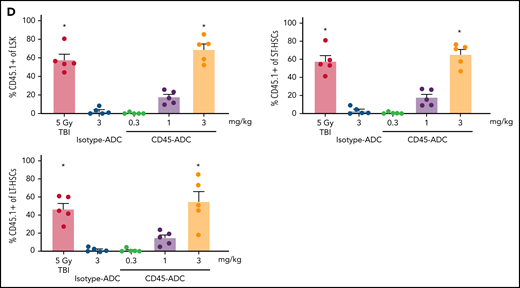
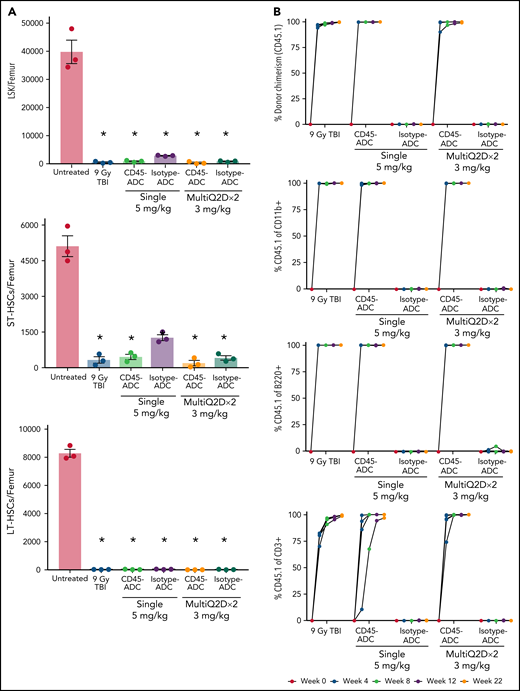
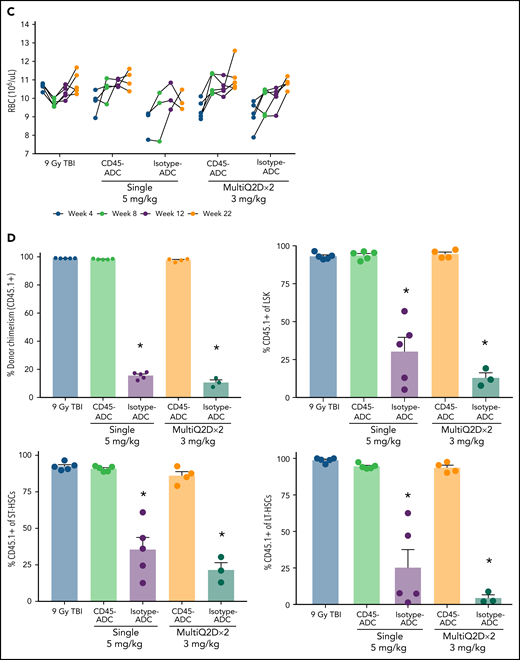
To assess the potential of a higher CD45-ADC dose as a monotherapy conditioning in the full mismatched model, B6 (H-2b) recipients were given a single 5 mg/kg dose of isotype-ADC or CD45-ADC on d-3 and allogeneic CByJ.SJL(B6)-Ptprca/J (H-2d,CD45.1) donor BM (40 × 106; d0). Additionally, we explored multidosing of Q2D × 2 of 3 mg/kg CD45-ADC on d-5 and d-3. Three mice per group were killed on day 0 before BMT to assess depletion of recipient HSPCs. The total number of LSKs and HSCs in BM was significantly reduced with CD45-ADC or isotype-ADC treatment vs untreated and comparable to depletion in lethal-TBI (9 Gy) conditioning (Figure 7A). PB time-course analyses revealed >95% donor chimerism at 4 weeks with CD45-ADC treatment (5 mg/kg single dose or 3 mg/kg Q2D × 2 totaling 6 mg/kg) and remained stable over 22 weeks (Figure 7B). Notably, PB T-, B-, and myeloid-cell donor chimerism were comparable between CD45-ADC and lethal-TBI conditioning. Time-course of PB RBC counts posttransplant was comparable between groups (Figure 7C). BM analysis at 22 weeks posttransplant revealed >90% donor chimerism in CD45-ADC groups in the total BM, LSK, ST-HSC, and LT-HSC populations, comparable to lethal-TBI conditioning (Figure 7D). In contrast, isotype-ADC groups showed moderate chimerism in these populations, significantly lower than lethal-TBI or CD45-ADC conditioning. Donor engraftment was not observed with isotype-ADC in PB, although moderate chimerism was observed in BM populations (Figure 7D).
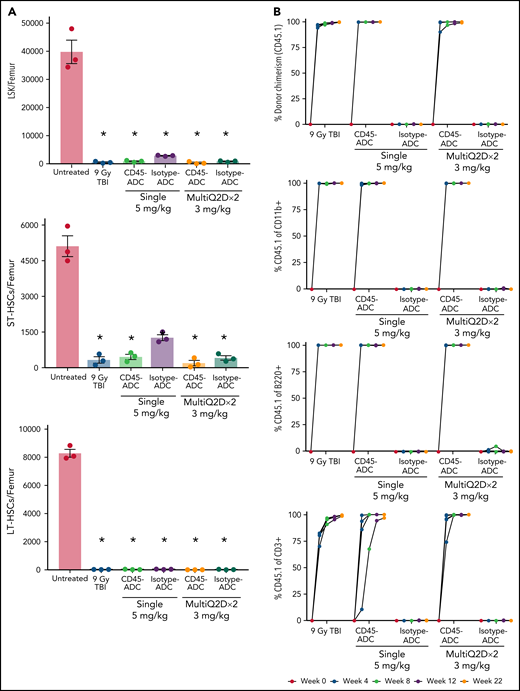

Conditioning of recipients with a higher dose of CD45-ADC can enable complete engraftment of donor cells as a single agent. B6 (H-2b, CD45.2) recipients (16- to 18-week-old female) were conditioned with a single dose of 5 mg/kg isotype-ADC or CD45-ADC on d-3, or multidose Q2D × 2 of 3 mg/kg isotype-ADC or CD45-ADC on d-5 and d-3, or 900 cGy (9 Gy) TBI on d-1, and transplanted with 40 × 106 CByJ.SJL(B6)-Ptprca/J (H-2d, CD45.1) BM on d0. Mice in multidose groups were transfused at 24 hours and 6 days post second dose with 300 μl of packed RBCs from B6 mice. One mouse from the CD45-ADC multidose group and 2 mice from the isotype-ADC multidose group died by 3 weeks posttransplant. (A) Depletion of BM hematopoietic progenitor cells (LSKs) and HSCs were assessed at d0 by flow cytometry (n = 3 mice per group). Nontreated mice served as control. Data represent mean ± SEM. *P < .05 vs nontreated mice via 1-way ANOVA posthoc Dunnett’s multiple comparisons test. (B) Engraftment of donor cells (H-2d, CD45.1) and lineage subsets in the PB of transplanted mice were assessed by flow cytometry. Each individual line represents a single mouse tracked over 22 weeks. (C) Peripheral RBC counts as a time-course posttransplant. Each individual line represents a single mouse tracked over 22 weeks, as indicated. (D) Overall donor (H-2d, CD45.1) chimerism and donor-derived hematopoietic progenitor cells (LSKs) and HSCs in BM of transplanted recipients were analyzed at 22 weeks posttransplant by flow cytometry. Data represent mean ± SEM (n = 5 mice per group). *P < .05 vs 9 Gy TBI via 1-way ANOVA posthoc Dunnett’s multiple comparisons test. (A-D), Experiments were performed twice, and data shown are from 1 representative experiment.
Conditioning of recipients with a higher dose of CD45-ADC can enable complete engraftment of donor cells as a single agent. B6 (H-2b, CD45.2) recipients (16- to 18-week-old female) were conditioned with a single dose of 5 mg/kg isotype-ADC or CD45-ADC on d-3, or multidose Q2D × 2 of 3 mg/kg isotype-ADC or CD45-ADC on d-5 and d-3, or 900 cGy (9 Gy) TBI on d-1, and transplanted with 40 × 106 CByJ.SJL(B6)-Ptprca/J (H-2d, CD45.1) BM on d0. Mice in multidose groups were transfused at 24 hours and 6 days post second dose with 300 μl of packed RBCs from B6 mice. One mouse from the CD45-ADC multidose group and 2 mice from the isotype-ADC multidose group died by 3 weeks posttransplant. (A) Depletion of BM hematopoietic progenitor cells (LSKs) and HSCs were assessed at d0 by flow cytometry (n = 3 mice per group). Nontreated mice served as control. Data represent mean ± SEM. *P < .05 vs nontreated mice via 1-way ANOVA posthoc Dunnett’s multiple comparisons test. (B) Engraftment of donor cells (H-2d, CD45.1) and lineage subsets in the PB of transplanted mice were assessed by flow cytometry. Each individual line represents a single mouse tracked over 22 weeks. (C) Peripheral RBC counts as a time-course posttransplant. Each individual line represents a single mouse tracked over 22 weeks, as indicated. (D) Overall donor (H-2d, CD45.1) chimerism and donor-derived hematopoietic progenitor cells (LSKs) and HSCs in BM of transplanted recipients were analyzed at 22 weeks posttransplant by flow cytometry. Data represent mean ± SEM (n = 5 mice per group). *P < .05 vs 9 Gy TBI via 1-way ANOVA posthoc Dunnett’s multiple comparisons test. (A-D), Experiments were performed twice, and data shown are from 1 representative experiment.
In a separate experiment using the BALB/c>B6 model, we directly compared the engraftment potential of CD45-ADC alone or combined with 50 cGy TBI. Depletion of HSCs, LSKs, and immune cell subsets in BM was analyzed on d0 (supplemental Figure S11A,B). PB analysis at 8 weeks showed a significant increase in the frequency of mice with >10% alloengraftment (43% vs 100%) and mean donor chimerism (21% vs 77%; P = .002) when the pretransplant CD45-ADC single dose was increased from 3 mg/kg to 5 mg/kg (supplemental Figure S11C). Notably, anti-CD40L mAb and 50 cGy TBI added to CD45-ADC (3 mg/kg) resulted in >87% mean donor PB chimerism in 100% of recipients, significantly higher (P < .0001) than in cohort not given anti-CD40L mAb. In ADC-conditioned mismatch recipients, clinical signs of GVHD were absent, and d62 histologic analysis (3 mice per group) in GVHD organs (spleen, lung, liver, colon, ileum) had scores49 ≤1 with 2 exceptions: 1 of 3 mice given CD45-ADC (5 mg/kg) had grade 2 ileum and 1 of 3 mice given isotype-ADC (5 mg/kg) had grade 4 ileum. Evidence of potential PBD-related cardiotoxicity was absent by histology (data not shown). Also, recipients in all groups remained healthy with almost 100% survival through the observation period following allogeneic BMT (supplemental Figure S11D,E). These results suggest that higher single doses of CD45-ADC can effectively condition recipients for allogeneic engraftment and are fully lymphoablative and myeloablative.
Discussion
Here we demonstrated the efficacy of CD45-ADC as a conditioning strategy for multilineage donor engraftment in 3 different mouse transplant models. CD45-ADC (3 mg/kg) mediated complete depletion of HSPCs in BM and hematopoietic cells and mature lymphocytes in peripheral organs, resulting in rapid engraftment in both congenic and minor mismatch transplant models as a single agent. CD45-ADC given as a primary agent resulted in stable mixed chimerism of fully allogeneic donor BM when combined with very low dose TBI (50 cGy) and tolerogenic anti-CD40L mAb. Adding JAK1/2 or 3 inhibitors further increased the degree of alloengraftment observed. CD45-ADC given at a ∼1.7- to 2-fold higher dose than used for multimodal therapy regimen provided for complete lymphoablation and enabled long-term stable allogeneic donor chimerism (>90%) as a single agent.
A saporin-based anti-CD117-ADC targeting CD117 (c-kit) receptor expressed on HSCs resulted in ∼50% engraftment of fully allogeneic donor cells.50,51 Promising results have been reported in a xenograft mouse model, nonhuman primates, and autologous gene therapy utilizing other CD117-ADC or CD117 mAb conditioning approaches.52-54 Although recipient HSPC depletion by unconjugated anti-CD117 mAb facilitates donor HSC engraftment by opening BM niches,13 host mature lymphoid cells are not depleted, setting up the possibility of donor graft rejection or host niche and cytokine competition with newly developed donor lymphocytes. A more effective and safer conditioning agent with improved immunodepletion was desirable. Because CD45-ADC permitted donor BM cells replacement of host hematopoietic and immune cells and complete alloengraftment as both a single agent or combined with low TBI dose (RIC) in preclinical models described here, CD45-ADC filled this gap and is a candidate for targeted conditioning for gene-modified autologous or allogeneic grafts.
Unconjugated anti-CD45 mAb can deplete lymphocytes but requires a high TBI dose or the addition of chemotherapy to achieve sufficient alloengraftment.55,56 Lytic anti-CD45 mAb or anti-CD45 mAb radioimmunotherapy in conjunction with standard chemotherapy have been tested in the clinic for conditioning in patients with hematological malignancies with the dual benefit that hematological malignancies are CD45+.57-59 While radiolabeled anti-CD45 mAbs have been shown to facilitate alloengraftment, logistical considerations and concerns for BM and organ toxicity in allo-HSCT warrant consideration.60,61
Alternatively, anti-CD45 mAb conjugated to saporin as a CD45-ADC showed efficient HSC depletion as a single agent in syngeneic immunocompetent mouse model17 or FA model,19 enabling stable multilineage engraftment with minimal organ toxicity. Although CD45-saporin and 200 cGy TBI conditioning resulted in efficient multilineage engraftment in Rag1 mutant mice given syngeneic donor BM that has no immune-mediated graft resistance,62 the more difficult allogeneic HSCT engraftment has yet to be reported.
A long-term goal in allo-HSCT is to sustain donor grafts without immune suppression. Recent findings suggest that recipient treatment with immunosuppressive or immunomodulatory reagents (eg, anti-CD40L mAb) and approaches to reduce host HSCs permit durable alloengraftment in an MHC-mismatch transplant model accompanied by donor-specific tissue tolerance.50,63 In an MHC-mismatch transplant model, we demonstrated CD45-ADC (3 mg/kg) could act as a primary agent when combined with low-dose TBI (50 cGy) and anti-CD40L mAb in a RIC regimen resulting in stable, long-term mixed chimerism (48%). Adding a clinically relevant pharmacological agent (rapamycin) or a JAK inhibitor (ruxolitinib, barcitinib, tofacitinib) significantly augmented alloengraftment to 78%, the latter associated with increased frequencies of donor-derived CD4 Tregs and decreased frequencies of effector/memory T cells. Rapamycin has a synergistic effect with anti-CD40L mAb for inducing allogeneic donor engraftment,39 posttransplant cyclophosphamide causes CD8 T-cell dysfunction and induces suppression,64 and JAK inhibitor can inhibit T- and myeloid-cell but not B-cell65 function. All these conditions favor donor cells and allogeneic engraftment. We experienced differences in CD45-ADC-mediated mean donor engraftment between experiments, most likely due to batch-to-batch variability in drug-antibody ratio as developed by the manufacturer.
Most strikingly, we observed that conditioning fully allogeneic recipients with a single dose of 5 mg/kg or 2 doses of 3 mg/kg CD45-ADC was sufficient for stable multilineage donor engraftment (>90%) without irradiation or additional immunosuppression, comparable to lethal TBI. To our knowledge, this is the first report of high, durable mean chimerism levels achieved using an ADC as a single agent in a major mismatch transplant model. Taken together, we have shown that CD45-ADC is a potent immunotoxin that can efficiently deplete HSPCs and lymphoid cells, allowing robust donor engraftment and immune reconstitution in 3 distinct preclinical mouse models. The shorter half-life along with superior HSC-depletion property makes CD45-ADC highly desirable for HSCT conditioning strategy with exciting clinical translational potential.
Acknowledgments
The authors thank members of B.R.B.’s laboratory and Magenta Therapeutics for technical assistance.
This work was supported in part by National Institutes of Health grants R01 HL147324 and R37 AI34495 and Kids' First Fund (to B.R.B.).
Authorship
Contribution: A.S. designed and performed experiments, analyzed data, and wrote the manuscript; S.H., T.L., K.H., N.C., L.L., P.B., R.P., and G.O.G. designed and performed experiments, discussed data, and edited the manuscript; R.P., G.O.G., J.P., and L.M.O. provided ADC reagents; M.J.R. and A.P.-M. provided data and edited the manuscript; J.P., M.L.M., J.E.W., H.-P.K., and L.M.O. discussed data and edited the manuscript; and B.R.B. designed experiments, discussed data, and edited the manuscript.
Conflict-of-interest disclosure: B.R.B. receives remuneration as an advisor to Magenta Therapeutics and BlueRock Therapeutics, research funding from BlueRock Therapeutics, Rheos Medicines, Childrens’ Cancer Research Fund, and Kids' First Fund, and is a cofounder of Tmunity Therapeutics. J.E.W. was an advisor at Magenta Therapeutics. S.H., T.L., K.H., N.C., L.L., P.B., R.P., G.O.G., J.P., and L.M.O. were employees and equity holders of Magenta Therapeutics at the time the work was completed.
Correspondence: Bruce R. Blazar, Division of Blood & Marrow Transplant & Cellular Therapy, Masonic Cancer Center and Department of Pediatrics, University of Minnesota, 420 Delaware St SE, Minneapolis, MN 55455; e-mail: blaza001@umn.edu.
Requests for data sharing may be submitted to Bruce R. Blazar (blaza001@umn.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.