

Key Points
Developed a targeted sequencing platform covering 63 genes linked to heritable bleeding, thrombotic, and platelet disorders.
The ThromboGenomics platform provides a sensitive genetic test to obtain molecular diagnoses in patients with a suspected etiology.
Abstract
Inherited bleeding, thrombotic, and platelet disorders (BPDs) are diseases that affect ∼300 individuals per million births. With the exception of hemophilia and von Willebrand disease patients, a molecular analysis for patients with a BPD is often unavailable. Many specialized tests are usually required to reach a putative diagnosis and they are typically performed in a step-wise manner to control costs. This approach causes delays and a conclusive molecular diagnosis is often never reached, which can compromise treatment and impede rapid identification of affected relatives. To address this unmet diagnostic need, we designed a high-throughput sequencing platform targeting 63 genes relevant for BPDs. The platform can call single nucleotide variants, short insertions/deletions, and large copy number variants (though not inversions) which are subjected to automated filtering for diagnostic prioritization, resulting in an average of 5.34 candidate variants per individual. We sequenced 159 and 137 samples, respectively, from cases with and without previously known causal variants. Among the latter group, 61 cases had clinical and laboratory phenotypes indicative of a particular molecular etiology, whereas the remainder had an a priori highly uncertain etiology. All previously detected variants were recapitulated and, when the etiology was suspected but unknown or uncertain, a molecular diagnosis was reached in 56 of 61 and only 8 of 76 cases, respectively. The latter category highlights the need for further research into novel causes of BPDs. The ThromboGenomics platform thus provides an affordable DNA-based test to diagnose patients suspected of having a known inherited BPD.
Introduction
High-throughput sequencing (HTS) of genomic DNA is being introduced into clinical practice1 and broadly falls into 2 categories: whole genome sequencing (WGS)2 and targeted sequencing of pre-specified regions of the genome by means of probe-based capture. These regions may include all exons (whole exome sequencing [WES]) and be sequenced to moderate depth, or they may comprise a much smaller fraction of the genome and be sequenced to high depth.3 Capture probes targeting regions that have been widely studied and implicated in a group of rare heritable disorders can turn HTS into a valuable tool for their affordable diagnosis.
In this study, we focus on the diagnosis of rare heritable bleeding, thrombotic, and platelet disorders (BPDs). Previously, we have defined a BPD case as a patient having an abnormal platelet count, volume, morphology, or function, or with a tendency to bleed abnormally.4 The abnormal phenotypes must furthermore be judged to have a genetic basis, thereby ruling out diseases that may have been acquired or thought to be caused by exposure to known environmental risk factors. For the purposes of this study, we also include patients with an abnormal tendency to thrombus formation in our definition. Currently, 90% of BPD cases that do not have hemophilia5 or von Willebrand disease6,7 (VWD) never receive a conclusive molecular diagnosis due to the unavailability of affordable genetic tests.8 Hence treatment is compromised in some cases and the rapid identification of affected relatives may be impeded.
The aims of the ThromboGenomics project are to develop a multi-gene HTS platform for the diagnosis of BPDs, to deposit knowledge about novel pathogenic variants in a sustainable and freely available database, and to leverage systematic Human Phenotype Ontology (HPO)-term based coding of patient phenotypes4 to improve our understanding of genotype-phenotype correlations in BPDs. To deliver the project to high scientific and ethical standards, a global ThromboGenomics network of clinicians and researchers with expertise in BPDs (see supplemental Figure 1, available on the Blood Web site) was formed. Currently, the ThromboGenomics HTS platform can detect variants in the exonic fraction of 63 BPD genes, and many of their introns and untranslated regions (UTRs). Multiplexing allows the sequencing of DNA samples from 24 cases simultaneously. A custom capture is used instead of WES because it provides deeper coverage of the regions of interest for a given number of sequencing reads and allows a higher grade of multiplexing, thereby reducing the cost per patient diagnosed.
Here, we describe the technical performance of the ThromboGenomics platform and its accuracy for detecting causal variants in a curated set of transcripts in 63 BPD genes. We sequenced 300 samples (Figure 1), of which 260 were unrelated, drawn from 4 groups of individuals having the following: diagnostic abnormalities on laboratory assays with previously ascertained pathogenic variants (“known” group; n = 159), phenotypes strongly indicative of a particular disorder on the basis of laboratory abnormalities but without knowledge of causal variants (“suspected” group; n = 61), phenotypes that could not be matched to any known BPD because the laboratory assays were either normal or not diagnostic of an established disorder (“uncertain” group; n = 76), and 4 samples from unaffected relatives.

Breakdown of the 300 samples sequenced with the ThromboGenomics platform. The width of each box is proportional to the number of individuals it represents. The 4 main categories are shown as labels in italics. The shaded area in each box shows the proportion of samples in which pathogenic or likely pathogenic variants were identified with the ThromboGenomics platform. Note that the mother of a hemophilia A patient from the “suspected” group appears in shading in the box representing the “unaffected” group.
Breakdown of the 300 samples sequenced with the ThromboGenomics platform. The width of each box is proportional to the number of individuals it represents. The 4 main categories are shown as labels in italics. The shaded area in each box shows the proportion of samples in which pathogenic or likely pathogenic variants were identified with the ThromboGenomics platform. Note that the mother of a hemophilia A patient from the “suspected” group appears in shading in the box representing the “unaffected” group.
We developed a variant filtering procedure, assessed the platform’s reproducibility, used HPO-based prioritization of candidate variants, and reviewed the quality of the variant pathogenicity literature. Finally, we discuss the rules used by the multidisciplinary team (MDT) to review the sequencing results and generate reports.
Methods
Enrollment, gene and transcript selection, platform design, and sequencing
Individuals were enrolled in 13 different countries. The gene list was established through discussion with experts in BPDs and taking into account the quality of the evidence in the peer-reviewed literature supporting the claim to pathogenicity of variants in each gene, such as the number of unrelated individuals carrying the variants and functional validation of their effects in vitro. Transcripts were selected by experts for each gene, where available. The platform was designed to target all exonic and many intronic, and UTRs of BPD genes. For further details on gene and transcript selection, platform design, sample preparation, and sequencing, see supplemental Methods.
Clinical bioinformatics
The reads in the de-multiplexed paired-end FASTQ files are processed as described in supplemental Materials. Briefly, reads are aligned using BWA9 0.7.10. Then, single nucleotide variants (SNVs) and insertions/deletions (indels) are called using GATK10 3.3 HaplotypeCaller, and large copy number variants (CNVs) are called using ExomeDepth 1.1.5.11 Because it is not possible to call inversions and complex structural rearrangements accurately with capture technology, they are not called.
We infer gender using two statistics based on sequence reads aligned to well-covered target regions (>95% of samples covered at 20X): the ratio between heterozygote and non-reference homozygote genotypes (het/hom) on the X chromosome sites, and the ratio between the median coverage on X and the median coverage on the autosomes (aut/X). The het/hom ratio is computed using heterozygote SNVs with an allele depth between 0.3 and 0.7 to guard against errors.
In order to estimate ethnic background, we project standardized genotypes onto the first two principal components obtained from the standardized genotypes of 2504 HapMap12 individuals sequenced by the 1000 Genomes project.13 We use SNVs falling within well-covered target regions (>95% of samples covered at 20X), having a minor allele frequency (MAF) >0.02 in 1000 Genomes, and pruned with PLINK14 to ensure that r2 < 0.2 between pairs of SNVs.
We annotate SNVs and indels with their predicted impact against Ensembl 75, presence in human gene mutation database (HGMD) 2015.2, and their MAFs in the exome aggregation consortium (ExAC) (http://biorxiv.org/content/early/2015/10/30/030338) release 0.3 and 1000 Genomes13 databases using SnpEff 4.0.15 If variants are in HGMD,16 then they are retained as long as their MAF in ExAC and in 1000 Genomes is <2.5%. Otherwise, variants must have a MAF <0.1% in ExAC and 1000 Genomes, have <4 alternate alleles (to guard against errors in repetitive regions), and have a predicted moderate or high impact on translation of one of the ThromboGenomics transcripts according to SnpEff. In this latter group, we also allow through variants predicted to affect splice regions if they do not fail quality control in ExAC. Finally, any variants with a MAF >10% within the entirety of the ThromboGenomics data set are filtered out to remove potential systematic artifacts. The filtering criteria were informed by the variants in the known category of samples but applied universally.
Our approach allows us to retain confidently called pathogenic variants which are regulatory or moderately common if they are already known to be pathogenic. Such variants include, for instance, the regulatory non-coding SNVs in RBM8A17,18 responsible for Thrombocytopenia-Absent Radius (TAR) syndrome in the presence of a loss-of-function variant on the alternate haplotype, moderately common variants in VWF linked to reduced levels of the VWF protein19-21 and the F5 Leiden22 variant.
HPO-based prioritization of variants
We compute the semantic similarity S of a case’s HPO-coded phenotype to that of the HPO profiles of a BPD gene using the “best-match average” metric23 and Lin’s24 measure of similarity between terms:
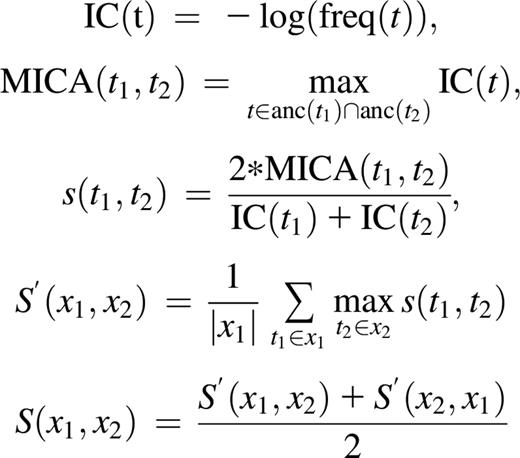
where freq(t) is the frequency of term t in OMIM, anc(t) refers to the union of term t and its ancestors in the HPO, and (x1, x2) refers to the pair of sets of HPO terms being compared.
Web application for variant assessment
Variant calls and phenotypes are visualized in the Sapientia web application (Congenica Inc., Cambridge, United Kingdom) during MDT meetings. Sapientia displays variant information such as predicted effect, MAFs in reference cohorts (eg, ExAC and UK10K25 ), and links to external resources (eg, HGMD, ClinVar, and PubMed), as well as showing case data such as phenotype information in the form of HPO terms. The web application allows the MDT to annotate each variant with respect to its predicted contribution to the disease phenotype and its likely pathogenicity. Following international guidelines,26 variants are marked as pathogenic (high impact or previously observed in at least 4 unrelated cases with a similar pathology), likely pathogenic (previously observed in <4 unrelated cases with a similar pathology), or as having unknown clinical significance. Considerations such as predicted impact (eg, missense or loss of function) and conservation/pathogenicity scores are used to inform how variants are categorized in the context of the observed phenotype. After deliberation, the MDT produces a research report for the referring clinician including information about variants declared pathogenic or likely pathogenic, but not usually about variants of unknown significance (VUS) (supplemental Figure 2). The typical overall turnaround time from sample submission to the production of a research report is less than 16 weeks.
Results
Coverage profile of the ThromboGenomics platform
We have assessed the sequencing coverage profile of the ThromboGenomics platform using data from 300 samples. The mean exonic coverage along the 58 autosomal genes (comprising 228 863 bp; supplemental Table 1) across samples from individuals of both sexes was on average 1178 (range, 123 to 2356) (Figure 2A). The mean fraction of exonic bases covered at 20X and 50X was 0.993 and 0.989, respectively (Figure 2B). We have produced individualized coverage profiles for each gene on the platform showing that virtually all exonic regions of the ThromboGenomics transcripts are covered sufficiently for sensitive variant calling (supplemental File 1). The profile of ITGA2B encoding αIIb of the major platelet integrin is shown in Figure 2C as an example. A small number of short regions that may potentially suffer from low coverage are also highlighted in 19 genes (supplemental Table 2). Overall, 613 bp overlapping coding regions and only 44 out of the 8294 HGMD variants (0.53%) for the 63 genes were covered <20X in 95% of samples. Of these, 22 variants lie in exon 26 of VWF, which is perfectly homologous with part of the von Willebrand pseudogene 1 and 20 HGMD variants are in the highly guanine-cytosine–rich exon 2 of GP1BB. However, a change in the polymerase enzyme used in the library preparation improved the coverage on this (supplemental Figure 3) and other guanine-cytosine–rich regions such that all HGMD variants except for those on exon 26 of VWF could subsequently be called with confidence.

Technical evaluation of the ThromboGenomics platform. (A) Histogram of mean autosomal target coverage for 300 samples. (B) Mean fraction of exonic (solid black) bases and HGMD variants (dashed red) covered at least at 0X, 1X, …, 50X, showing that 99% of the targeted exonic regions are covered by at least 50 sequencing reads. (C) Coverage profile for the ITGA2B gene.
Technical evaluation of the ThromboGenomics platform. (A) Histogram of mean autosomal target coverage for 300 samples. (B) Mean fraction of exonic (solid black) bases and HGMD variants (dashed red) covered at least at 0X, 1X, …, 50X, showing that 99% of the targeted exonic regions are covered by at least 50 sequencing reads. (C) Coverage profile for the ITGA2B gene.
Sample identity assurance
The MDT ensures that the gender (Figure 3A) and ethnic background (Figure 3B), including admixture, inferred from the genotype data match the information provided by the clinical care team. Individuals with European ancestry are over-represented in the large collections of samples used for allele frequency based filtering. Consistent with this notion, individuals with non-European ancestry (particularly East Asian or African) tend to have more candidate variants (after filtering) than European or South Asian individuals (Figure 3B).

Sample identity assurance. (A) The het/hom ratio vs the aut/X ratio is used to infer the gender of each individual. One sample from a male individual with an abnormally high aut/X ratio was substantially more degraded than all others. (B) A scatterplot of the first two principal components derived from the 1000 Genomes genotypes, with individuals colored by major population, and projected ThromboGenomics individuals shown as black circles if they have fewer than 7 candidate variants and triangles if they have at least 7 candidate variants. For clarity, admixed American HapMap individuals are not shown. There is a lower density of ThromboGenomics individuals with African or East Asian ancestry but they all have at least 7 variants, whereas ∼80% of ThromboGenomics individuals with European ancestry have fewer than 7 variants.
Sample identity assurance. (A) The het/hom ratio vs the aut/X ratio is used to infer the gender of each individual. One sample from a male individual with an abnormally high aut/X ratio was substantially more degraded than all others. (B) A scatterplot of the first two principal components derived from the 1000 Genomes genotypes, with individuals colored by major population, and projected ThromboGenomics individuals shown as black circles if they have fewer than 7 candidate variants and triangles if they have at least 7 candidate variants. For clarity, admixed American HapMap individuals are not shown. There is a lower density of ThromboGenomics individuals with African or East Asian ancestry but they all have at least 7 variants, whereas ∼80% of ThromboGenomics individuals with European ancestry have fewer than 7 variants.
Platform reproducibility
The ThromboGenomics platform increases the total length of genome sequenced per case over 80-fold compared with a typical Sanger sequencing-based test,27 which raises the concern that a false diagnosis may arise from a spurious match between phenotype and a falsely called genotype. It is not feasible to verify all variants (including nonpathogenic variants) systematically using an alternative gold standard genotyping method and as such we cannot obtain a direct estimate of the false discovery rate of the ThromboGenomics platform. However, any susceptibility toward spurious genotyping calls would likely manifest in low concordance rates between the variants obtained from different sequencing runs of the same sample. We thus assessed whether the false discovery rate is reasonably controlled by sequencing 6 DNA samples in two separate runs and compared the candidate variants obtained between replicates. We found that every one of the 22 candidates called in either replicate was found in both replicates for all 6 pairs of samples. These results indicate that the library preparation, sequencing, variant calling, and variant filtering altogether produce reproducible results and the risk of erroneous diagnoses due to falsely called variants is negligible.
Overall, 75 samples had at least one of the CNV calls produced by the ExomeDepth algorithm comprising 47 deletions and 28 amplifications. To assess the dependability of these calls, we focused on the 29 heterozygous autosomal deletions, found in 44 different samples, and expected to be significantly depleted of heterozygous SNV calls under the hypothesis that the samples are truly haploid. In 82% of cases, no heterozygous SNVs spanned the putative deleted regions at all and the remainder contained between one and three heterozygous SNVs (supplemental Figure 4). Although some of these heterozygote calls may be due to read misalignment, a small proportion of CNV calls are likely the result of artifactual changes in coverage. Nevertheless, the highly significant overall depletion (P <10−6), coupled with a proven sensitivity to identify pathogenic CNVs (see next section), indicates that a reasonable balance between sensitivity and specificity is achieved by ExomeDepth in the difficult task of identifying CNVs. Given the symmetric modeling of deletions and amplifications, we expect a similar performance in calling duplications.
Performance of variant calling and filtering
The DNA from 300 individuals was sequenced and, across the entire dataset, 20 039 variants were called, of which 520 SNVs, 47 indels, and 75 CNVs remained after automated filtering. The mean number of variants per individual before filtering is 2014.11 and this is reduced to 5.34 candidate causal variants (range, 2 to 12) by the filtering procedure. Assuming there are two causal variants per individual and the filtering method classifies variants into candidates and noncandidates for pathogenicity in the worst possible way gives a lower bound for the specificity of variant filtration of 99.73%. On average, the 5.34 candidate variants consisted of 4.74 SNVs, 0.35 indels, and 0.25 CNVs (Figure 4). The CNV reads ratios were clustered into groups corresponding to different zygosity (Figure 4D).

Candidate variants per sample. (A-C) Bar plots of the number of candidate SNVs, indels, and CNVs per individual. (D) Scatterplot of the Bayes factor vs the observed over expected reads ratio for each CNV called by ExomeDepth and the thresholds distinguishing different levels of changes in zygosity. Note that the number of called CNVs is slightly biased upwards relative to the number of true CNVs because a single underlying CNV can sometimes be coded as multiple adjacent calls by the ExomeDepth algorithm. The number of CNVs surviving filtering is slightly elevated relative to the number of surviving indels, because we include CNV calls with a Bayes factor down to 4.5 for maximum sensitivity and because CNVs do not undergo any external cohort-based frequency filtering. Het, heterozygous.
Candidate variants per sample. (A-C) Bar plots of the number of candidate SNVs, indels, and CNVs per individual. (D) Scatterplot of the Bayes factor vs the observed over expected reads ratio for each CNV called by ExomeDepth and the thresholds distinguishing different levels of changes in zygosity. Note that the number of called CNVs is slightly biased upwards relative to the number of true CNVs because a single underlying CNV can sometimes be coded as multiple adjacent calls by the ExomeDepth algorithm. The number of CNVs surviving filtering is slightly elevated relative to the number of surviving indels, because we include CNV calls with a Bayes factor down to 4.5 for maximum sensitivity and because CNVs do not undergo any external cohort-based frequency filtering. Het, heterozygous.
The candidate variants obtained from 159 individuals in the “known” group were used to assess the sensitivity of the platform. Members of this group had, or shared carrier status with, relatives who had one of 30 different BPDs. They included 19 with MYH9-related disorder, 11 with Glanzmann thrombasthenia, and 10 each with TAR syndrome, Wiskott-Aldrich syndrome (WAS), platelet-type VWD, fibrinogen deficiency, and autosomal dominant thrombocytopenia. A further 8 and 6 individuals had Gray platelet and Hermansky-Pudlak syndromes, respectively. The remaining 65 individuals in this group carried variants underlying other BPDs (Table 1). The previously known variants included 119 SNVs, 19 indels, and 7 large deletions in 37 different BPD genes. Of the 138 SNVs and indels, 102 (73.9%) were HGMD pathogenic variants and the remaining 36 variants were deemed to be causal by the clinician who submitted the sample and confirmed as likely pathogenic by the MDT. The longest indel detected in this group was called in DNA samples from 2 related patients diagnosed with platelet-type VWD due to a heterozygous 27 bp in-frame deletion in GP1BA that removes amino acids 459 to 467. The large deletions varied in size, ranging from at least one exon to entire genes. After initial tuning of the bioinformatics pipeline, all causal variants across known BPD cases were identified after filtering and the genotypes matched the previously determined zygosity of the corresponding samples. Thus, the ThromboGenomics platform has an empirical sensitivity based on these 159 samples harboring 145 causal variants (which do not include inversions) of 100% to detect known causal variants in BPD genes.
The 63 BPD genes present in the ThromboGenomics platform
| Coagulation Factor Disorders | Genes | Main MOI | Known (41) | Suspected (26) | Uncertain (2) |
|---|---|---|---|---|---|
| Combined V and VIII deficiency | LMAN1; MCFD2 | AR; AR | — | — | — |
| Factor V deficiency and factor V Leiden | F5 | AR | 4 | 5 | 1 |
| Factor VII deficiency | F7 | AR | 4 | 5 | — |
| Factor X deficiency | F10 | AR | 2 | — | — |
| Factor XI deficiency | F11 | AR | 2 | 1 | 1 |
| Factor XIII deficiency | F13A1; F13B | AR; AR | 2; 0 | 1; 0 | — |
| Fibrinogen deficiency | FGA; FGB; FGG | AD; AR; AR | 7; 1; 2 | 3; 0; 0 | — |
| Fletcher factor deficiency | KLKB1 | AR | — | — | — |
| Hemophilia A | F8 | XR | 5 | 3 | 0 |
| Hemophilia B | F9 | XR | 5 | — | — |
| Multiple coagulation factor deficiency type 1 | GGCX | AR | — | — | — |
| Multiple coagulation factor deficiency type 2 | VKORC1 | AR | — | — | — |
| Plasminogen activator inhibitor 1 deficiency | SERPINE1 | AR | — | — | — |
| Plasminogen deficiency | PLG | AR | — | 1 | — |
| Prothrombin deficiency | F2 | AR | 2 | 4 | — |
| Kininogen deficiency | KNG1 | AR | — | — | — |
| VWD | VWF | AD | 5 | 3 | — |
| Platelet disorders | Genes | Main MOI | Known(110) | Suspected(23) | Uncertain(4) |
| ADP receptor defect | P2RY12 | AR | 1 | — | — |
| Amegakaryocytic thrombocytopenia with radioulnar synostosis | HOXA11 | AD | — | — | — |
| Autosomal dominant thrombocytopenia | ANKRD26; CYCS; TUBB1 | AD; AD; AD | 1; 4; 5 | 0; 0; 1 | 0; 0; 1 |
| BSS | GP1BA; GP1BB; GP9 | AR; AR;AR | 3; 1; 0 | 0; 1; 0 | — |
| Bleeding diathesis due to glycoprotein VI deficiency | GP6 | AR | 1 | — | — |
| Chediak-Higashi syndrome | LYST | AR | — | — | — |
| CAMT | MPL | AR | 5 | — | — |
| Cyclic thrombocytopenia and thrombocythemia 1 | THPO | AD | — | — | — |
| Deficiency of phospholipase A2, group IVA | PLA2G4A | AR | — | — | — |
| Dense granule abnormalities | NBEA | AD | — | — | — |
| Familial platelet disorder with predisposition to AML | RUNX1 | AD | 2 | — | 2 |
| Filamin A-related disorders | FLNA | XR | 1 | — | — |
| Ghosal syndrome | TBXAS1 | AR | — | — | — |
| Glanzmann thrombasthenia | ITGA2B; ITGB3 | AR; AR | 5; 6 | 4; 0 | — |
| Gray platelet syndrome | NBEAL2 | AR | 8 | — | — |
| Hermansky-Pudlak syndrome | HPS1; AP3B1; HPS3; HPS4; HPS5; HPS6; DTNBP1; BLOC1S3 | AR; AR; AR; AR; AR; AR; AR; AR | 1; 0; 2; 0; 0; 2; 1; 0 | 4; 0; 0; 0; 2; 3; 0; 0 | — |
| May-Hegglin and other MYH9-related disorders | MYH9 | AD | 19 | 4 | 1 |
| Paris-Trousseau thrombocytopenia and Jacobson syndrome | FLI1 | AD | — | — | — |
| Platelet-type VWD | GP1BA | AD | 10 | — | — |
| Québec platelet disorder | PLAU | AD | — | 2 | — |
| TAR syndrome | RBM8A | AR | 10 | — | — |
| Thromboxane A2 receptor defect | TBXA2R | AR | 2 | 1 | — |
| WAS | WAS | XR | 16 | 1 | — |
| X-linked thrombocytopenia with dyserythropoiesis | GATA1 | XR | 4 | — | — |
| Thrombotic disorders | Genes | Main MOI | Known(8) | Suspected(7) | Uncertain(3) |
| Antithrombin deficiency | SERPINC1 | AR | 4 | — | 1 |
| Heparin co-factor 2 deficiency | SERPIND1 | AD | — | — | — |
| Histidine-rich glycoprotein deficiency | HRG | AD | — | — | — |
| Protein C deficiency | PROC | AR | 2 | 3 | 1 |
| Protein S deficiency | PROS1 | AR | 2 | 4 | 1 |
| Thrombomodulin deficiency | THBD | AD | — | — | — |
| Tissue plasminogen activator deficiency | PLAT | AD | — | — | — |
| Coagulation Factor Disorders | Genes | Main MOI | Known (41) | Suspected (26) | Uncertain (2) |
|---|---|---|---|---|---|
| Combined V and VIII deficiency | LMAN1; MCFD2 | AR; AR | — | — | — |
| Factor V deficiency and factor V Leiden | F5 | AR | 4 | 5 | 1 |
| Factor VII deficiency | F7 | AR | 4 | 5 | — |
| Factor X deficiency | F10 | AR | 2 | — | — |
| Factor XI deficiency | F11 | AR | 2 | 1 | 1 |
| Factor XIII deficiency | F13A1; F13B | AR; AR | 2; 0 | 1; 0 | — |
| Fibrinogen deficiency | FGA; FGB; FGG | AD; AR; AR | 7; 1; 2 | 3; 0; 0 | — |
| Fletcher factor deficiency | KLKB1 | AR | — | — | — |
| Hemophilia A | F8 | XR | 5 | 3 | 0 |
| Hemophilia B | F9 | XR | 5 | — | — |
| Multiple coagulation factor deficiency type 1 | GGCX | AR | — | — | — |
| Multiple coagulation factor deficiency type 2 | VKORC1 | AR | — | — | — |
| Plasminogen activator inhibitor 1 deficiency | SERPINE1 | AR | — | — | — |
| Plasminogen deficiency | PLG | AR | — | 1 | — |
| Prothrombin deficiency | F2 | AR | 2 | 4 | — |
| Kininogen deficiency | KNG1 | AR | — | — | — |
| VWD | VWF | AD | 5 | 3 | — |
| Platelet disorders | Genes | Main MOI | Known(110) | Suspected(23) | Uncertain(4) |
| ADP receptor defect | P2RY12 | AR | 1 | — | — |
| Amegakaryocytic thrombocytopenia with radioulnar synostosis | HOXA11 | AD | — | — | — |
| Autosomal dominant thrombocytopenia | ANKRD26; CYCS; TUBB1 | AD; AD; AD | 1; 4; 5 | 0; 0; 1 | 0; 0; 1 |
| BSS | GP1BA; GP1BB; GP9 | AR; AR;AR | 3; 1; 0 | 0; 1; 0 | — |
| Bleeding diathesis due to glycoprotein VI deficiency | GP6 | AR | 1 | — | — |
| Chediak-Higashi syndrome | LYST | AR | — | — | — |
| CAMT | MPL | AR | 5 | — | — |
| Cyclic thrombocytopenia and thrombocythemia 1 | THPO | AD | — | — | — |
| Deficiency of phospholipase A2, group IVA | PLA2G4A | AR | — | — | — |
| Dense granule abnormalities | NBEA | AD | — | — | — |
| Familial platelet disorder with predisposition to AML | RUNX1 | AD | 2 | — | 2 |
| Filamin A-related disorders | FLNA | XR | 1 | — | — |
| Ghosal syndrome | TBXAS1 | AR | — | — | — |
| Glanzmann thrombasthenia | ITGA2B; ITGB3 | AR; AR | 5; 6 | 4; 0 | — |
| Gray platelet syndrome | NBEAL2 | AR | 8 | — | — |
| Hermansky-Pudlak syndrome | HPS1; AP3B1; HPS3; HPS4; HPS5; HPS6; DTNBP1; BLOC1S3 | AR; AR; AR; AR; AR; AR; AR; AR | 1; 0; 2; 0; 0; 2; 1; 0 | 4; 0; 0; 0; 2; 3; 0; 0 | — |
| May-Hegglin and other MYH9-related disorders | MYH9 | AD | 19 | 4 | 1 |
| Paris-Trousseau thrombocytopenia and Jacobson syndrome | FLI1 | AD | — | — | — |
| Platelet-type VWD | GP1BA | AD | 10 | — | — |
| Québec platelet disorder | PLAU | AD | — | 2 | — |
| TAR syndrome | RBM8A | AR | 10 | — | — |
| Thromboxane A2 receptor defect | TBXA2R | AR | 2 | 1 | — |
| WAS | WAS | XR | 16 | 1 | — |
| X-linked thrombocytopenia with dyserythropoiesis | GATA1 | XR | 4 | — | — |
| Thrombotic disorders | Genes | Main MOI | Known(8) | Suspected(7) | Uncertain(3) |
| Antithrombin deficiency | SERPINC1 | AR | 4 | — | 1 |
| Heparin co-factor 2 deficiency | SERPIND1 | AD | — | — | — |
| Histidine-rich glycoprotein deficiency | HRG | AD | — | — | — |
| Protein C deficiency | PROC | AR | 2 | 3 | 1 |
| Protein S deficiency | PROS1 | AR | 2 | 4 | 1 |
| Thrombomodulin deficiency | THBD | AD | — | — | — |
| Tissue plasminogen activator deficiency | PLAT | AD | — | — | — |
BPDs targeted by the ThromboGenomics platform are grouped by disorder type and gene. For each gene and disease, the main MOI and the number of individuals in the “known,” “suspected,” and “uncertain” categories found to carry a pathogenic variant by the ThromboGenomics platform are shown, with sub-totals for each set of disorders shown in parentheses. One patient in the “uncertain” group is shown on 2 rows because she was given a digenic molecular diagnosis involving a likely pathogenic variant in SERPINC1 and another in PROC. GP1BA appears twice because variants therein may be implicated in disorders listed on 2 separate rows (BSS and platelet-type VWD). Note that gain-of-function variants in coagulation factor genes F2, F5, FGA, FGB, and FGG may be involved in thrombotic disorders but these are not shown, with the exception of factor V Leiden.
AD, autosomal dominant; ADP, adenosine diphosphate; AML, acute myeloid leukemia; AR, autosomal recessive; BSS, Bernard-Soulier syndrome; CAMT, congenital amegakaryocytic thrombocytopenia; MOI, mode of inheritance; XR, X-linked recessive.
Yield of the ThromboGenomics platform for cases with a “suspected” etiology
We evaluated the utility of the platform in a clinical diagnostic setting by processing 61 samples from the “suspected” group, of which 52 were unrelated, using the same bioinformatics parameter settings as above. This group included 28, 24, and 9 cases with a coagulation factor, platelet, or thrombotic disorder, respectively. The called variants were reviewed by the MDT in the context of the HPO terms annotated for each case.28 In all but 5 of the 61 cases (91.8%; 90.4% for probands only), the MDT identified pathogenic or likely pathogenic variants that fully or partially explained the disease phenotype (Table 1). Overall, we identified 29 pathogenic and 28 likely pathogenic variants, comprising 44 SNVs, 13 indels, and 1 duplication, of which 28 variants were novel and the remaining ones had previously been deposited in HGMD as BPD-causing variants.
A noteworthy example concerns 2 cases from the same pedigree who were coded with the HPO terms “impaired platelet aggregation,” “spontaneous, recurrent epistaxis,” and “intramuscular hematoma” (Figure 5A). During analysis, all SNVs and indels that passed filtering were determined to be VUS. However, a duplication spanning the entirety of the PLAU gene was uncovered in both pedigree members (Figure 5B), hence a positive diagnosis was reached for Québec platelet disorder.29 This example highlights the value of the ThromboGenomics platform because the standard laboratory marker of this condition, platelet aggregometry, is by no means conclusive.

Case study. (A) HPO encoded phenotype of a case in the “suspected” category, visualized as a graph using the hpoPlot package. Note that “abnormality of leukocytes” is also an “abnormality of the immune system” (not shown). (B) The ratio between observed and expected read depth over the PLAU gene for the case (red) and superimposed over the 95% confidence interval (gray shaded area). In the lower panel, the central position of each exon of the PLAU gene is shown as a vertical bar and the gene coordinates are provided on the horizontal axis. The data indicate that the case carries an additional copy of the PLAU gene (Bayes factor = 145), which is compatible with a diagnosis of suspected Québec platelet syndrome.
Case study. (A) HPO encoded phenotype of a case in the “suspected” category, visualized as a graph using the hpoPlot package. Note that “abnormality of leukocytes” is also an “abnormality of the immune system” (not shown). (B) The ratio between observed and expected read depth over the PLAU gene for the case (red) and superimposed over the 95% confidence interval (gray shaded area). In the lower panel, the central position of each exon of the PLAU gene is shown as a vertical bar and the gene coordinates are provided on the horizontal axis. The data indicate that the case carries an additional copy of the PLAU gene (Bayes factor = 145), which is compatible with a diagnosis of suspected Québec platelet syndrome.
The unexplained cases included 2 with suspected protein S deficiency, 1 with suspected BSS, and 1 with suspected type 1 VWD. We have not yet been able to determine whether this was due to a lack of sensitivity (eg, because of a complex rearrangement that cannot be detected by targeted sequencing), or because the cases had causative variants in relevant regulatory elements or in other trans-acting genes. In type 1 VWD, pathogenic variants are found in VWF in only ∼50% of cases28 using Sanger sequencing. Alternatively, the 3 cases may have an acquired BPD.
We note that several samples in the “suspected” group were submitted after a negative result had been returned by Sanger sequencing in genes thought to harbor causal variants. In 5 cases, this result was overturned by the detection of causal variants by ThromboGenomics sequencing, which was subsequently confirmed in a second round of Sanger sequencing. A noteworthy example concerns 2 members in the same pedigree, initially diagnosed with α/δ-storage pool disease. The ThromboGenomics test results revealed a mutation in the RUNX1 gene, changing the diagnosis to a RUNX-1–associated thrombocytopenia with increased risk of acute myeloid leukemia. A further example illustrating how the ThromboGenomics platform can deliver more pertinent information than a standard single gene-screening approach relates to a 1-year-old boy with thrombocytopenia, normal mean platelet volume, multiple hematomas after preterm birth without any other symptoms, and having parents with normal platelet counts. Variants were found in both MYH9 and WAS that were coded by the MDT as “likely pathogenic” and “VUS,” respectively. His mother carries a variant in WAS (located on the X chromosome) that was previously described for another male WAS patient,30 whereas his father has a variant in MYH9 variant that has not been described previously. Further studies are required to determine whether the co-inheritance of the variants in MYH9 and WAS are causal of the lack of effective hemostasis. This example highlights the advantage of an HTS strategy that can identify potential disease-modifying factors acting in trans over a single gene analysis strategy.
Yield of the ThromboGenomics platform for cases with a highly “uncertain” etiology
The third group of 76 “uncertain” cases, of which 62 are unrelated, is made up of a mixture of cases with unexplained BPDs that are not suggestive of a particular known pathology. This group mainly comprises patients with clinical bleeding problems, but having normal laboratory coagulation and platelet function tests, platelet storage pool disorder, or patients who have had thrombotic events and low protein S levels, although with a normal PROS1 gene. We detected pathogenic or likely pathogenic variants in 8 cases, corresponding to a sensitivity of 10.5% (9.68% for probands only). In 2 cases, the variants uncovered a contributory defect in a coagulation factor that explained the phenotype only partially (eg, due to reduced levels that did not explain the bleeding). In the remaining 6 cases, defects explaining the phenotype in full were found in MYH9, PROC, PROS1, RUNX1, SERPINC1, and TUBB1, including a digenic molecular diagnosis involving a “likely pathogenic” variant in SERPINC1 and another in PROC, possibly explaining the thrombotic phenotype observed in this patient.
Negative Sanger sequencing results were overturned in 3 cases within this group also, demonstrating that the ThromboGenomics platform can outperform Sanger sequencing in terms of sensitivity. However, the vast majority of cases in the “uncertain” group were given a negative result by the MDT, which underscores the need for further research into the molecular etiology of uncharacterized BPDs.
HPO-based prioritization of candidate variants
We transcribed phenotypes linked to the diseases associated with the 63 BPD genes into HPO terms (supplemental Table 3) and obtained HPO terms describing the phenotypes of a subset of the cases in our collection. To assess the potential utility of HPO methods for prioritizing candidate variants, we compared the HPO terms for 109 cases, previously determined to carry a pathogenic or likely pathogenic variant by the MDT, to the HPO profiles linked to the genes in which they carried candidate variants. For example, a case with BSS from the “suspected” group was coded with six HPO terms and subsequently found to carry candidate variants in 4 genes. The homozygous variant in GP1BB, identified independently by the MDT as likely pathogenic, was chosen by the prioritization algorithm as the top candidate because the HPO profile linked to GP1BB was more similar to the HPO profile of the case than the profiles of any of the other 3 genes in which the case had a candidate variant (Figure 6A). The overall results, shown in Figure 6B, indicate that in 85% (93/109) of cases, the correct gene, as identified by the MDT, scored the highest similarity to the case phenotype out of all the candidate variants (P < 10−6). Whenever the top-ranked gene did not correspond to the MDT-designated gene, the difference tended to be smaller than when there was concordance (supplemental Figure 5). Thus, HPO-based prioritization offers a promising route toward streamlining the review process by MDTs.

HPO-based prioritization. (A) HPO profile of a case with BSS encoded as a graph. Note atypical presence of hearing impairment, which is likely unrelated to the BSS. The plot beneath the graph shows the similarities between the patient profile and each gene in which the case has a candidate variant. The profile of GP1BB is the most similar out of the 4 genes with candidate variants. (B) For each of the 109 HPO-coded cases for which a causative variant was assigned by the MDT, the similarity is shown between the case profile and the profiles of the genes in which the case has a candidate variant. The similarity to the gene containing the variant(s) determined to be pathogenic or likely pathogenic in each case (red circles) and the similarity to other genes containing VUS (gray dashes) are shown. Case index 1 corresponds to the BSS case shown in (A).
HPO-based prioritization. (A) HPO profile of a case with BSS encoded as a graph. Note atypical presence of hearing impairment, which is likely unrelated to the BSS. The plot beneath the graph shows the similarities between the patient profile and each gene in which the case has a candidate variant. The profile of GP1BB is the most similar out of the 4 genes with candidate variants. (B) For each of the 109 HPO-coded cases for which a causative variant was assigned by the MDT, the similarity is shown between the case profile and the profiles of the genes in which the case has a candidate variant. The similarity to the gene containing the variant(s) determined to be pathogenic or likely pathogenic in each case (red circles) and the similarity to other genes containing VUS (gray dashes) are shown. Case index 1 corresponds to the BSS case shown in (A).
On the reliability of the variant pathogenicity literature
The presence of a candidate variant in HGMD is often considered a strong indicator of pathogenicity subject to a phenotype match between the disease linked with the variant, the patient’s phenotype, and a consistent mode of inheritance (Table 1). The variants in HGMD belong to different classes depending on whether they are considered disease-causing mutations (labeled “DM” if definitive and “DM?” if the curator had reservations), disease-associated polymorphisms, or other types of variants. The variant with dbSNP ID rs139428292 in the 5′ UTR of the RBM8A gene is causal of TAR if the alternate RBM8A allele harbors a loss-of-function variant. This UTR variant is listed in HGMD as an “in vitro or in vivo functional polymorphism” instead of as a cause of TAR. The factor V Leiden variant rs6025 is listed as a “disease-associated polymorphism with additional supporting functional evidence.” Consequently, we use HGMD variants in all classes in our MDT analysis.
The vast majority of HGMD variants in the 63 BPD genes (7320 out of 8294) have a MAF that is either zero or undetermined in the 60 706 controls from the ExAC database. However, 140 variants have a MAF >1/1000, of which 69 are listed as disease-causing (Figure 7). We reviewed the literature for these 140 variants and concluded that there is sufficient evidence supporting a claim to penetrant pathogenicity for only seven variants (supplemental Table 4). Historically, assignment of pathogenicity has sometimes been based on publications of variants in small pedigrees without large numbers of control samples, or supporting biochemical or cell biology data, such as expression studies. We considered such variants to be of unproven pathogenicity in a Mendelian disease setting in accordance with current standards. The genes F5,31,32 F8,33,34 F11,35 PROS1,35 FLNA,36 THBD,37,38 VWF,39 and WAS40 had the highest rates of these doubtfully annotated variants, with counts ranging between 3 (F5, F11, THBD, and WAS) and 17 (VWF). By way of example, for MYH9, a methionine 1651 threonine is classified in HGMD as DM. However, the MAF is 1.29 in 1000 in ExAC and therefore it cannot underlie a high-penetrance autosomal dominant disorder. Indeed, the authors41 reporting this mutation observed it in a single pedigree in which 2 cases and no unaffected relatives were screened, and decided that its absence in a mere 45 control samples was sufficient to infer causality for the child’s Alport syndrome and the mother’s hearing loss. Regarding F8, the doubtful DM variants are typically in the B-domain and may influence F8 levels but are unlikely to be causal of hemophilia A. With the above considerations in mind, the ThromboGenomics MDT critically assesses the evidence supporting claims to pathogenicity of each candidate variant in the context of allele frequencies in the major variant databases, even for variants that are present in HGMD.
![Figure 7. HGMD variants and corresponding MAFs in ExAC. (A) Truncated log-scale barplot showing the number of HGMD variants by HGMD phenotype (edited for improved legibility). (B) Log-scale barplot showing the number of HGMD variants binned by MAF in ExAC. (C) Histogram of the 140 variants in BPD genes with a MAF in ExAC exceeding 1/1000 (ie, belonging to the blue bins in panel [B]) broken down by HGMD phenotype (edited for improved legibility). The individual Phred-scaled MAFs of the variants (ie, such that 30 corresponds to 1/1000 and 20 to 1/100) are superimposed on the histogram and colored by whether they are classified as disease causing (DM and DM? categories). ADAMTS13, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; IgE, immunoglobulin E; IVF, in vitro fertilization.](/view-large/figure/7513635/2791f7.jpeg)
HGMD variants and corresponding MAFs in ExAC. (A) Truncated log-scale barplot showing the number of HGMD variants by HGMD phenotype (edited for improved legibility). (B) Log-scale barplot showing the number of HGMD variants binned by MAF in ExAC. (C) Histogram of the 140 variants in BPD genes with a MAF in ExAC exceeding 1/1000 (ie, belonging to the blue bins in panel [B]) broken down by HGMD phenotype (edited for improved legibility). The individual Phred-scaled MAFs of the variants (ie, such that 30 corresponds to 1/1000 and 20 to 1/100) are superimposed on the histogram and colored by whether they are classified as disease causing (DM and DM? categories). ADAMTS13, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; IgE, immunoglobulin E; IVF, in vitro fertilization.
HGMD variants and corresponding MAFs in ExAC. (A) Truncated log-scale barplot showing the number of HGMD variants by HGMD phenotype (edited for improved legibility). (B) Log-scale barplot showing the number of HGMD variants binned by MAF in ExAC. (C) Histogram of the 140 variants in BPD genes with a MAF in ExAC exceeding 1/1000 (ie, belonging to the blue bins in panel [B]) broken down by HGMD phenotype (edited for improved legibility). The individual Phred-scaled MAFs of the variants (ie, such that 30 corresponds to 1/1000 and 20 to 1/100) are superimposed on the histogram and colored by whether they are classified as disease causing (DM and DM? categories). ADAMTS13, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; IgE, immunoglobulin E; IVF, in vitro fertilization.
Discussion
We have described a comprehensive and cost-effective strategy for the diagnosis of BPDs. The HTS platform, and accompanying processing and filtering methods, have high sensitivity (100% based on 159 samples) to detect and shortlist causal variants (SNVs, indels, and CNVs) when the variants are known to be in a BPD gene on the ThromboGenomics platform. When the phenotype is strongly indicative of the presence of a particular disease etiology but the variants are unknown, sensitivity remains high (>90% based on 61 samples). Our variant filtering approach has high specificity (>99.5%) because it greatly reduces the number of candidates requiring consideration by the MDT and, as we have shown, HPO-based prioritization methods may reduce the burden on MDTs even further by highlighting pathogenic or likely pathogenic variants as the top candidate in ∼85% of cases. Sanger results have been overturned by results obtained by HTS, and the CNV-calling pipeline compares favorably with other assays such as multiplex ligation-dependent probe amplification.42 In order to facilitate the interpretation of genetic data, variants are annotated against clinically relevant transcripts, which were selected by experts and deposited in the locus reference genomic public reference database for use by clinical genetics laboratories.
At MDT meetings, we assume that truly pathogenic variants in BPD genes generally have a MAF <1/1000 for autosomal recessive disorders, and are likely much rarer for X-linked and dominant disorders. Decisions used to reach a diagnosis are guided by MAFs in major reference databases and data extracted from the literature, which has been deposited in the HGMD database. We have shown that variants in all HGMD classes must be considered as potentially pertinent, yet 140 variants have a MAF in ExAC >1/1000, only 4 of which are established as pathogenic and penetrant, whereas the others either exert small effects or have uncertain clinical significance. The VWF gene, for example, which has an open reading frame length in the top 1% of the genome-wide distribution, has 17 variants labeled as pathogenic or likely pathogenic with a MAF >1/1000 in ExAC. Reasons for this include low control sample numbers and an over-reliance on in vitro function tests, pathogenicity prediction algorithms, and crystallography data. Given the good performance of the sequencing, variant calling, and filtering procedures, the specificity of the ThromboGenomics test as a whole must be determined in large part by the rate at which nonpertinent variants are falsely declared pathogenic. Although we have not been able to measure specificity directly, careful adjudication of the results by the MDT along the lines we have discussed should ensure that false positive reports are rare.
Overall, we have identified 204 distinct pathogenic or likely pathogenic variants, of which 8 are CNVs and 64 are absent from HGMD. The 73 cases where no conclusive diagnosis could be reached will be considered for inclusion in the 100 000 Genomes projects to be analyzed by WGS. Aggregating these cases with the current set of ∼1000 BPD cases already analyzed by WES or WGS will improve the power to identify novel causes of BPDs. Meanwhile, the on-going work of the ThromboGenomics project will improve the catalogs of pathogenic variants for known BPD genes to aid future diagnoses. However, because the ThromboGenomics platform cannot currently identify inversions and ∼45% of severe hemophilia A cases are due to inversions, a simple polymerase chain reaction-based test can be performed to exclude them prior to HTS.
The clinical importance of an affordable HTS test to patient care should not be underestimated. For example, in the United Kingdom, sequencing of only HPS1 and HPS3 genes for patients with a suspected diagnosis of HPS is reimbursed. However, the precise genetic diagnosis of HPS cases is clinically relevant because those with causal variants in the HPS1, 2 and 4 genes may develop lung fibrosis, which requires monitoring, whereas this is not the case for variants in the remaining 6 HPS genes. Furthermore, the identification of variants in genes such as RUNX1 and ETV6 which are associated with a heightened risk of malignancy, would allow patients at risk to benefit from surveillance.
During the validation period of the ThromboGenomics platform, 13 new BPD genes have been reported, and these 13 and a further 25 genes (supplemental Table 5) for cerebral small vessel disease, hereditary hemorrhagic telangiectasia, arteriovenous malformations, and pulmonary arterial hypertension have been included in the next version of the ThromboGenomics platform. For the version reported in this study, we used one capture reaction for every 2 samples and multiplexed 24 samples on a single HiSeq lane. With improved reagents and protocols for multiplexing and the substantial increase in the number of reads per HiSeq lane, the capture of at least 4 samples per reaction will be feasible in the near future, while maintaining excellent coverage. As a result of this modification, the cost per sample tested can be reduced further.
HTS-based tests are rapidly becoming routine in clinical practice and the ThromboGenomics platform is an example of this transformation. The aim is for the ThromboGenomics test (available through www.thrombogenomics.org.uk) to become the first choice for hemostasis and thrombosis physicians and hematologists requiring a molecular diagnosis for BPD cases. This platform and the underpinning principle of freely accessible expert knowledge about genes, transcripts, and causal variants, and the approach of using HPO terms for coding phenotype can be used by reference laboratories to reduce the diagnostic delay in reaching a conclusive molecular diagnosis for BPD patients. We believe that, by facilitating provision of a definitive diagnosis, our platform will bring substantial benefits to the estimated 2 million BPD cases worldwide.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Roche NimbleGen and Beckman Coulter for their support in the initial stages of this project, Congenica Inc for adapting the Sapientia software to the needs of the MDT, and Jo Westmoreland from the Medical Research Council (MRC) Laboratory for Molecular Biology Visual Aids group for providing the world map picture to represent the ThromboGenomics network.
This study, including the enrollment of cases, sequencing, and analysis received support from the National Institute for Health Research (NIHR) BioResource–Rare Diseases. The NIHR BioResource is funded by the NIHR (http://www.nihr.ac.uk). Research in the Ouwehand Laboratory is also supported by grants from Bristol-Myers Squibb, the British Heart Foundation, the British Society of Haematology, the European Commission, the MRC, the NIHR, and the Wellcome Trust; the laboratory also receives funding from National Health Service Blood and Transplant (NHSBT). The clinical fellows received funding from the MRC (C.L. and S.K.W.); the NIHR–Rare Diseases Translational Research Collaboration (S. Sivapalaratnam); and the British Society for Haematology and National Health Service Blood and Transplant (T.K.B.).
Authorship
Contribution: I.S. developed and validated the ThromboGenomics platform, processed samples, sought transcript curators, fostered and coordinated the growth of the ThromboGenomics community, coordinated MDT meetings, and wrote the paper; J.C.S. performed experiments; F.H., S.V.V.D., and S.T. performed data analysis; K.M. maintained the database of transcripts and contributed to MDT meetings; T.K.B., C.L., and S.K.W. were part of the clinical care team and curated genes; T.K.B., C.L., S.K.W., S. Schulman, and M.J.A.V. provided HPO-coding of BPDs; D.G. performed HPO analysis; S.P. supervised ethics documentations; T.K.B., C.L., S.K.W., S. Schulman, S. Sivapalaratnam, M.J.A.V., M.-C.A., M. Ballmaier, G.B., E.B., M. Bertoli, L.B., P.C., R.F., B.F., M. Gattens, M. Germeshausen, D.P.H., Y.M.C.H., A.M.K., R.K., M.P.L., R.L., M.M., C.M.M., P.N., M.O., D.J.P., M.T.R., A.S., C.v.G., S.R.-V., P.G., M.A.L., A.D.M., K.G., and K.F. provided samples; A.P.A. developed database for phenotype collection and HPO coding; M.K. provided bioinformatics support for gene curation; P.F.B., D.B.B., K.F., W.H.O., A.C.G., M.P.L., and P.R. are Cochairs of the Genomics in Thrombosis and Hemostasis International Society on Thrombosis and Haemostatis–Scientific and Standardization Committee; M.C., D.L.F., C.G., J.A.G., D.J.H., J.W.M.H., M.H., N.H., W.H.K., S.K., J.L., A.N., M.N.-A., A.T.N., M.O., P.R., P.A.S., W.S., D.A.W., B.Z., P.G., D.B.B., A.D.M., and K.F. curated the BPD genes; L.C.D. maintained the gene list; J.D.J., R.P.M., and D.W. extracted DNA; K.P. enrolled cases in Belgium; P.P. helped prepare documentation for gene curation; C.J.P. managed the bioinformatics pipeline; A.R. developed the initial probe design; K.G. chaired the MDT meetings; K.G., K.F., S. Sivapalaratnam, and M.A.L. reviewed the paper; W.H.O. conceived and managed the project, wrote the paper, and chaired the Genomics in Thrombosis and Haemostasis International Society on Thrombosis and Haemostatis–Scientific and Standardization Committee; and E.T. supervised the data analysis and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Keith Gomez, The Katharine Dormandy Haemophilia Centre and Thrombosis Unit, Royal Free London National Health Service Foundation Trust, Royal Free Hospital, Pond St, Hampstead, London, NW3 2QG, United Kingdom; e-mail k.gomez@ucl.ac.uk; and Willem H. Ouwehand, University of Cambridge, Department of Haematology, Long Rd, Cambridge, CB2 0PT, United Kingdom; e-mail: who1000@cam.ac.uk.
