

Abstract
Increased expression levels of miR-181 family members have been shown to be associated with favorable outcome in patients with cytogenetically normal acute myeloid leukemia. Here we show that increased expression of miR-181a and miR-181b is also significantly (P < .05; Cox regression) associated with favorable overall survival in cytogenetically abnormal AML (CA-AML) patients. We further show that up-regulation of a gene signature composed of 4 potential miR-181 targets (including HOXA7, HOXA9, HOXA11, and PBX3), associated with down-regulation of miR-181 family members, is an independent predictor of adverse overall survival on multivariable testing in analysis of 183 CA-AML patients. The independent prognostic impact of this 4-homeobox-gene signature was confirmed in a validation set of 271 CA-AML patients. Furthermore, our in vitro and in vivo studies indicated that ectopic expression of miR-181b significantly promoted apoptosis and inhibited viability/proliferation of leukemic cells and delayed leukemogenesis; such effects could be reversed by forced expression of PBX3. Thus, the up-regulation of the 4 homeobox genes resulting from the down-regulation of miR-181 family members probably contribute to the poor prognosis of patients with nonfavorable CA-AML. Restoring expression of miR-181b and/or targeting the HOXA/PBX3 pathways may provide new strategies to improve survival substantially.
Introduction
MicroRNAs (miRNAs) are an abundant class of small noncoding RNAs of approximately 22 nucleotides (nt) that posttranscriptionally regulate expression of target genes via cleavage/degradation and/or translation repression of the encoded mRNAs.1 Altered miRNA expression has been associated with various types of cancers,1-3 including acute myeloid leukemia (AML),4,5 and changes in the expression of certain miRNAs has been shown to be associated with prognosis in AML patients.6-9 The miR-181 family was reported first as an important regulator of B-cell development10 and T cell–related immune response.11 Subsequently, miR-181 family members were identified as either tumor suppressors or oncogenes in various cancers depending on tissue types.12-14 Increased expression levels of miR-181 family members have been shown to be associated with favorable outcome in patients with cytogenetically normal AML (CN-AML),7-9 whereas the prognostic impact of miR-181 in cytogenetically abnormal AML (CA-AML) remains to be investigated.
Homeobox genes encode transcription factors that contain homeodomain(s), which can bind DNA in a sequence-specific manner. Homeotic or Hox genes are highly conserved in most animals, and in mammals, there are 4 (ie, A, B, C, and D) clusters and 39 Hox genes.15,16 Hox proteins can form heterodimers or heterotrimers with members of the 3-amino-acid loop extension family of cofactors, including PBX and MEIS proteins to regulate the transcription of downstream targets directly.15-18 The aberrant overexpression of these homeobox genes (particularly HOXA5, HOXA7, HOXA9, HOXA10, MEIS1, and PBX3) has been frequently reported previously in various subtypes of intermediate or poor prognosis CA-AML, including those bearing MLL (mixed lineage leukemia) rearrangements19-27 or trisomy 8.28 However, although the expression profile of HOXA7, HOXA9, or PBX3 has been implied to be associated with poor survival of AML patients in several individual studies,20,29-31 their prognostic impact has not been tested in multivariable models or been validated by further large-scale independent studies. In addition, the molecular mechanism(s) underlying their differential expression among various subtypes of CA-AML with different prognosis remains unclear.
In the present study, we tested the prognostic impact of all 4 members of the miR-181 family (ie, miR-181a, miR-181b, miR-181c, and miR-181d) in 2 sets of patients with CA-AML. We show first that the increased expression level of miR-181a or miR-181b is significantly (P < .05) associated with longer overall survival (OS) in both patient sets we tested. Among the putative target genes that exhibited a significantly (P < .05) inverse correlation of expression with miR-181b in a patient set with both miRNA and mRNA expression profiles available, we identified a signature composed of 4 homeobox genes (including HOXA7, HOXA9, HOXA11, and PBX3) whose increased expression level was significantly (P < .05) associated with shorter OS in 183 CA-AML patients of 3 independent sets in both univariable and multivariable model analyses. The prognostic impact of this 4-gene signature was further confirmed in a validation set of 271 CA-AML patients. We further used both in vitro and in vivo models to validate the biologic function of miR-181a/miR-181b and one of its target genes (ie, PBX3) in a subtype of CA-AML carrying MLL rearrangements.
Methods
Additional information about methods is provided in supplemental Methods (available on the Blood Web site; the Supplemental Materials link at the top of the online article). Supplemental Figure 1 illustrates the entire study design.
Patient samples
All of the CA-AML patient samples were obtained before treatment and with informed consent at the corresponding hospitals in accordance with the Declaration of Helsinki; study protocols were approved by the respective institutional review boards. All patients were treated according to the protocols of the corresponding institutes/hospitals (supplemental Methods). The samples were collected from the University of Chicago Hospital (USA-set I and USA-set II), the Cancer and Leukemia Group B (CALGB; USA-set III), the AML Study Group (Germany-set I),20 and the Dutch-Belgian Hematology-Oncology Cooperative group (Netherlands-set I),32 respectively. The clinical and molecular characteristics of these patients are shown in Table 1.
Clinical and molecular characteristics of CA-AML patients
| Characteristic | miRNA array sets (n = 86) | mRNA array sets (n = 454) | ||||
|---|---|---|---|---|---|---|
| Training set (n = 183) | Validation set (n = 271) | |||||
| USA-set-I (n = 33)33 | USA-set-II (n = 53) | USA-set-II-35S (n = 35)* | USA-set-III (n = 87) | Germany-set-I20 (n = 61) | Netherlands-set-I32 (n = 271) | |
| Sex, no. (%) | ||||||
| Male | 11 (33) | 27 (51) | 20 (57) | 46 (53) | 35 (57) | 138 (51) |
| Female | 22 (67) | 26 (49) | 15 (43) | 41 (47) | 26 (43) | 133 (49) |
| Age, y | ||||||
| Median | 41 | 56 | 44 | 41 | 48 | 43 |
| Range | 5-87 | 0-84 | 0-84 | 0-74 | 19-74 | 15-74 |
| OS, y | ||||||
| Median | 3 | 2 | 2 | 6 | 1 | 2 |
| Range | 0-25 | 0-16 | 0-16 | 0-13 | 0-4 | 0-19 |
| White cell count, 1 × 10−3/mm3 | ||||||
| Median | NA | 30 | 24 | 20 | 29 | 21 |
| Range | NA | 0.5-182 | 0.8-182 | 0.3-206 | 1-189 | 1.3-510 |
| Blast cell count, % | ||||||
| Median | 95 | 44 | 35 | 65 | NA | 62 |
| Range | 10-100 | 0-94 | 0-88 | 3-95 | NA | 0-98 |
| Platelet count, 1 × 10−3/mm3 | ||||||
| Median | NA | 48 | 47 | 30 | NA | 50 |
| Range | NA | 13-263 | 16-164 | 6-138 | NA | 3-998 |
| FAB type, no. (%) | ||||||
| M0 | 0 | 1 (2) | 1 (3) | 0 | 2 (3) | 13 (5) |
| M1 | 2 (6) | 4 (8) | 2 (6) | 0 | 6 (10) | 39 (14) |
| M2 | 5 (15) | 8 (15) | 6 (17) | 29 (33) | 14 (23) | 71 (26) |
| M3 | 8 (24) | 9 (17) | 8 (23) | 31 (36) | 9 (15) | 18 (7) |
| M4 | 8 (24) | 11 (21) | 8 (23) | 23 (26) | 18 (30) | 58 (21) |
| M5 | 8 (24) | 7 (13) | 5 (14) | 4 (5) | 8 (13) | 49 (18) |
| M6 | 0 | 0 | 0 | 0 | 0 | 4 (1) |
| Not determined | 2 (6) | 13 (24) | 5 (14) | 0 | 4 (7) | 19 (7) |
| Cytogenetic abnormalities, no. (%) | ||||||
| t(15;17) | 7 (21) | 9 (17) | 8 (23) | 31 (36) | 9 (15) | 19 (7) |
| t(8;21) | 7 (21) | 9 (17) | 5 (14) | 29 (33) | 9 (15) | 37 (14) |
| inv(16)/t(16;16) | 4 (12) | 9 (17) | 7 (20) | 22 (25) | 15 (25) | 38 (14) |
| t(11q23) | 15 (45) | 10 (19) | 7 (20) | 5 (6) | 7 (11) | 16 (6) |
| +8 | 0 | 10 (19) | 5 (14) | 0 | 2 (3) | 37 (14) |
| −7/del(7q) | 0 | 2 (4) | 1 (3) | 0 | 10 (16) | 35 (13) |
| −3/inv(3)/t(3;3) | 0 | 0 | 0 | 0 | 0 | 20 (7) |
| t(6;9) | 0 | 0 | 0 | 0 | 0 | 5 (2) |
| t(9;22) | 0 | 0 | 0 | 0 | 0 | 5 (2) |
| Complex karyotype | 0 | 0 | 0 | 0 | 4 (7) | 31 (11) |
| Other abnormal karyotype | 0 | 4 (8) | 2 (6) | 0 | 5 (8) | 80 (30) |
| Not determined | 0 | 0 | 0 | 0 | 0 | 0 |
| Molecular abnormalities, no. (%) | ||||||
| FLT3-ITD or -TKD | NA | NA | NA | NA | 12 (20) | 63 (23) |
| N- or K-RAS | NA | NA | NA | NA | NA | 31 (11) |
| NPM1 | NA | NA | NA | NA | NA | 24 (9) |
| CEBPA | NA | NA | NA | NA | NA | 11 (4) |
| MLL-PTD | NA | NA | NA | NA | 2 (3) | NA |
| Characteristic | miRNA array sets (n = 86) | mRNA array sets (n = 454) | ||||
|---|---|---|---|---|---|---|
| Training set (n = 183) | Validation set (n = 271) | |||||
| USA-set-I (n = 33)33 | USA-set-II (n = 53) | USA-set-II-35S (n = 35)* | USA-set-III (n = 87) | Germany-set-I20 (n = 61) | Netherlands-set-I32 (n = 271) | |
| Sex, no. (%) | ||||||
| Male | 11 (33) | 27 (51) | 20 (57) | 46 (53) | 35 (57) | 138 (51) |
| Female | 22 (67) | 26 (49) | 15 (43) | 41 (47) | 26 (43) | 133 (49) |
| Age, y | ||||||
| Median | 41 | 56 | 44 | 41 | 48 | 43 |
| Range | 5-87 | 0-84 | 0-84 | 0-74 | 19-74 | 15-74 |
| OS, y | ||||||
| Median | 3 | 2 | 2 | 6 | 1 | 2 |
| Range | 0-25 | 0-16 | 0-16 | 0-13 | 0-4 | 0-19 |
| White cell count, 1 × 10−3/mm3 | ||||||
| Median | NA | 30 | 24 | 20 | 29 | 21 |
| Range | NA | 0.5-182 | 0.8-182 | 0.3-206 | 1-189 | 1.3-510 |
| Blast cell count, % | ||||||
| Median | 95 | 44 | 35 | 65 | NA | 62 |
| Range | 10-100 | 0-94 | 0-88 | 3-95 | NA | 0-98 |
| Platelet count, 1 × 10−3/mm3 | ||||||
| Median | NA | 48 | 47 | 30 | NA | 50 |
| Range | NA | 13-263 | 16-164 | 6-138 | NA | 3-998 |
| FAB type, no. (%) | ||||||
| M0 | 0 | 1 (2) | 1 (3) | 0 | 2 (3) | 13 (5) |
| M1 | 2 (6) | 4 (8) | 2 (6) | 0 | 6 (10) | 39 (14) |
| M2 | 5 (15) | 8 (15) | 6 (17) | 29 (33) | 14 (23) | 71 (26) |
| M3 | 8 (24) | 9 (17) | 8 (23) | 31 (36) | 9 (15) | 18 (7) |
| M4 | 8 (24) | 11 (21) | 8 (23) | 23 (26) | 18 (30) | 58 (21) |
| M5 | 8 (24) | 7 (13) | 5 (14) | 4 (5) | 8 (13) | 49 (18) |
| M6 | 0 | 0 | 0 | 0 | 0 | 4 (1) |
| Not determined | 2 (6) | 13 (24) | 5 (14) | 0 | 4 (7) | 19 (7) |
| Cytogenetic abnormalities, no. (%) | ||||||
| t(15;17) | 7 (21) | 9 (17) | 8 (23) | 31 (36) | 9 (15) | 19 (7) |
| t(8;21) | 7 (21) | 9 (17) | 5 (14) | 29 (33) | 9 (15) | 37 (14) |
| inv(16)/t(16;16) | 4 (12) | 9 (17) | 7 (20) | 22 (25) | 15 (25) | 38 (14) |
| t(11q23) | 15 (45) | 10 (19) | 7 (20) | 5 (6) | 7 (11) | 16 (6) |
| +8 | 0 | 10 (19) | 5 (14) | 0 | 2 (3) | 37 (14) |
| −7/del(7q) | 0 | 2 (4) | 1 (3) | 0 | 10 (16) | 35 (13) |
| −3/inv(3)/t(3;3) | 0 | 0 | 0 | 0 | 0 | 20 (7) |
| t(6;9) | 0 | 0 | 0 | 0 | 0 | 5 (2) |
| t(9;22) | 0 | 0 | 0 | 0 | 0 | 5 (2) |
| Complex karyotype | 0 | 0 | 0 | 0 | 4 (7) | 31 (11) |
| Other abnormal karyotype | 0 | 4 (8) | 2 (6) | 0 | 5 (8) | 80 (30) |
| Not determined | 0 | 0 | 0 | 0 | 0 | 0 |
| Molecular abnormalities, no. (%) | ||||||
| FLT3-ITD or -TKD | NA | NA | NA | NA | 12 (20) | 63 (23) |
| N- or K-RAS | NA | NA | NA | NA | NA | 31 (11) |
| NPM1 | NA | NA | NA | NA | NA | 24 (9) |
| CEBPA | NA | NA | NA | NA | NA | 11 (4) |
| MLL-PTD | NA | NA | NA | NA | 2 (3) | NA |
NA indicates information not available; and FAB, French-American-British.
USA-set-II-35S patients were included in both miRNA and mRNA microarrays, and this set is a part of the USA-set-II.
RNA preparation
Blasts and mononuclear cells were purified by use of NycoPrep 1.077A (Axis-Shield) according to the manufacturer's manual, and then total RNA was isolated by use of miRNeasy Mini Kit (QIAGEN; for USA set I,33 -II, and -III), Trizol reagent (Invitrogen; for Netherlands set I),32 or guanidinium isothiocyanate followed by cesium chloride-gradient purification (for Germany set I).20
miRNA and mRNA expression profiling assays
The miRNA expression profiling assays of USA-set I and USA-set II were conducted by use of a bead-based method33,34 and Exiqon miRCURY LNA arrays Version 10.0 (covering 757 human miRNAs; Exiqon), respectively.
The mRNA microarrays of Germany-set I20 and Netherlands-set I32 were conducted by use of Stanford cDNA arrays and Affymetrix U133 Plus Version 2.0 arrays (Affymetirx), respectively. Two novel datasets, including USA-set II-35S and USA-set III, were analyzed by use of Agilent's custom-design microarrays (Agilent Technologies) and Affymetrix GeneChip Human Exon Version 1.0 ST arrays, respectively. Data analyses are described in supplemental data.
All the microarray data have been deposited in the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo), and the accession numbers include GSE425, GSE14468, GSE30285, and GSE30258.
Statistical analyses
OS was measured from the date the patient was enrolled onto the study until the date of death, and patients alive at last follow-up were censored. Survival was estimated according to the method of Kaplan and Meier. The log-rank test was used to assess statistical significance. Cox regression was used to assess the association of a given variable with OS. For a given gene signature composed of 2 or more genes, a compound covariate35 was derived for each patient sample by computing a linear combination of expression values of all genes in that signature. Multivariable testing was performed using Cox proportional hazards models. P values < .05 were considered statistically significant.
Identification of the 9 poor survival–associated miR-181 potential target genes via a meta-analysis
In each dataset of USA-set II-35S, USA-set III, and Germany-set I, Cox regression was used to estimate the prognostication of OS for every gene. Then a meta-analysis was conducted on the Cox regression P values of OS from the 3 CA-AML training sets using the Stouffer method36 :
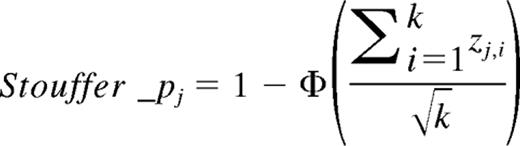
where Zi = Φ−1(1 − pi), pi is a P value for the ith study of k studies in total, and Φ and Φ−1 denote the standard normal cumulative distribution function and its inverse.
A total of 9 (Table 2) of the 159 potential target genes of miR-181 (supplemental Table 2) remained as significant predictors after FDR adjustment for multiple comparisons of meta-analysis P values across the 3 training sets (FDR < 0.05; R packages stats, Hommel adjustment37 ) and therefore were selected for further studies.
Nine candidate genes of miR-181 that are significantly associated with poor OS
| Gene | Training sets (n = 183) | Validation set (n = 271) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coefficient average | FDR | USA-set-II-35S (n = 35) | USA-set-III (n = 87) | Germany-set-I (n = 61) | Netherlands-set-I (n = 271) | |||||||||
| HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | |||
| HOXA7 | 0.477 | 0.03 | 2.09 | 1.36-3.19 | < .001 | 1.22 | 0.80-1.85 | .36 | 1.65 | 1.15-2.36 | .01 | 1.34 | 1.14-1.57 | < .001 |
| HOXA9 | 0.483 | 0.02 | 2.12 | 1.38-3.25 | < .001 | 1.18 | 0.79-1.76 | .42 | 1.70 | 1.16-2.47 | .01 | 1.41 | 1.21-1.64 | < .001 |
| HOXA11 | 0.433 | 0.03 | 1.47 | 1.06-2.04 | .02 | 1.70 | 1.18-2.43 | .004 | 1.47 | 0.83-2.59 | .18 | 1.00 | 0.85-1.17 | .99 |
| PBX3 | 0.546 | 0.02 | 2.06 | 1.42-2.97 | < .001 | 1.61 | 1.19-2.18 | .002 | 1.55 | 1.06-2.27 | .02 | 1.17 | 1.00-1.36 | .04 |
| MRPS18B | 0.407 | 0.03 | 1.53 | 1.06-2.23 | .02 | 1.74 | 1.27-2.39 | < .001 | 1.27 | 0.81-2.00 | .29 | 1.04 | 0.89-1.22 | .59 |
| PLA2G4A | 0.531 | < .001 | 1.79 | 1.21-2.64 | .003 | 1.60 | 1.16-2.22 | .005 | 1.72 | 1.11-2.65 | .01 | 1.31 | 1.11-1.54 | .002 |
| TRPS1 | 0.395 | 0.05 | 1.53 | 1.08-2.18 | .02 | 1.41 | 1.02-1.95 | .04 | 1.52 | 1.02-2.25 | .04 | 1.37 | 1.18-1.59 | < .001 |
| GNS | 0.258 | < .001 | 1.71 | 1.02-2.88 | .04 | 1.69 | 1.21-2.36 | .002 | 0.75 | 0.49-1.15 | .18 | 1.02 | 0.85-1.21 | .87 |
| PGPEP1 | 0.488 | 0.03 | 2.68 | 1.38-5.22 | .004 | 1.30 | 0.93-1.80 | .12 | 1.25 | 0.87-1.78 | .23 | 1.14 | 0.99-1.32 | .07 |
| Gene | Training sets (n = 183) | Validation set (n = 271) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coefficient average | FDR | USA-set-II-35S (n = 35) | USA-set-III (n = 87) | Germany-set-I (n = 61) | Netherlands-set-I (n = 271) | |||||||||
| HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | |||
| HOXA7 | 0.477 | 0.03 | 2.09 | 1.36-3.19 | < .001 | 1.22 | 0.80-1.85 | .36 | 1.65 | 1.15-2.36 | .01 | 1.34 | 1.14-1.57 | < .001 |
| HOXA9 | 0.483 | 0.02 | 2.12 | 1.38-3.25 | < .001 | 1.18 | 0.79-1.76 | .42 | 1.70 | 1.16-2.47 | .01 | 1.41 | 1.21-1.64 | < .001 |
| HOXA11 | 0.433 | 0.03 | 1.47 | 1.06-2.04 | .02 | 1.70 | 1.18-2.43 | .004 | 1.47 | 0.83-2.59 | .18 | 1.00 | 0.85-1.17 | .99 |
| PBX3 | 0.546 | 0.02 | 2.06 | 1.42-2.97 | < .001 | 1.61 | 1.19-2.18 | .002 | 1.55 | 1.06-2.27 | .02 | 1.17 | 1.00-1.36 | .04 |
| MRPS18B | 0.407 | 0.03 | 1.53 | 1.06-2.23 | .02 | 1.74 | 1.27-2.39 | < .001 | 1.27 | 0.81-2.00 | .29 | 1.04 | 0.89-1.22 | .59 |
| PLA2G4A | 0.531 | < .001 | 1.79 | 1.21-2.64 | .003 | 1.60 | 1.16-2.22 | .005 | 1.72 | 1.11-2.65 | .01 | 1.31 | 1.11-1.54 | .002 |
| TRPS1 | 0.395 | 0.05 | 1.53 | 1.08-2.18 | .02 | 1.41 | 1.02-1.95 | .04 | 1.52 | 1.02-2.25 | .04 | 1.37 | 1.18-1.59 | < .001 |
| GNS | 0.258 | < .001 | 1.71 | 1.02-2.88 | .04 | 1.69 | 1.21-2.36 | .002 | 0.75 | 0.49-1.15 | .18 | 1.02 | 0.85-1.21 | .87 |
| PGPEP1 | 0.488 | 0.03 | 2.68 | 1.38-5.22 | .004 | 1.30 | 0.93-1.80 | .12 | 1.25 | 0.87-1.78 | .23 | 1.14 | 0.99-1.32 | .07 |
Coefficient average indicates the average correlation coefficient from the 3 training sets.
Derivation of the 9-gene and 4-gene signatures
The derivation was performed as described previously7 with some modifications. As shown in Table 2, we identified 9 genes whose expression was significantly associated (P < .05) with poor OS in at least 2 of the 3 training sets (ie, USA-set II-35S, USA-set III, and Germany-set I; Table 2) and were potential targets of miR-181 that exhibited a significantly (P < .05) inverse correlation of expression with miR-181b (supplemental Table 2). Among them, 4 genes belong to the homeobox gene superfamily, including HOXA7, HOXA9, HOXA11, and PBX3. Then, a compound covariate35 was derived for each patient sample by computing a linear combination of expression values of the whole set of the 9 genes or a subset of the 4 homeobox genes. The value of the compound covariate for patient i was ci = Σ wj xij, where xij is the log-transformed expression value for probe set j in patient i and wj is the weight assigned to probe set j (here wj was set equal to the average Cox regression correlation coefficient from the 3 training sets for each gene/probe in the signature; Table 2). The sum was overall 9 or 4 genes. Then, the derived sum values in a given patient set were dichotomized on the basis of the median value to separate patients into 2 groups. Kaplan-Meier curves were made to estimate actuarial probabilities of OS, and the log-rank test was used to evaluate the significance.
Retroviral constructs
Precursor sequence of miR-181a and miR-181b were PCR amplified from human normal bone marrow mononuclear cells and were then cloned into MSCVneo (Clontech) or MSCV-PIG vector that contains a PGK-puromycin-IRES-GFP (PIG),3 and labeled as MSCVneo-miR-181a or miR-181b, or MSCV-PIG-miR-181a or miR-181b. MSCVneo-MLL-AF9 is a kind gift from Dr Scott Armstrong. Coding region (CDS) of PBX3 that has no target site for miR-181a/miR-181b and, thus, would not be negatively regulated by miR-181a/miR-181b, was synthesized by GenScript USA and was then cloned into MSCV-PIG and labeled as MSCV-PIG-PBX3-CDS. All inserts have been confirmed by sequencing. The primers are shown in supplemental Table 5.
Luciferase reporter and mutagenesis assays
Cell apoptosis, viability, and proliferation assays
miR-181a (MSCVneo-miR-181a or miR-181a mimics), miR-181b (MSCVneo-miR-181b or miR-181b mimics), or miRNA control (MSCVneo empty vector or scrambled oligos) were cotransfected with MSCV-PIG-PBX3-CDS or MSCV-PIG empty vector into MONOMAC-6/t(9,11), THP-1/t(9,11), or KOCL-48/t(4,11) cells using the Amaxa Nucleofector Technology (Lonza Walkersville) as described previously.33 The miRNA mimics and scrambled oligos were ordered from Dharmacon RNAi Technologies. Forty-eight hours later, apoptosis was assessed through analyzing caspase-3 and caspase-7 activation using ApoONE Homogeneous Caspase 3/7 Assay (Promega) or through checking annexin V expression by flow cytometry; cell viability was assessed through analyzing metabolic activity of the cells using CellTiter-Blue Reagent (Promega) following the corresponding manufacturer's manuals. To monitor cell proliferation/growth, MONOMAC-6 cells were seeded into 96-well plate with 0.02 million cells per well in triplicates 24 hours after transfection with various plasmids, and then cell numbers were counted every day for 5 days.
Retrovirus preparation
In vitro colony-forming selection
To select double-transduction-positive cells, we conducted colony-forming assays first.
For primary bone marrow transplantation (BMT) assays, hematopoietic progenitor (ie, lineage negative, Lin−) cells were obtained from a cohort of 4- to 6-week-old B6.SJL (CD45.1) mice 5 days after 5-fluorouracil treatment (150 mg/kg) using the Mouse Lineage Cell Depletion Kit (Miltenyi Biotec), and were then cotransduced with MSCV-PIG + MSCVneo-MLL-AF9 (negative control), MSCV-PIG-miR-181a + MSCVneo-MLL-AF9 (MA9 + miR181a), and MSCV-PIG-miR-181b + MSCVneo-MLL-AF9 (MA9 + miR181b), respectively, through “spinoculation.”33,38 Then, 4 aliquots of 1 × 104 of the transfected cells were plated into 4 35-mm Nunc Petri dishes in 1.1 mL of Methocult M3230 methylcellulose medium (StemCell Technologies) containing 10 ng/mL each of murine recombinant IL-3, IL-6, and GM-CSF and 30 ng/mL of murine recombinant stem cell factor (R&D Systems), along with 1.0 mg/mL of G418 (Invitrogen) and 2.5 μg/mL of puromycin (Sigma-Aldrich). Cultures were incubated at 37°C in a humidified atmosphere of 5% CO2 in air for 7 days. Then, colonies cells were collected and replated in methylcellulose dishes every 7 days with 1 × 104 cells as input for up to 4 passages.
Primary BMT assays and characterization of leukemia
Colony cells were collected from the colony-forming assays, and after PBS washing twice, the cells were transplanted via tail vein injection into lethally irradiated (960 cGy, 96 cGy/min, γ-rays) 8- to 10-week-old C57BL/6 (CD45.2) recipient mice. For each recipient mouse, a total of 1.5 × 106 (for primary BMT) donor cells and a radioprotective dose of whole bone marrow cells (1 × 106) freshly harvested from a C57BL/6 mouse were transplanted. The maintenance, monitoring, and endpoint treatment of the transplanted recipient mice are described in supplemental Methods.
Secondary BMT assays
For secondary BMT assays, MA9 and MA9 + miR181b leukemic bone marrow blast cells were collected from primary transplantation mice, and then retrovirally transduced with either MSCV-PIG (still labeled as MA9 and MA9 + miR181b, respectively) or MSCV-PIG-PBX3-CDS (labeled as MA9 + PBX3-CDS and MA9 + miR181b+PBX3-CDS, respectively) through “spinoculation.”33,38 The transduced cells were then plated in methylcellulose dishes for colony-forming for 7 days, except for the selection with puromycin alone. Colony cells were collected and washed with PBS, and then transplanted into sublethally irradiated (480 cGy) 8- to 10-week-old C57BL/6 (CD45.2) secondary recipient mice via tail vein injection, with 5 × 105 donor cells plus a radioprotective dose of whole bone marrow cells (1 × 106; freshly harvested from a C57BL/6 mouse) per secondary recipient mouse. The recipient mice were then maintained and monitored for leukemogenesis.
Flow cytometry
Cells from peripheral blood, BM, spleen, or liver were harvested for analysis of immunophenotypes. After washing with PBS, blocking nonspecific binding with affinity-purified anti–mouse CD16/32 (eBioscience), cells were stained at 4°C with various antibodies diluted in Flow Cytometry Staining Buffer (eBioscience) for 30 minutes. Subsequently, cells were washed with PBS and resuspended in IC Fixation Buffer (eBioscience) for flow cytometry analysis. Antibodies (eBioscience) used for our flow cytometric analysis include PE anti–mouse CD11b (12-0112; ie, Mac-1), PE anti–mouse Ly-6G (Gr-1, Gr1; 12-5931), PE anti–mouse/human CD45R (B220; 12-0452), allophycocyanin (APC) anti–mouse/human CD45R (B220; 17-0452), allophycocyanin anti–mouse CD4 (L3T4; 17-0041), allophycocyanin anti–mouse CD8 (β-subunit; CD8b, Ly-3; 17-0083), PE anti–mouse CD45.1 (12-0453), and allophycocyanin anti–mouse CD45.2 (17-0454).
Results
Increased expression of both miR-181a and miR-181b is significantly associated with favorable outcome in 2 independent sets of CA-AML patients
In our previous bead-based miRNA expression profiling assay, we observed that miR-181a, miR-181b, miR-181c, and miR-181d were all expressed at a higher level in the favorable prognosis subtypes of CA-AML carrying t(8;21), inv(16), or t(15;17) than in an intermediate- to poor-risk subtype of CA-AML harboring MLL rearrangements.33 We therefore speculated that increased expression of miR-181 family members might also be associated with favorable prognosis in CA-AML patients. Indeed, we found that increased expression of miR-181a and miR-181b was significantly (P < .05; Cox regression; supplemental Table 1; Figure 1A) associated with longer OS in a set of 33 untreated primary CA-AML patients (ie, USA-set I; Table 1) that were studied in our previous bead-based miRNA profiling assay.33 To validate this finding, we then analyzed an independent set of 53 CA-AML patients (USA-set II; Table 1) using Exiqon miRNA array platform. Patients expressing higher levels of miR-181a, miR-181b, or miR-181d had a significantly longer OS than those expressing lower levels (P < .05; Figure 1B; supplemental Table 1).

Increased expression of miR-181 family members is associated with favorable OS. (A) USA-set I (n = 33). (B) USA-set II (n = 53). The patients in each set were dichotomized into 2 groups based on the median value of expression signature of a given miR-181 family member. Kaplan-Meier curves were generated to depict outcomes. The P value was determined by log-rank test. | indicates censored. In USA-set II, miR-181c and miR-181d did not have reliable expression value in 1 patient sample.
Increased expression of miR-181 family members is associated with favorable OS. (A) USA-set I (n = 33). (B) USA-set II (n = 53). The patients in each set were dichotomized into 2 groups based on the median value of expression signature of a given miR-181 family member. Kaplan-Meier curves were generated to depict outcomes. The P value was determined by log-rank test. | indicates censored. In USA-set II, miR-181c and miR-181d did not have reliable expression value in 1 patient sample.
In USA-set I, we showed that increased expression of miR-181a or miR-181b was significantly (P < .05) associated with longer OS in univariable testing (supplemental Table 1), although only that of miR-181b remained significant (P < .05) on multivariable testing (Table 3), after adjusting for other variables that have a P less than .1 in the univariable analyses (supplemental Table 1). Similar results were observed in the validation set (ie, USA-set II; Table 3; supplemental Table 1), except for that increased expression of miR-181d also exhibited a significant (P < .05) association with longer OS in univariable testing in USA-set II (supplemental Table 1).
Multivariable analyses of OS of CA-AML patients
| Patient set | Variables | HR | 95% CI | P | |
|---|---|---|---|---|---|
| USA-set-I (n = 33) | miR-181a (high vs low) | 0.70 | 0.48-1.02 | .06 | |
| miR-181b (high vs low) | 0.65 | 0.43-0.98 | .04 | ||
| miR-181c (high vs low) | 0.79 | 0.57-1.09 | .15 | ||
| miR-181d (high vs low) | 0.74 | 0.49-1.11 | .15 | ||
| Age, years ( ≥ 60 vs < 60) | 3.20 | 0.98-10.45 | .05 | ||
| t(11q23) vs others | 8.17 | 1.65-40.45 | .01 | ||
| t(15;17) vs others | 0.12 | 0.02-0.61 | .01 | ||
| USA-set-II (n = 53) | miR-181a (high vs low) | 0.49 | 0.23-1.05 | .07 | |
| miR-181b (high vs low) | 0.41 | 0.20-0.85 | .02 | ||
| miR-181c (high vs low) | 0.56 | 0.29-1.09 | .09 | ||
| miR-181d (high vs low) | 0.49 | 0.23-1.03 | .06 | ||
| Age, years ( ≥ 60 vs < 60) | 2.68 | 1.30-5.55 | .008 | ||
| t(11q23) vs others | 3.03 | 1.34-6.87 | .007 | ||
| Training sets (n = 183) | USA-set-II-35S (n = 35) | 4-gene signature (high vs low) | 4.81 | 1.52-15.29 | .008 |
| 9-gene signature (high vs low) | 6.21 | 1.95-19.80 | .002 | ||
| Age (each 10-year increase) | 1.46 | 1.20-1.80 | < .001 | ||
| USA-set-III (n = 87) | 4-gene signature (high vs low) | 2.66 | 1.03-6.88 | .04 | |
| 9-gene signature (high vs low) | 1.80 | 0.63-5.15 | .27 | ||
| PB blasts (high vs low) | 5.66 | 1.87-17.16 | .002 | ||
| t(15;17) vs others | 0.38 | 0.15-0.97 | .04 | ||
| Germany Set-I (n = 106) | 4-gene signature (high vs low) | 2.62 | 1.04-6.58 | .04 | |
| 9-gene signature (high vs low) | 1.95 | 0.84-4.55 | .12 | ||
| del(7q)/−7 vs others | 4.82 | 1.57-14.81 | .006 | ||
| Complex vs others | 11.73 | 2.62-52.49 | .001 | ||
| log2(WBC) (each 2-unit increase) | 2.94 | 1.37-6.31 | .006 | ||
| Validation set (n = 271) | Netherlands-set-I (n = 271) | 4-gene signature (high vs low) | 1.66 | 1.03-2.70 | .04 |
| 9-gene signature (high vs low) | 1.54 | 1.00-2.37 | .05 | ||
| Age (each 10-year increase) | 3.06 | 1.17-7.99 | .02 | ||
| Complex vs others | 2.03 | 1.25-3.29 | .004 | ||
| 7q vs others | 2.23 | 1.42-3.51 | .0005 | ||
| 3q vs others | 1.76 | 1.00-3.11 | .05 | ||
| t(8;21) vs others | 0.37 | 0.19-0.71 | .003 | ||
| inv(16) vs others | 0.41 | 0.21-0.79 | .008 | ||
| CBF AML vs others | 0.41 | 0.21-0.79 | .008 | ||
| FLT3-ITD vs others | 1.74 | 1.09-2.79 | .02 | ||
| FLT3-TKD vs others | 0.44 | 0.21-0.90 | .02 | ||
| CEBPA mutation vs wild-type | 0.26 | 0.08-0.85 | .03 |
| Patient set | Variables | HR | 95% CI | P | |
|---|---|---|---|---|---|
| USA-set-I (n = 33) | miR-181a (high vs low) | 0.70 | 0.48-1.02 | .06 | |
| miR-181b (high vs low) | 0.65 | 0.43-0.98 | .04 | ||
| miR-181c (high vs low) | 0.79 | 0.57-1.09 | .15 | ||
| miR-181d (high vs low) | 0.74 | 0.49-1.11 | .15 | ||
| Age, years ( ≥ 60 vs < 60) | 3.20 | 0.98-10.45 | .05 | ||
| t(11q23) vs others | 8.17 | 1.65-40.45 | .01 | ||
| t(15;17) vs others | 0.12 | 0.02-0.61 | .01 | ||
| USA-set-II (n = 53) | miR-181a (high vs low) | 0.49 | 0.23-1.05 | .07 | |
| miR-181b (high vs low) | 0.41 | 0.20-0.85 | .02 | ||
| miR-181c (high vs low) | 0.56 | 0.29-1.09 | .09 | ||
| miR-181d (high vs low) | 0.49 | 0.23-1.03 | .06 | ||
| Age, years ( ≥ 60 vs < 60) | 2.68 | 1.30-5.55 | .008 | ||
| t(11q23) vs others | 3.03 | 1.34-6.87 | .007 | ||
| Training sets (n = 183) | USA-set-II-35S (n = 35) | 4-gene signature (high vs low) | 4.81 | 1.52-15.29 | .008 |
| 9-gene signature (high vs low) | 6.21 | 1.95-19.80 | .002 | ||
| Age (each 10-year increase) | 1.46 | 1.20-1.80 | < .001 | ||
| USA-set-III (n = 87) | 4-gene signature (high vs low) | 2.66 | 1.03-6.88 | .04 | |
| 9-gene signature (high vs low) | 1.80 | 0.63-5.15 | .27 | ||
| PB blasts (high vs low) | 5.66 | 1.87-17.16 | .002 | ||
| t(15;17) vs others | 0.38 | 0.15-0.97 | .04 | ||
| Germany Set-I (n = 106) | 4-gene signature (high vs low) | 2.62 | 1.04-6.58 | .04 | |
| 9-gene signature (high vs low) | 1.95 | 0.84-4.55 | .12 | ||
| del(7q)/−7 vs others | 4.82 | 1.57-14.81 | .006 | ||
| Complex vs others | 11.73 | 2.62-52.49 | .001 | ||
| log2(WBC) (each 2-unit increase) | 2.94 | 1.37-6.31 | .006 | ||
| Validation set (n = 271) | Netherlands-set-I (n = 271) | 4-gene signature (high vs low) | 1.66 | 1.03-2.70 | .04 |
| 9-gene signature (high vs low) | 1.54 | 1.00-2.37 | .05 | ||
| Age (each 10-year increase) | 3.06 | 1.17-7.99 | .02 | ||
| Complex vs others | 2.03 | 1.25-3.29 | .004 | ||
| 7q vs others | 2.23 | 1.42-3.51 | .0005 | ||
| 3q vs others | 1.76 | 1.00-3.11 | .05 | ||
| t(8;21) vs others | 0.37 | 0.19-0.71 | .003 | ||
| inv(16) vs others | 0.41 | 0.21-0.79 | .008 | ||
| CBF AML vs others | 0.41 | 0.21-0.79 | .008 | ||
| FLT3-ITD vs others | 1.74 | 1.09-2.79 | .02 | ||
| FLT3-TKD vs others | 0.44 | 0.21-0.90 | .02 | ||
| CEBPA mutation vs wild-type | 0.26 | 0.08-0.85 | .03 |
Variables for which P < .1 in univariable models (supplemental Tables 1 or 4) were included in multivariable analyses. Hazard ratios (HR) > 1 or < 1 indicate, respectively, a higher or lower risk of an event (death) for higher values of continuous variables and for the first category listed for categorical variables in OS models.
Up-regulation of a gene signature composed of 9 potential targets of miR-181 is associated with poor prognosis in CA-AML patients
Of the 53 samples in USA-set II, we conducted Agilent custom-design microarray assay for 35 samples (termed as USA-set II-35S; Table 1). Because increased expression of miR-181b remains significantly (P < .05) associated with favorable OS in multivariable testing (Table 3) and its expression is very significantly positively correlated (r > 0.7; P < .0001; Pearson correlation) with that of miR-181a or miR-181d (supplemental Table 3), we chose miR-181b as the representative of the miR-181 family to identify potential target genes in CA-AML. Through correlating expression of miR-181b with those of miR-181 putative target genes, we identified 159 genes that exhibited a significantly inverse (P < .05; Pearson correlation) correlation of expression with miR-181b (supplemental Table 2).
Meanwhile, we have also conducted Affymetrix Exon array assay of a set of 87 CA-AML cases (USA-set III; Table 1). In addition, we extracted gene expression profiles of 61 CA-AML samples (termed as Germany-set I; Table 1) from a previous study by Bullinger et al.20 We then performed Cox regression analyses to assess the association of expression levels of the 159 candidate target genes with OS in patients of USA-set II-35S, USA-set III, and Germany-set I, respectively. We identified 9 candidate targets (Table 2) whose increased expression levels were significantly associated with a worse OS after FDR adjustment for multiple tests across the 3 patient sets (FDR < 0.05; Hommel adjustment37 ). Notably, among the 9 genes, there were 4 homeobox genes, including 3 HOXA genes (ie, HOXA7, HOXA9, and HOXA11) and a HOXA cofactor gene (ie, PBX3). As shown in Figure 2, patients in intermediate- to poor-prognosis subtypes of AML carrying t(11q23) or trisomy 8 expressed significantly (P < .05; t test) lower levels of miR-181 and higher levels of the 4 homeobox genes compared with those in the favorable subtypes of AML bearing t(15;17), t(8;21), or inv(16).

Expression levels of miR-181 family members and the 4 homeobox genes can separate favorable subtypes of CA-AML from unfavorable ones. The favorable subtypes of CA-AML carrying t(15;17), t(8;21), or inv(16) exhibited an increased expression of miR-181 family members and a decreased expression of the 4 homeobox genes compared with the poor prognosis subtypes of CA-AML bearing t(11q23) or (+8).
Expression levels of miR-181 family members and the 4 homeobox genes can separate favorable subtypes of CA-AML from unfavorable ones. The favorable subtypes of CA-AML carrying t(15;17), t(8;21), or inv(16) exhibited an increased expression of miR-181 family members and a decreased expression of the 4 homeobox genes compared with the poor prognosis subtypes of CA-AML bearing t(11q23) or (+8).
Increased level of a 4-gene signature, but not the 9-gene signature, is an independent predictor of poor prognosis in CA-AML patients
We further derived 2 gene sum-expression-value signatures, which were a linear combination of the expression values of the 4 (ie, HOXA7, HOXA9, HOXA11, and PBX3) and 9 (ie, the 4 plus MRPS18B, PLA2G4A, TRPS1, GNS, and PGPEP1) genes, respectively, weighted by the average correlation coefficient (supplemental Methods). As shown in supplemental Table 4, increased expression levels of both the 4-gene signature and 9-gene signature were significantly (P < .05) associated with poor survival in all the 3 training sets, including USA-set II-35S, USA-set III, and Germany-set I. When patients in a given set were divided into 2 equal-size groups based on the median sum value of the 4- or 9-gene signature, patients with higher values exhibited significantly (P < .05) shorter OS than those with lower values in each training set (supplemental Figure 2A-C). Through multivariable testing, we found that the increased level of the 4-gene signature, but not that of the 9-gene signature, remained significantly (P < .05) associated with shorter OS in all 3 training sets (Table 3), after adjusting for all other variables that have a P less than .1 in univariable analyses (supplemental Table 4).
To validate these findings, we also extracted gene expression profiles of 271 CA-AML samples (termed as Netherlands-set I) from a previous study by Wouters et al.32 The significance of the association of increased level of the 4-gene signature with shorter OS was confirmed in this validation set, and the 9-gene signature exhibited a similar prognostic impact (supplemental Table 4; Table 3; Figure 3). Considering multivariable testing results from both training and validation sets, our data suggest that up-regulation of the 4-gene signature is an independent predictor of poor prognosis in CA-AML patients.

Increased levels of the 4- and 9-gene signatures are associated with adverse OS. The patients in the validation set (ie, Netherlands-set I; n = 271) were dichotomized into 2 groups based on the median value of the 4-gene (4G signature; left panel) or the 9-gene (9G signature; right panel) signature, and then Kaplan-Meier curves were generated to depict outcomes. The P value was determined by log-rank test. | indicates censored.
Increased levels of the 4- and 9-gene signatures are associated with adverse OS. The patients in the validation set (ie, Netherlands-set I; n = 271) were dichotomized into 2 groups based on the median value of the 4-gene (4G signature; left panel) or the 9-gene (9G signature; right panel) signature, and then Kaplan-Meier curves were generated to depict outcomes. The P value was determined by log-rank test. | indicates censored.
Ectopic expression of miR-181b significantly decreased cell viability/growth and increased apopotosis of human MLL-rearranged leukemic cells, and significantly inhibited MLL-fusion-mediated cell transformation in vitro and leukemogenesis in vivo
As increased expression of miR-181a and, particularly of miR-181b, is significantly (P < .05) associated with favorable prognosis in CA-AML patients (Figure 1; Table 3; supplemental Figure 1), we further investigated their biologic function in CA-AML in vitro and in vivo.
We chose MLL-rearranged AML as a model because it is a subtype of CA-AML in which miR-181b was expressed at a low level33 (Figure 2; supplemental Figure 3). We cloned the precusor sequence of miR-181a and miR-181b, respectively, into MSCV-PIG, a retroviral vector containing a PGK-puromycin-IRES-GFP (PIG) cassette.3 Then, we transfected MSCV-PIG-miR-181a, MSCV-PIG-miR-181b, and MSCV-PIG (as a negative control), respectively, into 3 MLL-rearranged AML cell lines, including MONOMAC-6/t(9,11), THP-1/t(9,11), and KOCL-48/t(4,11). We found that ectopic expression of both miR-181a and miR-181b could decrease cell viability (Figure 4A left panel) and growth (Figure 4B), and increase apopotosis (Figure 4A right panel; Figure 5D) of the MLL-rearranged leukemic cells of each cell line, although miR-181b exhibited more evident effects than miR-181a. Similar effects were observed when we used miR-181a or miR-181b mimic oligos instead of the vector-based ones (supplemental Figure 4).

Ectopic expression of miR-181a and, particularly, miR-181b exhibits anti–tumor effects in MLL-rearranged AML in vitro and in vivo. *P < .05 (2-tailed t test). (A) Effect of ectopic expression of miR-181a and miR-181b on viability (left panel) or apoptosis (right panel) of MONOMAC-6, THP-1, and KOCL-48 cells, respectively. The controls are cells transfected with empty plasmids. Data are mean ± SD from 3 independent experiments. (B) Cell growth/proliferation analysis of MONOMAC-6 cells transfected with MSCV-PIG (ie, control), MSCV-PIG-miR-181a, or MSCV-PIG-miR-181b. Cell growth is significantly (P < .05) slower in leukemic cells transduced with miR-181a or miR-181b than those transduced with empty vector after 3 days of culture. (C) Colony-forming/replating assay of mouse normal BM progenitor cells transduced with MSCVneo + MSCV-PIG (ie, control), MSCVneo + MSCV-PIG-miR-181a (ie, miR-181a), MSCVneo + MSCV-PIG-miR-181b (ie, miR-181b), MSCVneo-MLL-AF9 + MSCV-PIG (ie, MA9), MSCVneo-MLL-AF9 + MSCV-PIG-miR-181a (ie, MA9 + miR181a), or MSCVneo-MLL-AF9 + MSCV-PIG-miR-181b (ie, MA9 + miR181b). Duplicates were plated for each combination with 1 × 104 cells per dish, and every 7 days the cells were replated for up to 4 passages. Two independent experiments were conducted. Data are mean ± SD. (D) In primary mouse bone marrow transplantation (BMT) assay, both MA9 + miR181a mice (n = 5) and MA9 + miR181b mice (n = 5) developed leukemia slower than MA9 (ie, MLL-AF9 alone) mice (n = 5), although only the difference between MA9 + miR181b and MA9 mice is statistically significant (P = .004; log-rank test). (E) Relative expression of miR-181a or miR-181b expression in MA9, MA9 + miR181a, or MA9 + miR181b leukemic mouse BM cells (samples from 3 mice in each cohort were analyzed). The level in MA9 was set as 1.
Ectopic expression of miR-181a and, particularly, miR-181b exhibits anti–tumor effects in MLL-rearranged AML in vitro and in vivo. *P < .05 (2-tailed t test). (A) Effect of ectopic expression of miR-181a and miR-181b on viability (left panel) or apoptosis (right panel) of MONOMAC-6, THP-1, and KOCL-48 cells, respectively. The controls are cells transfected with empty plasmids. Data are mean ± SD from 3 independent experiments. (B) Cell growth/proliferation analysis of MONOMAC-6 cells transfected with MSCV-PIG (ie, control), MSCV-PIG-miR-181a, or MSCV-PIG-miR-181b. Cell growth is significantly (P < .05) slower in leukemic cells transduced with miR-181a or miR-181b than those transduced with empty vector after 3 days of culture. (C) Colony-forming/replating assay of mouse normal BM progenitor cells transduced with MSCVneo + MSCV-PIG (ie, control), MSCVneo + MSCV-PIG-miR-181a (ie, miR-181a), MSCVneo + MSCV-PIG-miR-181b (ie, miR-181b), MSCVneo-MLL-AF9 + MSCV-PIG (ie, MA9), MSCVneo-MLL-AF9 + MSCV-PIG-miR-181a (ie, MA9 + miR181a), or MSCVneo-MLL-AF9 + MSCV-PIG-miR-181b (ie, MA9 + miR181b). Duplicates were plated for each combination with 1 × 104 cells per dish, and every 7 days the cells were replated for up to 4 passages. Two independent experiments were conducted. Data are mean ± SD. (D) In primary mouse bone marrow transplantation (BMT) assay, both MA9 + miR181a mice (n = 5) and MA9 + miR181b mice (n = 5) developed leukemia slower than MA9 (ie, MLL-AF9 alone) mice (n = 5), although only the difference between MA9 + miR181b and MA9 mice is statistically significant (P = .004; log-rank test). (E) Relative expression of miR-181a or miR-181b expression in MA9, MA9 + miR181a, or MA9 + miR181b leukemic mouse BM cells (samples from 3 mice in each cohort were analyzed). The level in MA9 was set as 1.

Forced expression of PBX3 coding region exhibits opposite functions than miR-181a/miR-181b. (A) Forced expression of PBX3-CDS could reverse the effects of miR-181a/miR-181b on viability (left panel) and apoptosis (right panel) of MONOMAC-6, THP-1, and KOCL-48 cells, respectively. (B) Cell growth/proliferation analysis of MONOMAC-6 cells transfected with MSCV-PIG (ie, control), MSCV-PIG-miR-181a, MSCV-PIG-miR-181b, MSCV-PIG-PBX3 (CDS), MSCV-PIG-PBX3 (CDS)–miR181a, or MSCV-PIG-PBX3 (CDS)–miR181b. *P < .05. (C) Western blot assay of expression of PBX3 at the protein level in MONOMAC-6 cells 96 hours after transfection with more than 6 individual constructs. (D) Apoptosis analysis by flow cytometry with anti–annexin V antibody staining in MONOMAC-6 cells 72 hours after transfection with more than 6 individual constructs. PI indicates propidium iodide. Red rectangle indicates apoptotic cells. (E) In secondary BMT assay, mice with MA9 + miR181b (n = 10) developed leukemia significantly (P = .02) slower than those of MA9 alone (n = 5). Mice with MA9 + miR181b + PBX3-CDS (n = 6) developed leukemia at a similar speed as those with MA9 + PBX3-CDS (n = 5), and both faster, although not significantly (P = .06), than MA9 mice (n = 5). (F) Quantitative PCR analysis of miR-181b and PBX3 expression levels in secondary BMT mouse leukemic BM cells. The quantitative PCR primers of PBX3 were designed to detect expression of both human and mouse PBX3. Data are mean ± SD values of 3 mouse BM samples per cohort.
Forced expression of PBX3 coding region exhibits opposite functions than miR-181a/miR-181b. (A) Forced expression of PBX3-CDS could reverse the effects of miR-181a/miR-181b on viability (left panel) and apoptosis (right panel) of MONOMAC-6, THP-1, and KOCL-48 cells, respectively. (B) Cell growth/proliferation analysis of MONOMAC-6 cells transfected with MSCV-PIG (ie, control), MSCV-PIG-miR-181a, MSCV-PIG-miR-181b, MSCV-PIG-PBX3 (CDS), MSCV-PIG-PBX3 (CDS)–miR181a, or MSCV-PIG-PBX3 (CDS)–miR181b. *P < .05. (C) Western blot assay of expression of PBX3 at the protein level in MONOMAC-6 cells 96 hours after transfection with more than 6 individual constructs. (D) Apoptosis analysis by flow cytometry with anti–annexin V antibody staining in MONOMAC-6 cells 72 hours after transfection with more than 6 individual constructs. PI indicates propidium iodide. Red rectangle indicates apoptotic cells. (E) In secondary BMT assay, mice with MA9 + miR181b (n = 10) developed leukemia significantly (P = .02) slower than those of MA9 alone (n = 5). Mice with MA9 + miR181b + PBX3-CDS (n = 6) developed leukemia at a similar speed as those with MA9 + PBX3-CDS (n = 5), and both faster, although not significantly (P = .06), than MA9 mice (n = 5). (F) Quantitative PCR analysis of miR-181b and PBX3 expression levels in secondary BMT mouse leukemic BM cells. The quantitative PCR primers of PBX3 were designed to detect expression of both human and mouse PBX3. Data are mean ± SD values of 3 mouse BM samples per cohort.
To investigate the effect of forced expression of miR-181a/miR-181b in primary cell transformation and leukemogenesis, we cotransduced MSCV-PIG-miR-181a, MSCV-PIG-miR-181b, and MSCV-PIG, respectively, together with MSCVneo-MLL-AF9 into mouse normal bone marrow progenitor (ie, lineage-negative) cells and plated onto methylcellulose medium with G418 and puromycin selection. Colony cells were replated every 7 days for up to 4 passages. As shown in Figure 4C, cells transduced with empty vector (as negative control), miR-181a, or miR-181b could not form colonies after 2 passages of plating. Although cotransduction of miR-181a or miR-181b with MLL-AF9 still could form hundreds of colonies after 2 passages of plating, the colony numbers were significantly (P < .05) lower than that of transduction of MLL-AF9 alone (Figure 4C), indicating that miR-181a/miR-181b can significantly inhibit cell transformation (ie, immortalization) mediated by MLL fusions.
Meanwhile, we have also collected colony cells from the first passage and then transplanted them into lethally irradiated recipient mice (“Primary bone marrow transplantation (BMT) assays and characterization of leukemia”). As shown in Figure 4D, forced expression of both miR-181a and miR-181b could delay leukemogenesis mediated by MLL-AF9, although only miR-181b exhibited a statistically significant (P < .05; log-rank test) effect. The forced expression of miR-181a or miR-181b was confirmed in leukemic BM cells by quantitative PCR (Figure 4E). As expected, all recipient mice of MLL-AF9 or miR-181 (a or b) + MLL-AF9 developed myeloid leukemia (supplemental Figure 5; supplemental Table 6).
Endogenous expression of the 4 HOXA-PBX3 target genes was significantly repressed by forced expression of miR-181b in vitro and in vivo
As the 4 homeobox genes (ie, HOXA7, HOXA9, HOXA11, and PBX3) exhibited a significantly (P < .05; Pearson correlation) inverse correlation of expression with (Table 2; supplemental Table 2), and an opposite prognostic impact than, miR-181 family members, particularly miR-181b (Figures 1 and 3; Table 3), we checked endogenous expression changes of these 4 genes on manipulating expression of miR-181a/miR-181b in vitro and in vivo. As expected, we found that expression of all the 4 homeobox genes was significantly repressed by ectopically expressed miR-181b both in vitro (Figure 6A) and in vivo (Figure 6B). Ectopic expression of miR-181a has a similar, though milder, effect compared with that of miR-181b (Figure 6A-B).

Forced expression of miR-181a/miR-181b represses endogenous expression of the 4 homeobox target genes, and PBX3 is a direct target of miR-181a/miR-181b. (A-B) Quantitative PCR analyses of effects of the ectopic expression of miR-181a or miR-181b on the expression of the 4 homeobox genes (ie, HOXA7, HOXA9, HOXA11, and PBX3) in human MLL-rearranged leukemic cells (A, using MONOMAC-6 cells as a representative) or in MLL-fusion-mediated mouse primary leukemic BM cells (B). Data are mean ± SD. *P < .05. (C) Luciferase reporter and mutagenesis assays. Left panel: Predicted miR-181a/miR-181b target sites and corresponding mutants in the 3′-UTR of PBX3. Right panel: Forced expression of miR-181a or miR-181b can significantly repress luciferase activity of the reporter gene bearing 3′-UTR of PBX3 in human 293T cells, whereas mutation at the putative target site of miR-181 in the 3′-UTR can abrogate the inhibition. The normalized luciferase activities represent the firefly: β-Galactosidase ratios normalized to the control sample. Error bars present SD obtained from 3 independent experiments. (D) Quantitative PCR (left panel) and Western blot (right panel) assays of the effect of ectopic expression of miR-181a/mIR-191b on the endogenous expression of PBX3. MONOMAC-6 cells served as a representative.
Forced expression of miR-181a/miR-181b represses endogenous expression of the 4 homeobox target genes, and PBX3 is a direct target of miR-181a/miR-181b. (A-B) Quantitative PCR analyses of effects of the ectopic expression of miR-181a or miR-181b on the expression of the 4 homeobox genes (ie, HOXA7, HOXA9, HOXA11, and PBX3) in human MLL-rearranged leukemic cells (A, using MONOMAC-6 cells as a representative) or in MLL-fusion-mediated mouse primary leukemic BM cells (B). Data are mean ± SD. *P < .05. (C) Luciferase reporter and mutagenesis assays. Left panel: Predicted miR-181a/miR-181b target sites and corresponding mutants in the 3′-UTR of PBX3. Right panel: Forced expression of miR-181a or miR-181b can significantly repress luciferase activity of the reporter gene bearing 3′-UTR of PBX3 in human 293T cells, whereas mutation at the putative target site of miR-181 in the 3′-UTR can abrogate the inhibition. The normalized luciferase activities represent the firefly: β-Galactosidase ratios normalized to the control sample. Error bars present SD obtained from 3 independent experiments. (D) Quantitative PCR (left panel) and Western blot (right panel) assays of the effect of ectopic expression of miR-181a/mIR-191b on the endogenous expression of PBX3. MONOMAC-6 cells served as a representative.
PBX3 is a validated target of miR-181a/miR-181b
PBX3 was the only gene associated with poor survival in all 4 training and validation sets (Table 2), and it was repressed by miR-181a/miR-181b to a greater degree than the other 3 homeobox target genes (Figure 6A-B); we therefore chose PBX3 as an important candidate target of miR-181 for further studies. We first conducted luciferase reporter and mutagenesis assays and showed that PBX3 is a direct target of miR-181a/miR-181b (Figure 6C). Furthermore, forced expression of miR-181a/miR-181b significantly down-regulated endogenous expression of PBX3 in MLL-rearranged leukemic cells at both the RNA and protein levels (Figure 6D).
Forced expression of PBX3 can reverse the effects of miR-181a/miR-181b in vitro and in vivo
To investigate whether down-regulation of PBX3 is required for the function of forced expression of miR-181a/miR-181b, we synthesized human PBX3 coding region (ie, CDS, which does not contain 3′-UTR and thus would not be repressed by miR-181 family members) and then cloned it into MSCV-PIG (termed as MSCV-PIG-PBX3-CDS). We found that forced expression of the PBX3 coding region could reverse the effects of miR-181a/miR-181b on cell viability (Figure 5A; supplemental Figure 4 left panel), growth (Figure 5B), and apoptosis (Figure 5A,D; supplemental Figure 4 right panel) in MLL-rearranged leukemic cells. Ectopic expression of PBX3 at the protein level was validated by Western blot (Figure 5C). Knocking-down expression of PBX3 in MLL-rearranged leukemic cells by siRNAs exhibited similar effects (supplemental Figure 6) to those of the ectopic expression of miR-181a/miR-181b (Figure 4A).
To determine whether the effects of miR-181a/miR-181b on cell growth and apoptosis of leukemic cells are dependent on PBX3 signaling, we transfected MSCV-PIG, MSCV-PIG-miR-181a, or MSCV-PIG-miR-181b into KASUMI-1 cells, which bear AML1-ETO fusions resulting from t(8;21). Consistent with primary t(8;21) leukemic cells (Figure 2; supplemental Figure 3), KASUMI-1 cells have a very low level of endogenous expression of PBX3 and a relatively high level of miR-181a/miR-181b expression.33 As expected, forced expression of miR-181a and miR-181b has no significant effects on cell growth (supplemental Figure 7A) and apoptosis (supplemental Figure 7B) of KASUMI-1 cells, implying that effects of miR-181a/miR-181b do rely on their repression of PBX3 signaling.
To investigate whether forced expression of the PBX3 coding region can reverse effect of miR-181b in vivo, we collected MLL-AF9 (ie, MA9) and MLL-AF9 + miR-181b (ie, MA9 + miR181b) leukemic bone marrow blast cells from primary transplantation recipient mice (Figure 4D), and then retrovirally transduced with either MSCV-PIG (still labeled as MA9 and MA9 + miR181b, respectively) or MSCV-PIG-PBX3-CDS (labeled as MA9 + PBX3-CDS and MA9 + miR181b + PBX3-CDS, respectively) and selected for transduction-positive cells in methylcellulose dishes for 7 days. Then, the colony cells were collected and transplanted into sublethally irradiated secondary recipient mice. As expected, MA9 + miR181b leukemic cells caused a much slower leukemogenesis in secondary recipient mice than MA9 leukemic cells, and such delay could be rescued by forced expression of PBX3-CDS in both types of leukemic cells (Figure 5E; supplemental Table 6). The levels of ectopic and endogenous expression of miR-181b and PBX3 were confirmed by quantitative PCR (Figure 5F).
Discussion
Here we show that increased expression of both miR-181a and miR-181b is significantly (P < .05) associated with favorable OS in 2 sets of CA-AML patients (supplemental Table 1; Figure 1), although only that of miR-181b remained significant on multivariable testing (Table 3). Furthermore, we show that miR-181 family members (particularly, miR-181b) may target a group of homeobox superfamily genes (ie, HOXA7, HOXA9, HOXA11, and PBX3; Figure 6; Table 2). Increased expression of this 4-gene signature was significantly (P < .05) associated with shorter OS in all 4 training and validation sets (supplemental Table 4; Figure 3; supplemental Figure 2), and this association remained significant (P < .05) on multivariable testing after adjusting for other variables (Table 3). Notably, the gene expression data in different patient sets were derived from distinct microarray platforms (supplemental Figure 1), implying that the prognostic impact of this 4-gene signature is robust.
Moreover, our functional studies showed that ectopic expression of miR-181b significantly promoted cell apoptosis and proliferation, and inhibited viability of MLL-rearranged AML cells (Figures 4A-C and 5D; supplemental Figure 4). MLL-rearranged AML accounts for more than 10% of CA-AML and is associated with an intermediate to poor prognosis.26,39,40 We further showed that miR-181b could significantly inhibit MLL-fusion-mediated leukemogenesis in both primary and secondary BMT assays (Figures 4D and 5E). miR-181a exhibited a relatively weaker effect compared with miR-181b both in vitro (Figures 4A and 5D) and in vivo (Figure 4D), although their ectopic expression levels were roughly similar (data not shown). The relatively stronger biologic effects of miR-181b (Figures 4A,D and 5D), compared with miR-181a, might in part be the result of its relatively stronger repression on expression of the 4 homeobox target genes (Figure 6A-B). Nonetheless, it is possible that miR-181b and miR-181a (and probably also other miR-181 family members) orchestrate the regulation of target genes in a synergistic manner.
Although expression signature of HOXA7, HOXA9, or PBX3 has been reported to be associated with poor survival of AML patients in several individual studies,20,29-31 their prognostic impact has not been tested in multivariable models or been validated by further large-scale independent studies, and some controversial data were reported.15,16 Indeed, we found that none of the individual 4 homeobox genes (ie, HOXA7, HOXA9, HOXA11, and PBX3) can serve as an independent predictor of prognosis; instead, only the signature of the entire group of these 4 genes is an independent predictor of prognosis on multiple testing in all datasets we analyzed (supplemental Table 4).
HOXA7 and particularly HOXA9 have been shown to be required for the development and maintenance of MLL-associated leukemia,21,24,27,40-44 whereas the role of HOXA11 in leukemogenesis is elusive. As a cofactor of HOX proteins, PBX3 is a member of the PBX family of 3-amino-acid loop extension homeobox proteins,17 which can form stable heterocomplexes with HOX and MEIS proteins to regulate the transcription of downstream targets.18 However, although aberrant overexpression of PBX3 in CA-AML, such as MLL-associated leukemia, has been observed frequently,20,21,23-25 its biologic function remains unexplored. A previous study suggested that PBX3 and PBX2, but not PBX1, were required for MLL fusion- or HOXA9-mediated leukemogenesis.45 However, because PBX2 is not significantly up-regulated in MLL-associated leukemia45 (supplemental Figure 3), PBX3 would appear to be the most critical member of PBX family contributing to the development of MLL-associated leukemia. Here we demonstrated that PBX3 is a direct target of miR-181a/miR-181b in CA-AML (Figure 6C-D). More importantly, we also showed that PBX3 played a significant oncogenic role that could reverse the effects of miR-181b both in vitro and in vivo (Figure 5), suggesting that PBX3 may also play an important role in the development and maintenance of MLL-rearranged AML.
Despite the heterogeneous genetic backgrounds of human CA-AML, different treatment protocols applied (supplemental data), and different microarray platforms used, we identified a robust prognostic gene signature composed of 4 homeobox genes that are targeted by miR-181 family members (particularly miR-181b). The up-regulation of this gene signature is probably a consequence of the down-regulation of miR-181 family members, which in turn contributes to the poor prognosis of CA-AML patients. In the future, it would be important to identify the mechanism(s) underlying the down-regulation of the miR-181 family. Our data suggest that restoring expression of miR-181b or targeting HOXA/PBX pathways directly may help to improve the outcome of patients with unfavorable CA-AML. Although not feasible in clinical use yet, previous studies46,47 have demonstrated the promise of miRNA replacement therapies; thus, miR-181b mimics may be clinically applicable in the future. Interestingly, antagonists (eg, HXR9; a small peptide) that disrupt the formation of HOX/PBX heterodimers have also shown promising anti–tumor effects in vitro and in vivo in various aggressive cancers, such as melanoma,48 non-small-cell lung cancer,49 and ovarian cancer.50 Therefore, such antagonists might also be clinically appropriate for the treatment of poor-prognosis CA-AML, alone or in combination with other effective therapeutic drug(s), to substantially improve the survival of the patients.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Drs Masha Kocherginsky, Theodore Karrison, and Dezheng Huo for data coordination and advice on statistical analyses, and Gregory Hannon, Scott Hammond, Lin He, and Scott Armstrong for providing retroviral vectors.
This work was supported in part by the Leukemia & Lymphoma Society (Translational Research Grant, J.D.R. and J.C.), CALGB (pilot grant 20801, J.D.R. and J.C.), National Institutes of Health (R01 grant CA127277, J.C.), American Cancer Society (Research Scholar grant, J.C.), Leukemia & Lymphoma Society (Special Fellowship, Z.L.), Gabrielle's Angel Foundation for Cancer Research (J.C., Z.L., and H.H.), the Spastic Paralysis Foundation of the Illinois, Eastern Iowa Branch of Kiwanis International (J.D.R.), the Fidelity Foundation (J.D.R. and J.C.), the U10 of the CALGB LCSC committee (U10 CA101140, G.M.), the P50 of OSU Leukemia SPORE (P50CA140158 Project 2, C.D.B. and G.M.), Leukemia Clinical Research Foundation grant (C.D.B. and G.M), the Intramural Research Program of National Human Genome Research Institute, National Institutes of Health (A.E. and P.P.L.), the National Natural Science Foundation of China (60971099, X.Y.), the Deutsche José Carreras Leukämie Stiftung (grant R 06/41v, L.B.), the Deutsche Forschungsgemeinschaft (Heisenberg-Stipendium BU 1339/3-1, L.B), and National Institutes of Health (P01 CA40046, M.M.L.B.; and P30 CA014599, M.M.L.B.).
National Institutes of Health
Authorship
Contribution: J.C. conceived and designed the study; K.D., M.B.N., Y.Z., R.A.L., M.M.L.B., M.A.C., L.B., P.J.M.V., R.D., B.L., P.P.L., G.M., C.D.B., J.D.R., and J.C. provided study materials or patients; Z.L., M.D.R., K.M., X.Y., C.H., Y.A.L., L.B., P.J.M.V., R.D., B.L., G.M., C.D.B., J.D.R., and J.C. analyzed and/or interpreted data; Z.L., H.H., Y.L., X.J., P.C., S.A., A.E., M.H., C.P., Z.Z., P.P.L., and J.C. conducted experiments; Z.L. and J.C. prepared and wrote the manuscript; and all authors collected and assembled data and helped to revise and gave final approval of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jianjun Chen, Department of Medicine, University of Chicago, 900 E 57th St, KCBD Rm 7134, Chicago, IL 60637; e-mail: jchen@medicine.bsd.uchicago.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal