

Key Points
North American ATLL has a distinct genomic landscape with a high frequency of prognostic epigenetic mutations, including EP300 mutations.
ATLL samples with mutated EP300 have compromised p53 function and are selectively sensitive to decitabine treatment.

Abstract
Adult T-cell leukemia lymphoma (ATLL) is a rare T cell neoplasm that is endemic in Japanese, Caribbean, and Latin American populations. Most North American ATLL patients are of Caribbean descent and are characterized by high rates of chemo-refractory disease and worse prognosis compared with Japanese ATLL. To determine genomic differences between these 2 cohorts, we performed targeted exon sequencing on 30 North American ATLL patients and compared the results with the Japanese ATLL cases. Although the frequency of TP53 mutations was comparable, the mutation frequency in epigenetic and histone modifying genes (57%) was significantly higher, whereas the mutation frequency in JAK/STAT and T-cell receptor/NF-κB pathway genes was significantly lower. The most common type of epigenetic mutation is that affecting EP300 (20%). As a category, epigenetic mutations were associated with adverse prognosis. Dissimilarities with the Japanese cases were also revealed by RNA sequencing analysis of 9 primary patient samples. ATLL samples with a mutated EP300 gene have decreased total and acetyl p53 protein and a transcriptional signature reminiscent of p53-mutated cancers. Most importantly, decitabine has highly selective single-agent activity in the EP300-mutated ATLL samples, suggesting that decitabine treatment induces a synthetic lethal phenotype in EP300-mutated ATLL cells. In conclusion, we demonstrate that North American ATLL has a distinct genomic landscape that is characterized by frequent epigenetic mutations that are targetable preclinically with DNA methyltransferase inhibitors.
Introduction
Adult T-cell leukemia lymphoma (ATLL) is a rare aggressive T-cell neoplasm, which is caused by a retrovirus (human T-cell lymphotropic virus [HTLV]-1), and carries a dismal prognosis. HTLV-1 has low sequence variability, enabling its sequence to be used as a molecular tool to follow migrations. The most common subtype of HTLV-1 is the cosmopolitan subtype A, which is endemic to Japan, the Caribbean, Central and South America, north and west Africa, and parts of the Middle East.1 Consequently, ATLL is diagnosed most frequently in the Japanese and Caribbean populations2,3 but is likely underreported in Africa, Latin America,4 and the Middle East.5
Despite similar viral subtypes, the prognosis of ATLL is worse in the Caribbean and American populations than in the Japanese population. In our retrospective analysis of a single-center cohort of Caribbean/American ATLL (n = 53), the median overall survival (OS) was only 6.9 months,3,6 similar to other reported North American studies.2,7 The outcomes in Japanese cohorts of ATLL are more favorable, with median survival times for acute, lymphomatous, chronic, and smoldering subtypes of 8.3, 10.6, 31.5, and 55.0 months, respectively.8 The percentage of patients with aggressive subtypes (acute and lymphomatous) in the American cohorts is ∼91%,2,3,7 compared with 78% in the Japanese population.8 Thus, despite a younger age at diagnosis, North American ATLL patients appear to present with significantly more aggressive subtypes of the disease than their Japanese counterparts. The majority of research on this disease is from Japan, where it is endemic, and most preclinical models used in published ATLL studies are Japanese-derived cell lines and xenografts. It is not known whether North American ATLL is genotypically distinct from the Japanese cases and whether such differences, if present, can be correlated with clinical outcomes. Furthermore, understanding the mutational landscape of North American ATLL is critical in an effort to develop new targeted therapies for these patients.
Epidemiology of ATLL in the United States reflects emigration patterns from endemic areas, especially the Caribbean region, and is characterized by an increasing incidence in New York City and Miami.9,10 Montefiore Medical Center treats a significant proportion of ATLL patients in the United States as a result of the large number of Caribbean immigrants in the Bronx, NY.3,6 Here, we present the mutational and transcriptional landscape of Caribbean ATLL and demonstrate that it is characterized by a distinct mutation pattern that targets epigenetic pathways more frequently than that reported for Japanese ATLL. We also demonstrate that, although ATLL samples with EP300 mutations have compromised p53 function, they are hypersensitive to DNA methyltransferase inhibitors (DNMTIs) and, thus, provide a preclinical rationale for treating North American ATLL with epigenetic therapies.
Patients, materials, and methods
Patient samples and cell lines
Specimens were obtained from patients diagnosed with ATLL after Institutional Review Board approval by the Albert Einstein College of Medicine, in accordance with the Declaration of Helsinki. Patient characteristics, including demographics, laboratory parameters, cytogenetics and treatment course, and survival, were collected using retrospective chart review (supplemental Table 1). To establish long-term cultures, CD4 T cells were sorted from patient peripheral blood mononuclear cells using fluorescence-activated cell sorting (FACS) and expanded in Iscove modified Dulbecco medium supplemented with 20% human serum and 100 unit/mL interleukin-2 (IL-2). All short-term and long-term ATLL cultures have been characterized by multiparameter immunophenotyping with a panel of 17 markers, including CD3, CD4, CD25, CD44, FoxP3, CTLA-4, ICOS, and CCR4. The Japanese patient–derived ATLL cell lines ATL43Tb−, Su9T01, ED40515−, and ED41214− were maintained in RPMI 1640 supplemented with 10% fetal bovine serum, whereas the IL-2–dependent cell lines AT55T+, ED40515+, ED41214+, and AT43T+ were maintained in RPMI 1640 plus 10% fetal bovine serum supplemented with IL-2.
Genetic sequencing and clinical characteristics
Genomic DNA was extracted from peripheral blood, bone marrow, or formalin-fixed paraffin-embedded solid tumor samples from 30 ATLL patients and used to identify single nucleotide variants (SNVs), insertion/deletions, copy number variations, and translocation fusion genomic alterations in a panel of up to 173 reportable genes (patients 1-23) and up to 236 genes (patients 24-30) (supplemental Table 2). We also sequenced 8 cell lines (ATL43T+, ATL43Tb−, Su9T01, ATL55T+, ED40515−, ED40515+, ED41214−, and ED41214+) derived from Japanese ATLL patients. Targeted genomic regions were sequenced using next-generation sequencing by Genoptix (https://genoptix.com/test-menu/nexcourse-complete/). The presence or absence of genomic alterations within each of the genes is determined through bioinformatic analysis and comparison with databases (eg, COSMIC and dbSNP). Quality-control metrics included a minimum of 200 ng of genomic DNA and average mean sequencing depth of 500× coverage to give a limit of detection of 5% for SNVs, 10% for insertions/deletions and translocation fusions, gene amplifications ≥ 6 copies, and homozygous gene deletions < 0.3 copies. These mutational data were compared with clinical characteristics and patient outcomes to determine the prognostic impact of particular mutations.
Patient records were queried using Clinical Looking Glass software to identify all cases of HTLV positivity and ATLL by searching pathology and laboratory reports of patients who presented to Montefiore Medical Center between 2003 and 2017.6 The diagnosis of ATLL was confirmed based on clinical history, pathological findings, and HTLV-1 antibody positivity.6 Cases were classified by subtype using Shimoyama criteria.11 The index date was defined as the date on which a diagnosis of ATLL was made. Mortality data included death records within our institution, as well as those reported in Social Security records. For patients who were discharged to hospice with confirmed documentation of refractory disease and unknown date of death (n = 5), the date of discharge to palliative care was used as the date of death.
Statistical analysis
Data generated by Clinical Looking Glass software and complemented with individual chart review were transferred to a computer spreadsheet (Microsoft Excel; Microsoft, Redmond, WA). For the analysis of categorical variables, we reported proportions and P values calculated using the Pearson χ2 test or Fisher exact test, as appropriate. Kaplan-Meier curves were used to compare survival, and statistical significance was examined using the Wilcoxon rank-sum test. Statistical analyses were performed with Stata v12 (StataCorp, College Station, TX), and a 2-tailed α of 0.05 was used to denote significance.
Detailed procedures on in vitro cell cytotoxicity assays, RNA sequencing (RNA-seq), bioinformatics analysis, analysis of drug interaction, mass spectrometry, Western blot, and proviral load analysis are described in supplemental Methods, which are available on the Blood Web site.
Results
Mutational landscape of North American ATLL patients reveals a high frequency of prognostic epigenetic gene alterations
Targeted exon sequencing was performed on samples from 30 North American ATLL patients. They were diagnosed according to the World Health Organization classification12 and subclassified using Shimoyama criteria.11 Of these, 43.3% (n = 13) were acute, 43.3% (n = 13) were lymphomatous, and 13.4% (n = 4) were chronic/smoldering ATLL. Baseline demographic characteristics are shown in supplemental Table 1. For the 173 cancer-related genes analyzed in the assay format, 152 mutations in 80 genes were detected, with an average of 5 identified mutations per patient (Figure 1; Table 1; supplemental Table 2). The acute/lymphomatous cases and the chronic/smoldering cases had an average of 5.2 and 3.75 identified mutations per sample, respectively. Genes most commonly mutated in North American ATLL were TP53 (7/30, 23%), FAT1 (7/30, 23%), EP300 (6/30, 20%), NOTCH1 (6/30, 20%), APC (5/30, 17%), TBL1XR1 (4/30, 13%), and KDR, ALK, and PTCH1 (3/30, 10% each) (Figure 1A; supplemental Tables 3 and 4). In addition to inactivating mutations in the histone acetyltransferase EP300 (Figure 1C-D; supplemental Table 5), mutations were detected in other epigenetic and histone-modifying genes (TET2, EZH2, MED12, PBRM1, DNMT3A, KMT2A, HIST1H1E, SPEN, IDH1, SMARCB1, and ASXL1), resulting in a combined mutational frequency of 57% (17/30).
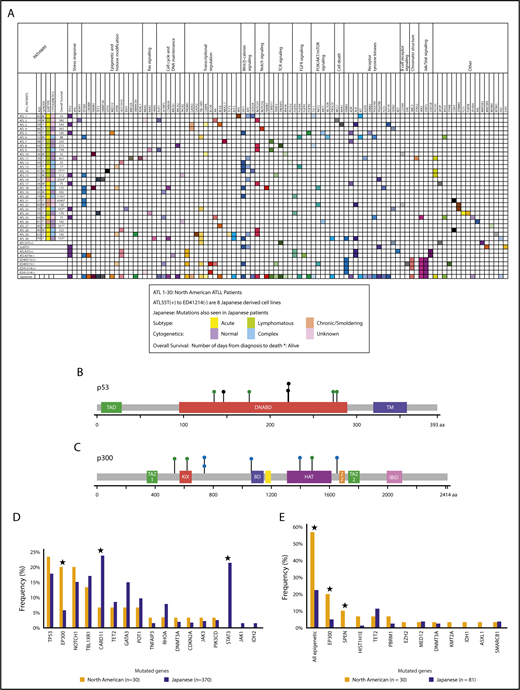
North American ATLL has a distinct mutational landscape. (A) North American ATLL samples (n = 30) and Japanese ATLL cell lines (n = 8) were sequenced by targeted deep next-generation sequencing. Identified mutations are grouped into various functional categories. Corresponding mutations reported in Japanese ATLL are marked in the last row. (B) Location of point mutations in the p53 protein structure. (C) Location of point mutations in the p300 protein structure. Green, blue, and black circles depict missense single nucleotide polymorphisms, splice site single nucleotide polymorphisms, and truncating mutations, respectively. (D) Frequency of mutations in North American and Japanese ATLL. (E) Frequency of epigenetic mutations in North American ATLL and whole-exome sequenced Japanese ATLL cases. *P < .05, North American vs Japanese.
North American ATLL has a distinct mutational landscape. (A) North American ATLL samples (n = 30) and Japanese ATLL cell lines (n = 8) were sequenced by targeted deep next-generation sequencing. Identified mutations are grouped into various functional categories. Corresponding mutations reported in Japanese ATLL are marked in the last row. (B) Location of point mutations in the p53 protein structure. (C) Location of point mutations in the p300 protein structure. Green, blue, and black circles depict missense single nucleotide polymorphisms, splice site single nucleotide polymorphisms, and truncating mutations, respectively. (D) Frequency of mutations in North American and Japanese ATLL. (E) Frequency of epigenetic mutations in North American ATLL and whole-exome sequenced Japanese ATLL cases. *P < .05, North American vs Japanese.
Subtype, OS, and mutational profile of North American ATLL
| ATL no. | Sex | Age at diagnosis, y | Subtype | OS, d | Mutations | Mutations, n | Treatment |
|---|---|---|---|---|---|---|---|
| 1 | Male | 83 | A | 33 | KIT, APC, MYC, TP53, DDX3X, TSC1 | 5 | Unknown |
| 2 | Male | 59 | L | 382 | NRAS, POT1, CEBPA, KEAP1, AR, NOTCH1, MCL1 | 5 | Etoposide, cyclophosphamide, cytarabine, cisplatin |
| 3 | Female | 28 | A | 543 | ALK, BCL6, RIPK1, GATA3, IGF1R, TP53, CEBPA | 7 | CHOP, cytarabine, denileukin diftitox, IT MTX |
| 4 | Female | 66 | L | 363 | NOTCH2, DDR2, GATA3 | 3 | EPOCH |
| 5 | Female | 74 | L | 160 | KDR, FAT1, FGFR2, AKT1, TP53, ERBB2, MED12 | 7 | EPOCH |
| 6 | Male | 53 | A | 88 | XPO1, TBL1XR1, PDGFRA, KIT, FGFR4, EP300, RICTOR | 6 | EPOCH (died after cycle 1) |
| 7 | Female | 48 | L | 225 | KDR, CARD11, NKX2-1(2) | 4 | Cyclophosphamide, doxorubicin, etoposide, MTX |
| 8 | Female | 64 | L | 215 | TNFAIP3, TSC1, BRCA2, NOTCH1 | 3 | Cyclophosphamide, doxorubicin, etoposide |
| 9 | Female | 63 | L | 739 | CARD11, NOTCH1, KRAS, POT1 | 3 | EPOCH |
| 10 | Male | 36 | L | 52 | CEBPA, RHOA, PBRM1, CD79A, STAG2 | 5 | Bortezomib, cytarabine, doxorubicin, etoposide |
| 11 | Female | 70 | L | 451 | NRAS, VHL, APC, EGFR, PTCH1, ASXL1, KMT2A | 6 | DA EPOCH bortezomib raltegravir |
| 12 | Female | 54 | L | 23 | FAT1, HIST1H1E, PLCG2 | 3 | CHOP died after cycle 1 |
| 13 | Female | 37 | A | 17 | FGFR4, APC, FAT1, SPEN | 3 | EPOCH |
| 14 | Male | 41 | L | 771* | EZH2, DDX3X, FAT1, PTCH1 | 4 | EPOCH + allogeneic transplant |
| 15 | Male | 51 | L | 312 | TP53, EP300, PALB2, PTCH1, TRAF3 | 5 | EPOCH |
| 16 | Female | 57 | C | 2554* | APC, PDGFRB, TET2, DNMT3A | 3 | IFN-AZT |
| 17 | Male | 36 | A | 75 | KDR, NOTCH1, TP53, SPOP | 3 | HyperCVAD, CHOP, EPOCH (1 cycle each) |
| 18 | Female | 67 | A | 175 | AKT2, CDKN2A, EP300, FAT1, FBXW7, FLT3, PBRM1, SPEN | 7 | EPOCH + raltegravir + bortezomib + IFN |
| 19 | Male | 65 | A | 392 | SETBP1, EP300 | 2 | EPOCH |
| 20 | Male | 74 | A | 1191* | FGFR3, FAT1 | 2 | EPOCH 1 cycle, IFN |
| 21 | Female | 37 | C/S | 6345* | CDH1, EP300 | 2 | Treated initially as mycosis fungoides, followed by brentuximab |
| 22 | Male | 60 | C/S | 126 | AKT2, ALK, CD79A, EP300, ZMYM3, AR | 6 | None, due to poor performance status and comorbidities |
| 23 | Female | 54 | A | 503* | DDR2, IDH1, TBL1XR1, TP53, ZMYM3 | 5 | ESHAP 1 cycle, followed by EPOCH |
| 24 | Female | 59 | L | 176 | FGFR3, FLT1, KRAS, P2RY8, TET2 | 5 | EPOCH + raltegravir + bortezomib, palliative after cycle |
| 25 | Male | 64 | A | 73 | AR, JAK3, PDGFRA, SETBP1, TP53, XPO1, ZFHX4† | 7 | EPOCH + raltegravir + bortezomib, died after cycle 2 |
| 26 | Female | 61 | S | 102 | AKT1, AXL,†PALB2 | 2 | None |
| 27 | Female | 40 | L | 351* | BCL6, MDM4† | 2 | EPOCH + raltegravir + bortezomib |
| 28 | Male | 40 | A | 322 | CDH1, HIST1H1E, NOTCH1 | 3 | EPOCH + raltegravir + bortezomib |
| 29 | Female | 63 | A | 192 | NOTCH1, APC, APC, GATA2, KLF2,†NTRK1, SMARCB1, SPEN, TBL1XR1, TCF3 | 10 | IFN-AZT, followed by 2 cycles ESHAP, followed by ICE + bortezomib, followed by a clinical trial |
| 30 | Female | 70 | A | 153* | ALK, ALK, CDKN2A, ERBB3, FAS,†FAT1, HRAS, KLF2,†PIK3CD, PIK3CD, TBL1XR1 | 11 | EPOCH |
| ATL no. | Sex | Age at diagnosis, y | Subtype | OS, d | Mutations | Mutations, n | Treatment |
|---|---|---|---|---|---|---|---|
| 1 | Male | 83 | A | 33 | KIT, APC, MYC, TP53, DDX3X, TSC1 | 5 | Unknown |
| 2 | Male | 59 | L | 382 | NRAS, POT1, CEBPA, KEAP1, AR, NOTCH1, MCL1 | 5 | Etoposide, cyclophosphamide, cytarabine, cisplatin |
| 3 | Female | 28 | A | 543 | ALK, BCL6, RIPK1, GATA3, IGF1R, TP53, CEBPA | 7 | CHOP, cytarabine, denileukin diftitox, IT MTX |
| 4 | Female | 66 | L | 363 | NOTCH2, DDR2, GATA3 | 3 | EPOCH |
| 5 | Female | 74 | L | 160 | KDR, FAT1, FGFR2, AKT1, TP53, ERBB2, MED12 | 7 | EPOCH |
| 6 | Male | 53 | A | 88 | XPO1, TBL1XR1, PDGFRA, KIT, FGFR4, EP300, RICTOR | 6 | EPOCH (died after cycle 1) |
| 7 | Female | 48 | L | 225 | KDR, CARD11, NKX2-1(2) | 4 | Cyclophosphamide, doxorubicin, etoposide, MTX |
| 8 | Female | 64 | L | 215 | TNFAIP3, TSC1, BRCA2, NOTCH1 | 3 | Cyclophosphamide, doxorubicin, etoposide |
| 9 | Female | 63 | L | 739 | CARD11, NOTCH1, KRAS, POT1 | 3 | EPOCH |
| 10 | Male | 36 | L | 52 | CEBPA, RHOA, PBRM1, CD79A, STAG2 | 5 | Bortezomib, cytarabine, doxorubicin, etoposide |
| 11 | Female | 70 | L | 451 | NRAS, VHL, APC, EGFR, PTCH1, ASXL1, KMT2A | 6 | DA EPOCH bortezomib raltegravir |
| 12 | Female | 54 | L | 23 | FAT1, HIST1H1E, PLCG2 | 3 | CHOP died after cycle 1 |
| 13 | Female | 37 | A | 17 | FGFR4, APC, FAT1, SPEN | 3 | EPOCH |
| 14 | Male | 41 | L | 771* | EZH2, DDX3X, FAT1, PTCH1 | 4 | EPOCH + allogeneic transplant |
| 15 | Male | 51 | L | 312 | TP53, EP300, PALB2, PTCH1, TRAF3 | 5 | EPOCH |
| 16 | Female | 57 | C | 2554* | APC, PDGFRB, TET2, DNMT3A | 3 | IFN-AZT |
| 17 | Male | 36 | A | 75 | KDR, NOTCH1, TP53, SPOP | 3 | HyperCVAD, CHOP, EPOCH (1 cycle each) |
| 18 | Female | 67 | A | 175 | AKT2, CDKN2A, EP300, FAT1, FBXW7, FLT3, PBRM1, SPEN | 7 | EPOCH + raltegravir + bortezomib + IFN |
| 19 | Male | 65 | A | 392 | SETBP1, EP300 | 2 | EPOCH |
| 20 | Male | 74 | A | 1191* | FGFR3, FAT1 | 2 | EPOCH 1 cycle, IFN |
| 21 | Female | 37 | C/S | 6345* | CDH1, EP300 | 2 | Treated initially as mycosis fungoides, followed by brentuximab |
| 22 | Male | 60 | C/S | 126 | AKT2, ALK, CD79A, EP300, ZMYM3, AR | 6 | None, due to poor performance status and comorbidities |
| 23 | Female | 54 | A | 503* | DDR2, IDH1, TBL1XR1, TP53, ZMYM3 | 5 | ESHAP 1 cycle, followed by EPOCH |
| 24 | Female | 59 | L | 176 | FGFR3, FLT1, KRAS, P2RY8, TET2 | 5 | EPOCH + raltegravir + bortezomib, palliative after cycle |
| 25 | Male | 64 | A | 73 | AR, JAK3, PDGFRA, SETBP1, TP53, XPO1, ZFHX4† | 7 | EPOCH + raltegravir + bortezomib, died after cycle 2 |
| 26 | Female | 61 | S | 102 | AKT1, AXL,†PALB2 | 2 | None |
| 27 | Female | 40 | L | 351* | BCL6, MDM4† | 2 | EPOCH + raltegravir + bortezomib |
| 28 | Male | 40 | A | 322 | CDH1, HIST1H1E, NOTCH1 | 3 | EPOCH + raltegravir + bortezomib |
| 29 | Female | 63 | A | 192 | NOTCH1, APC, APC, GATA2, KLF2,†NTRK1, SMARCB1, SPEN, TBL1XR1, TCF3 | 10 | IFN-AZT, followed by 2 cycles ESHAP, followed by ICE + bortezomib, followed by a clinical trial |
| 30 | Female | 70 | A | 153* | ALK, ALK, CDKN2A, ERBB3, FAS,†FAT1, HRAS, KLF2,†PIK3CD, PIK3CD, TBL1XR1 | 11 | EPOCH |
A, acute; AZT, azidothymidine; CHOP, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin hydrochloride), vincristine (oncovin), and prednisone; DA EPOCH, dose-adjusted etoposide, prednisone, vincristine, cyclophopshamide, doxorubicin; EPOCH, etoposide, prednisone, vincristine (oncovin), cyclophosphamide, and doxorubicin hydrochloride (hydroxydaunorubicin hydrochloride); ESHAP, etoposide, methylprednisone, cytarabine, cisplatin; ICE, ifosfamide, carboplatin, etoposide; IFN, interferon; IT, intrathecal; L, lymphomatous; MTX, methotrexate.
Alive.
Genes that were identified as part of the extended spectrum of 236 genes in patients ATL24 to ATL30.
The cadherin-related tumor suppressor FAT1, mutated in other malignancies,13 including T acute lymphoblastic leukemia,14 showed SNVs in 7 of 30 (23%) of our patients. Nevertheless, germline sequencing revealed that the FAT1 variants in 2 of the samples (ATL14 and ATL18) were of germline origin. On the other hand, comparison with the germline profiles demonstrated that the majority of epigenetic and TP53 mutations were somatic in nature (Figure 1B,D; supplemental Tables 6-8).
North American ATLL is characterized by significantly more epigenetic mutations and fewer JAK/STAT and T-cell receptor/NF-κB pathway mutations compared with Japanese ATLL
We next compared mutation patterns of North American ATLL with a Japanese cohort of 370 patients who underwent targeted capture sequencing of 88 candidate genes selected from a discovery cohort of 83 patient samples that were studied with whole-genome or whole-exome sequencing.15 There were 17 genes that were common to these 2 sequencing panels (173 genes in North American and 88 genes in Japanese) (Figure 1D; supplemental Tables 9-11).
Although many of the mutations identified in this study have been previously reported for Japanese ATLL, several differences were apparent. Because the large Japanese cohort of 370 cases was sequenced for some, but not all, epigenetic genes in our panel, we also compared our findings with their whole-exome data from 81 Japanese cases. More than half of our patients (17/30, 57%) had mutations in epigenetic or histone modifier genes, most notably inactivating mutations in the histone acetyltransferase EP300 (Figure 1C; supplemental Table 5); mutations were also detected in other epigenetic and histone modifying genes (TET2, EZH2, MED12, PBRM1, DNMT3A, KMT2A, HIST1H1E, SPEN, IDH1, SMARCB1, and ASXL1) (Figure 2C). In comparison, these mutations were seen at much lower frequencies in the Japanese cohort (18/81, 22.2%) (Figure 1E; supplemental Table 11). Of note, the frequency of EP300 mutations in the North American cohort (20%) was about 3 times greater than that seen in the Japanese cohort (5.7%; P = .01) (Figure 1E).
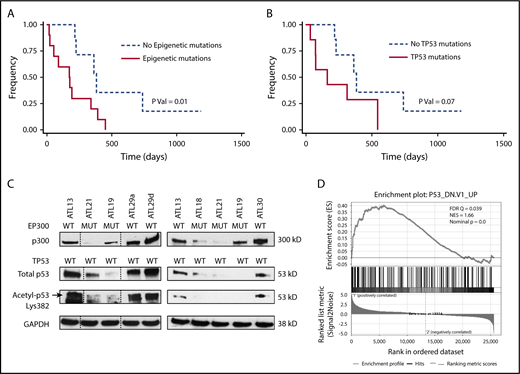
North American ATLL features a high frequency of epigenetic mutations that are prognostic. (A) OS of North American ATLL patients with epigenetic mutations is worse than those without these mutations. (B) OS of North American ATLL patients with TP53 mutations shows a trend toward worse survival, but it is not statistically significant. (C) EP300 mutations are associated with reduced p300 levels, as well as total and acetylated p53 levels, in primary patient samples. (D) Gene set enrichment analysis revealed enrichment of the P53_DN.V1_UP signature in the 2 EP300-mutated samples (ATL21 and ATL18) compared with 2 EP300 WT samples (ATL29 and ATL30).
North American ATLL features a high frequency of epigenetic mutations that are prognostic. (A) OS of North American ATLL patients with epigenetic mutations is worse than those without these mutations. (B) OS of North American ATLL patients with TP53 mutations shows a trend toward worse survival, but it is not statistically significant. (C) EP300 mutations are associated with reduced p300 levels, as well as total and acetylated p53 levels, in primary patient samples. (D) Gene set enrichment analysis revealed enrichment of the P53_DN.V1_UP signature in the 2 EP300-mutated samples (ATL21 and ATL18) compared with 2 EP300 WT samples (ATL29 and ATL30).
Integrated molecular analysis of the Japanese patients showed alterations that are highly enriched for T-cell receptor–NF-κB signaling (including JAK/STAT signaling), T-cell trafficking, and other T-cell–related pathways, as well as immunosurveillance genes.15 These trends were not seen in the North American ATLL cohort. For example, mutations in CARD11 were significantly less common in the North American cohort compared with Japanese cases (6.6% vs 23.8%). In addition, 21.4%, 2.2%, and 1.4% of Japanese patients had mutations in STAT3, JAK3, and JAK1, respectively, whereas in the North American cohort there was no STAT3 or JAK1 mutation, and only 1 patient showed a JAK3 (3.3%) alteration (Figure 1D; supplemental Tables 9 and 11).
Other mutations detected in both groups at comparable frequencies were TP53, NOTCH1, TBL1XR1, TET2, GATA3, POT1, TNFAIP3, DNMT3A, CDKN2A, and JAK3 (Figure 1D). Of note, TP53 was mutated at similar frequencies in these 2 patient cohorts. In the Japanese discovery cohort, 13.6% (11/81) of patients had TP53 mutations, among which acute, lymphomatous, and chronic cases accounted for 55%, 27%, and 18%, respectively. We also performed targeted exon sequencing on 8 Japanese-derived cell lines, because they are frequently used by ATLL investigators. Six of them had mutations in JAK3, STAT3, or STAT5B, consistent with the higher frequency reported for the Japanese cohort (Figure 1A; supplemental Table 14). Mutations identified in these cell lines, but not detected in Japanese or North American patients, include BRCA1, BCOR, FGFR1, RET, SYK, STAT5B, ITPKB, MAP2K2, MAP3K9, and ESR1.
Because the North American cohort has a higher percentage of aggressive cases compared with the Japanese cohort, we performed a subtype-specific comparison after separating the cases into aggressive (acute/lymphomatous) and indolent (chronic/smoldering) subtypes. The result showed that, even when the comparison was restricted to the aggressive cases, epigenetic and EP300 mutations were still detected at higher frequencies in the North American cohort relative to the Japanese cohort (supplemental Table 12).
EP300 mutations correlate with adverse prognosis and are associated with reduced p300 and p53 expression
Next, we evaluated the prognostic significance of the major mutation groups in North American ATLL. TP53 mutations are generally accepted as a marker of adverse prognosis in ATLL, and all 7 TP53 mutations were seen in acute (n = 5) or lymphomatous disease (n = 2) and were absent in patients with chronic/smoldering ATLL (Figure 1B; Table 2; supplemental Table 8). Even though patients with TP53 mutations presented with features of aggressive disease (higher white blood cell [WBC] count and serum calcium) (Tables 2 and 3), TP53 mutation was associated with a nonsignificant trend toward worse OS (P = .07, log-rank test; Figure 2B). Interestingly, as a group, epigenetic mutations did correlate with a significantly worse prognosis (P = .01, log-rank test; Figure 2A). The median survival of patients with and without any epigenetic mutations was 176 and 382 days, respectively.
Clinical presentation by ATLL subtype
| Patient characteristics | Acute (n = 13) | Lymphomatous (n = 13) | Chronic/smoldering (n = 4) |
|---|---|---|---|
| Age, y | 63 | 59 | 58 |
| WBC, 109/ L | 23.1 | 6.4 | 12.25 |
| Corrected calcium, mg/dl | 13.88 | 11.52 | 9.88 |
| Albumin, g/dl | 3.6 | 3.5 | 3.9 |
| Lactate dehydrogenase, U/L | 676 | 561 | 280 |
| TP53 mutation, % (n) | |||
| Mutated | 38 (5) | 15 (2) | 0 (0) |
| Unmutated | 62 (8) | 85 (11) | 100 (4) |
| Any epigenetic mutation, % (n) | |||
| Mutated | 54 (7) | 54 (7) | 75 (3) |
| Unmutated | 46 (6) | 46 (6) | 25 (1) |
| Patient characteristics | Acute (n = 13) | Lymphomatous (n = 13) | Chronic/smoldering (n = 4) |
|---|---|---|---|
| Age, y | 63 | 59 | 58 |
| WBC, 109/ L | 23.1 | 6.4 | 12.25 |
| Corrected calcium, mg/dl | 13.88 | 11.52 | 9.88 |
| Albumin, g/dl | 3.6 | 3.5 | 3.9 |
| Lactate dehydrogenase, U/L | 676 | 561 | 280 |
| TP53 mutation, % (n) | |||
| Mutated | 38 (5) | 15 (2) | 0 (0) |
| Unmutated | 62 (8) | 85 (11) | 100 (4) |
| Any epigenetic mutation, % (n) | |||
| Mutated | 54 (7) | 54 (7) | 75 (3) |
| Unmutated | 46 (6) | 46 (6) | 25 (1) |
Clinical presentation based on mutational status in acute/lymphomatous ATLL
| TP53 (n = 7) | EPI (n = 11) | None (n = 8) | P | |
|---|---|---|---|---|
| Age, y | 54 | 54 | 63 | .51 (between groups) |
| WBC | 19.2 | 14.9 | 5.2 | .04 (TP53 vs none) |
| Albumin | 3.4 | 4.1 | 3.5 | .13 (between groups) |
| Corrected calcium | 13.8 | 11.5 | 11.8 | .02 (TP53 vs none) |
| Lactate dehydrogenase | 2022 | 579 | 586 | .20 (between groups) |
| Median OS, d | 160 | 176 | 382 | |
| Progression-free survival, d | 98 | 128 | 240.5 |
| TP53 (n = 7) | EPI (n = 11) | None (n = 8) | P | |
|---|---|---|---|---|
| Age, y | 54 | 54 | 63 | .51 (between groups) |
| WBC | 19.2 | 14.9 | 5.2 | .04 (TP53 vs none) |
| Albumin | 3.4 | 4.1 | 3.5 | .13 (between groups) |
| Corrected calcium | 13.8 | 11.5 | 11.8 | .02 (TP53 vs none) |
| Lactate dehydrogenase | 2022 | 579 | 586 | .20 (between groups) |
| Median OS, d | 160 | 176 | 382 | |
| Progression-free survival, d | 98 | 128 | 240.5 |
EPI, any epigenetic mutation.
Among the epigenetic alterations detected in our cohort, mutations in the EP300 gene are the most common. Therefore, we examined functional consequences of these mutations in cultured leukemic cells isolated from our patients. As shown in Figure 2C, among the 7 samples examined, the 3 samples with a mutated EP300 status (ATL18, ATL19, and ATL21) expressed notably lower levels of p300 protein, as well as total p53 protein, compared with the 4 EP300 wild-type (WT) samples (ATL13, ATL29a, ATL29d, and ATL30). Because p53 is a protein substrate of p300 lysine acetyltransferase activity,16,17 we also measured acetyl p53 and found reduced acetyl p53 signals in EP300-mutated samples, as predicted. Because acetylation of p53 is indispensable for its in vivo function,18 these results suggest impaired p53 tumor-suppressive activity as a result of EP300 mutation. To substantiate this notion, we performed RNA-seq analysis of 2 pairs of EP300-mutated and WT samples. Gene set enrichment analysis of the differentially expressed genes (supplemental Figure 1) revealed enrichment of genes typically associated with a mutated TP53 status in the NCI-60 tumor cell line panel (Figure 2D), confirming attenuated p53 activity in the EP300-mutated samples.
North American ATLL has a distinct transcriptomic profile and differs from Japanese ATLL
To determine the transcriptomic characteristics of North American ATLL, we performed RNA-seq on primary North American ATLL samples (n = 9), as well as CD4 T cells and peripheral blood mononuclear cells from healthy controls (n = 4) (Figure 3; supplemental Figure 2). Unsupervised clustering analysis of the whole transcriptomes revealed that the ATLL samples had a distinct gene-expression profile compared with normal CD4 T cells (Figure 3A). The overexpressed and underexpressed genes in ATLL (Figure 3B) were enriched in many functionally important pathways (Figure 3C-D), including those associated with cancer, apoptosis, and immune cell–related functions (Figure 3D).
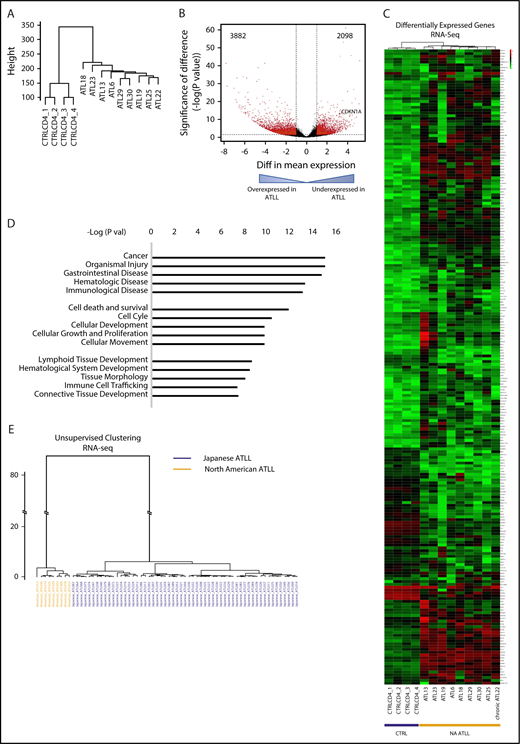
Distinct transcriptomic features are seen in North American ATLL. (A) Unsupervised clustering analysis of RNA-seq profiles of healthy CD4 controls and ATLL samples. (B) Volcano plot shows aberrantly expressed genes in North American ATLL compared with healthy controls. (C) Supervised clustering analysis reveals genes that are aberrantly expressed in North American ATLL. (D) Functional pathways enriched in differentially expressed genes. (E) Unsupervised clustering based on RNA-seq profiles shows transcriptomic differences between Japanese (blue, n = 57) and North American (orange, n = 9) ATLL samples.
Distinct transcriptomic features are seen in North American ATLL. (A) Unsupervised clustering analysis of RNA-seq profiles of healthy CD4 controls and ATLL samples. (B) Volcano plot shows aberrantly expressed genes in North American ATLL compared with healthy controls. (C) Supervised clustering analysis reveals genes that are aberrantly expressed in North American ATLL. (D) Functional pathways enriched in differentially expressed genes. (E) Unsupervised clustering based on RNA-seq profiles shows transcriptomic differences between Japanese (blue, n = 57) and North American (orange, n = 9) ATLL samples.
Given the differences in mutational patterns between the North American and Japanese ATLL cohorts, we also compared the transcriptional profiles. Unsupervised hierarchical clustering revealed that all North American samples clustered in a distinct group that is well separated from the Japanese cohort, suggesting significant differences in transcriptomic features between these 2 patient populations (Figure 3E).
Treatment with the DNMTI decitabine leads to reduced proliferation and increased apoptosis in ATLL
To evaluate the therapeutic usefulness of targeting such epigenetic marks, we tested the sensitivity of North American and Japanese ATLL samples to a DNMTI (decitabine). Among the 6 Japanese-derived cell lines tested, the cell line with mutated EP300 (ATL43Tb−) was highly sensitive (50% inhibitory concentration [IC50] < 1 μM), showing a pronounced decrease in viability after 5 days, whereas IC50 was not reached in the 5 resistant samples with a WT EP300 status (Figure 4A,C).
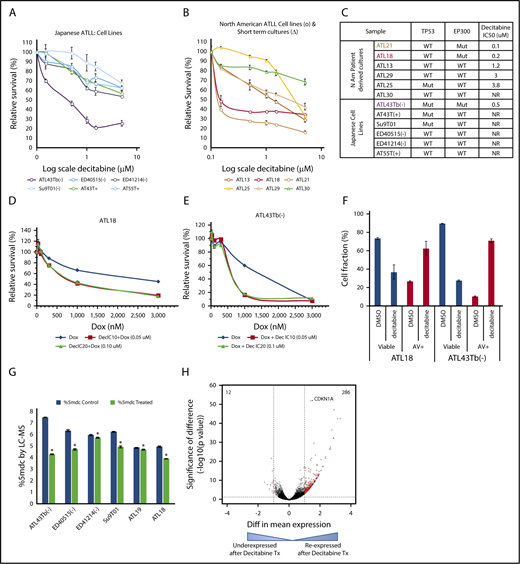
Decitabine has cytotoxic activity in ATLL. Results from cell-viability assays following 5 days of decitabine treatment in Japanese (A) and North American (B) ATLL samples. (C) Summary of decitabine IC50 doses for all samples tested with EP300 and TP53 mutation status. (D-E) Doxorubicin and decitabine combination showed synergistic efficacy in 1 ATLL cell line and 1 North American ATLL culture. (F) Apoptosis measured by Annexin V/propidium iodide staining in decitabine-treated samples. Results shown are representative of 2 (ATL43Tb−) and 3 (ATL18) independent experiments. (G) Reduction in total 5-methylcytosine content after 48 hours of decitabine treatment at 0.6 μM was quantified by mass spectrometry. *P < .05. (H) Volcano plot. RNA-seq analysis revealed many re-expressed genes in 4 primary samples (ATL18, ATL21, ATL29, ATL30) following decitabine treatment.
Decitabine has cytotoxic activity in ATLL. Results from cell-viability assays following 5 days of decitabine treatment in Japanese (A) and North American (B) ATLL samples. (C) Summary of decitabine IC50 doses for all samples tested with EP300 and TP53 mutation status. (D-E) Doxorubicin and decitabine combination showed synergistic efficacy in 1 ATLL cell line and 1 North American ATLL culture. (F) Apoptosis measured by Annexin V/propidium iodide staining in decitabine-treated samples. Results shown are representative of 2 (ATL43Tb−) and 3 (ATL18) independent experiments. (G) Reduction in total 5-methylcytosine content after 48 hours of decitabine treatment at 0.6 μM was quantified by mass spectrometry. *P < .05. (H) Volcano plot. RNA-seq analysis revealed many re-expressed genes in 4 primary samples (ATL18, ATL21, ATL29, ATL30) following decitabine treatment.
Because there are no established cell lines for North American ATLL, we derived short- and long-term CD4 cultures from circulating ATLL cells of our patients and tested their decitabine sensitivity. Six patient-derived ATLL cultures with known EP300 mutational and protein status were treated with decitabine in vitro. Two samples with EP300 mutations (ATL18 and ATL21) were found to be sensitive to decitabine treatment, whereas 4 EP300 WT samples were resistant to decitabine treatment (Figure 4B-C). Annexin V/propidium iodide–based staining confirmed that decitabine triggered apoptosis in the sensitive samples (Figure 4F).
Because the majority of North American ATLL cases present in the aggressive stages2,3,7 and eventually require cytotoxic chemotherapy, we also tested the efficacy of combining decitabine and doxorubicin, a major component of the combination chemotherapy for lymphoid malignancies. In Japanese and North American ATLL samples, this combination synergistically reduced total cell viability (Figure 4D-E). Decitabine treatment led to on-target decreases in total cytosine methylation, as measured by sensitive mass spectrometry analysis (Figure 4G). RNA-seq analysis of 4 pairs of decitabine-treated North American ATLL cells (ATL18, ATL21 ATL29, and ATL30) demonstrated 286 probes (red dots) that passed both thresholds (absolute log 2-fold change > 1.0 and adjusted P value < .05) and were upregulated following decitabine treatment (Figure 4H). Among the decitabine-induced genes was CDKN1A, a p53 target gene that encodes the cell cycle inhibitor p21/WAF. Other re-expressed genes mapped to important pathways, such as cellular development and growth, cell death and survival, cell cycle, cancer, organismal injury and abnormalities, tissue development, and cell morphology (supplemental Table 13).
Discussion
North American patients have a more aggressive clinical course and present at a younger age compared with Japanese patients.3,8 The differences in aggressiveness, despite the same HTLV-1 serotype, may be accounted for by different genetic alterations. Our study examines the mutational landscape and transcriptomics of North American ATLL. We report that, compared with Japanese ATLL patients, North American ATLL is characterized by a higher frequency of prognostic epigenetic mutations and fewer T-cell receptor/NF-κB and JAK/STAT mutations.15 We also characterized functional consequences of EP300 mutations, the most common epigenetic alteration identified in our patient cohort. Our results suggest that ATLLs with a mutated EP300 gene have compromised p53 activity, which could account, at least in part, for their therapy-resistant characteristics, and are hypersensitive to DNMTIs, such as decitabine.
This is the first North American cohort studied using a next-generation sequencing approach. The JAK/STAT signaling pathway is constitutively active in ATLL and has been documented in in vitro19,20 and in vivo studies.21 IL-2 is an autocrine growth factor for activated T cells. In vitro, HTLV-1–infected cultures become IL-2 independent with time, and demonstrate constitutive activation of the JAK/STAT signaling pathway.19,20 As such, it is not surprising that 23.8% of the Japanese cases carry activating mutations in STAT3, JAK3, or JAK1. However, these mutations were rarely detected in North American ATLL (Figure 1D). This may be accounted for by the lower numbers of chronic/smoldering cases in our cohort, because STAT3 mutations are preferentially detected among indolent cases in the Japanese cohort.22 The gene encoding CARD11, a cytoplasmic scaffolding protein required for T-cell receptor– and B-cell receptor–mediated activation of the NF-κB signaling pathway,23 was also mutated much more frequently in Japanese patients compared with North American cases (Figure 1D). It is possible that these differences in mutational profile reflect different pathogenic mechanisms, including the relative importance of dysregulated epigenetic vs cell signaling programs.
We identified a high number of epigenetic mutations in our patients. Epigenetic mutations in TET2, DNMT3A, and IDH have been well described in T-cell lymphomas.24,25 Mutations in TET2 (10/31, 32%) and MLL3 have also been described in ATLL.26 EP300, a histone acetyltransferase, had a significantly higher rate of mutation in North American ATLL patients. This gene has been characterized as a tumor suppressor and plays an important role in cell proliferation and differentiation via transcriptional regulation by chromatin remodeling.27 Mutations in EP300 have been identified in diffuse large B cell lymphoma,28,29 epithelial cancers,27 and ATLL.15 In our patient cohort, 6 (20%) of the cases carry inactivating mutations in EP300, the functional loss of which has been shown to be associated with a gain of repressive histone marks and gene silencing.30 Additionally, 2 (6.7%) of our cases harbor inactivating mutations in TET2. Such mutations are associated with increased DNA methylation and aberrant gene silencing in myeloid leukemias.31 Recently, it was also reported that p300-dependent acetylation stabilizes TET2, which, in turn, inhibits cancer-associated CpG island hypermethylation.32 If this p300–Tet2–CpG island methylation connection functions in ATLL as well, TET2 inactivation and reduced p300 activity (due to p300 mutation/underexpression) are expected to impair this regulatory pathway, leading to the CpG island hypermethylation phenotype, a hallmark of ATLL.15
TP53 mutations are known to be associated with worse prognoses; however, the prognostic impact of epigenetic mutations has not been described before for ATLL. In our cohort of 30 cases, mutations in TP53 and EP300 were mutually exclusive with 1 exception. Although TP53 mutations alone did not reach statistical significance due to cohort size (Figure 2B), cases carrying mutations in any of the epigenetic modifier genes had adverse survival relative to those with WT genes (Figure 2A).
Our observation of reduced p300 and acetylated p53 in EP300-mutated primary samples (Figure 2C) is consistent with a published study in diffuse large B-cell lymphoma, in which mutated EP300/CREBBP was found to cause defective p53 acetylation,33 as well as with the fact that acetylated p53 is more active as a transcription activator that can upregulate its own expression.17 Other evidence supporting the biological significance of EP300 mutations is our RNA-seq analysis, which shows enrichment of a mutated p53 transcriptional signature in the EP300-mutated samples (Figure 2D).
Prior reports have shown that belinostat, a histone deacetylase inhibitor, along with azidothymidine, can trigger apoptosis of ATLL cell lines derived from North American patients.34 Previous studies have shown that ATLL has a polycomb repressive complex 2–mediated trimethylation at histone H3Lys27 in about half of the genes in ATLL cells; these global alterations involved ATLL-specific gene-expression changes that included several tumor suppressors, transcription factors, epigenetic modifiers, microRNAs, and developmental genes.35 Combined with these previous reports, our findings strengthen the notion that the dysregulated epigenetic program plays an important role in ATLL pathogenesis, as well as the therapeutic response.
Aberrant DNA hypermethylation is associated with inappropriate transcriptional silencing of many important genes, including tumor suppressors; thus, it leads to unregulated cell growth and development of malignancies.36 It has been implicated in the pathogenesis of ATLL,15 as well as many other malignancies.37 Hypermethylation of numerous genes has been described in the pathogenesis of ATLL. PDLIM2 (a potent suppressor of Tax),38 CDKN2A (a cell cycle regulatory gene),39,40 C2H2 zinc-finger genes and MHC class I genes,15 BMP-6 (a regulator of cell growth),41 SHP1, HCAD, and DAPK,42 and APC43 have been found to be hypermethylated in ATLL, with increasing incidence as the disease progresses. Because most chemotherapeutic agents depend on the same apoptosis and differentiation pathways of cells for their action, hypermethylation and inactivation of tumor suppressors are often associated with marked chemoresistance, resulting in failure of chemotherapy.36
Chemosensitization with DNMTIs has been successfully demonstrated in patients with diffuse large B cell lymphoma,36 ovarian cancer,44 and breast cancer.45 It was noted that demethylation, followed by reprogramming at cancer-critical loci, is key to chemosensitization. In this regard, we also observed a significant synergy between doxorubicin and decitabine in North American ATLL cells. Of note, decitabine used as single agent caused significant cytotoxicity, as well as demethylation, which is infrequently seen in acute myeloid leukemia. Most importantly, the close correlation between EP300 mutation status and decitabine sensitivity suggests that decitabine treatment induced a synthetic lethal phenotype in EP300-mutated cases. Taken together, our findings show that North American ATLL has a distinct set of genetic alterations that may cause the chemorefractoriness seen in these patients. This study also provides a preclinical rationale for the use of epigenetic therapies in the treatment of North American ATLL with epigenetic alterations.
Part of the exon sequencing data was presented in poster form at the 58th annual meeting of the American Society of Hematology, San Diego, CA, 3 December 2016. Some of the data were presented in poster form at the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 9 December 2017.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Thomas Waldmann and Michael Petrus (National Institutes of Health/National Cancer Institute, Bethesda, MD), Naomichi Arima (Kagoshima University, Kagoshima, Japan) for the gift of the Japanese ATLL cell lines, and Naomichi Arima for valuable advice on establishing long-term ATLL cultures. Flow cytometry analyses were performed at the Albert Einstein College of Medicine Flow Cytometry facility, which is supported by National Institutes of Health National Cancer Institute Cancer Center Support grant P30CA013330. FACS sorting was performed at the Einstein Human Stem Cell FACS and Xenotransplantation Facility, which is supported by a grant from the New York State Department of Health (NYSTEM Program) for the shared facility (C029154). The authors also acknowledge the Einstein Epigenomics Core for assistance with RNA-seq analysis. The authors thank Xiaoxin Ren for technical assistance with isolating CD4 cells from control samples, as well as our ATLL patients and their families without whose support and cooperation this work would not have been possible.
This work was supported by Leukemia & Lymphoma Society Translational Research grant 6471-15 (B.H.Y.) and AIDS Malignancy Consortium grant UM1CA121947 (M.J.). E.Y.C. is supported by the Harry Eagle Scholarship from the Department of Cell Biology, Albert Einstein College of Medicine. U.A.S. is supported by the hematology/oncology fellowship program, Montefiore Medical Center and Jacobi Medical Center.
Authorship
Contribution: U.A.S. designed the study, collected and analyzed data, performed the experiments, designed the figures, and wrote the manuscript. E.Y.C., S.G.-M., O.G., T.D.B., Y.M., Y. Wei, E.I., G.S.C., N.G., C.P., and M.B. performed experiments. K.P. performed biostatistical and RNA-seq analysis. K.K. and S.O. collaborated and shared the Japanese targeted gene and RNA-seq data. R.R. and Y. Wang diagnosed and collected patient samples. S.G., I.M., A.S., O.D., A.B., K.G., N.K., I.B., A.V., M.J., and U.A.S. diagnosed and treated ATLL patients and collected samples. C.B., A.G., and J.H. assisted in gene sequencing for ATLL patients. A.J. and L.R. performed proviral load assays. A.V., B.H.Y., and M.J. designed the study, analyzed data, designed the figures, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Amit Verma, Albert Einstein College of Medicine, 1300 Morris Park Ave, Chanin Building, Room 302B, Bronx, NY 10461; e-mail: amit.verma@einstein.yu.edu; B. Hilda Ye, Albert Einstein College of Medicine, 1300 Morris Park Ave, Chanin Building, Room 302C, Bronx, NY 10461; e-mail: hilda.ye@einstein.yu.edu; Murali Janakiram, 111 E 210 ST, Hoffheimer 100, Bronx, NY, 10467; e-mail: mjanakir@montefiore.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal