

Key Points
Mass spectrometry–based HLA ligandome analysis of primary CML patient samples revealed a panel of novel CML-associated target antigens.
These antigens induced multifunctional T-cell responses and may be used as targets for T-cell–based immunotherapeutic approaches.

Abstract
Antileukemia immunity plays an important role in disease control and maintenance of tyrosine kinase inhibitor (TKI)-free remission in chronic myeloid leukemia (CML). Thus, antigen-specific immunotherapy holds promise for strengthening immune control in CML but requires the identification of CML-associated targets. In this study, we used a mass spectrometry–based approach to identify naturally presented HLA class I– and class II–restricted peptides in primary CML samples. Comparative HLA ligandome profiling using a comprehensive dataset of different hematological benign specimens and samples from CML patients in deep molecular remission delineated a panel of novel frequently presented CML-exclusive peptides. These nonmutated target antigens are of particular relevance because our extensive data-mining approach suggests the absence of naturally presented BCR-ABL– and ABL-BCR–derived HLA-restricted peptides and the lack of frequent tumor-exclusive presentation of known cancer/testis and leukemia-associated antigens. Functional characterization revealed spontaneous T-cell responses against the newly identified CML-associated peptides in CML patient samples and their ability to induce multifunctional and cytotoxic antigen-specific T cells de novo in samples from healthy volunteers and CML patients. Thus, these antigens are prime candidates for T-cell–based immunotherapeutic approaches that may prolong TKI-free survival and even mediate cure of CML patients.
Introduction
Chronic myeloid leukemia (CML) is characterized by the translocation t(9;22) that leads to the formation of the BCR-ABL fusion transcript.1,2 To inhibit the resulting fusion protein, which mediates constitutive tyrosine kinase activity, 5 approved tyrosine kinase inhibitors (TKIs) are available that have led to an impressive improvement in the prognosis of CML patients.3-7 Currently, the main treatment goal in CML is the achievement of a so-called “deep molecular response” (MR), in which discontinuation of TKI therapy can be considered. However, only few patients are able to permanently stop TKI therapy without suffering from molecular relapse.8,9 Thus, lifelong TKI therapy is the standard of care for most CML patients, but it can be associated with significant side effects and the risk of developing resistance to TKIs.10,11 Several studies provided evidence that immunological control may contribute to and even represent a marker for the achievement of deep MR in CML patients under TKI treatment (CMLTKI patients) and treatment-free remission (TFR). The restoration of immune responses is characterized by increased natural killer (NK)-cell and T-cell responses,12 reduced PD-1 expression on T cells,12 the correlation of CD62L expression on T cells13 in patients with MR, and the association between increased NK-cell count14 and CD86+ plasmacytoid dendritic cell count and function15 with TFR.
In turn, reinforcing CML-specific immune responses by T-cell–based immunotherapy may serve to enlarge the fraction of patients achieving long-term TFR or even cure. It has been shown that “nonspecific” immunotherapy approaches, such as allogeneic stem cell transplantation or interferon-α (IFN-α) therapy, enable long-lasting remissions in CML patients after discontinuation of TKI therapy.16-20 Immune checkpoint inhibitors, which have revolutionized the treatment of many solid tumors in recent years,21-23 are currently being evaluated in CML therapy.24 More advanced strategies to treat CML patients comprise agents inducing an immune response specifically directed against the leukemic cells, such as vaccines,25-27 T-cell receptor mimic antibodies,28-30 or engineered T cells.31,32 The prerequisite for such T-cell–based immunotherapeutic approaches is the identification of targets for CML-specific T-cell responses, which, in general, are represented by tumor-associated HLA-presented peptides on malignant cells.33,34 Several studies have suggested neoepitopes arising from tumor-specific mutations as central specificities of checkpoint inhibitor–induced T-cell responses in solid tumors with high mutational burden.33,35 However, the role of neoantigens for T-cell responses in cancer entities with low mutational burden, including CML, remains unclear. In addition to neoantigens, we and other investigators identified nonmutated tumor-associated HLA peptides that are able to induce peptide-specific T-cell responses and can serve as targets for T-cell–based immunotherapy approaches.36-39 In recent years, we implemented the characterization of such tumor-associated antigens in hematological malignancies (HMs) based on the direct isolation of naturally presented HLA ligands from leukemia cells and their subsequent identification by mass spectrometry (MS). Thus far, for acute myeloid leukemia, chronic lymphocytic leukemia (CLL), and multiple myeloma, we identified >100 tumor-exclusive highly frequent antigens that were validated as immunogenic targets for T-cell–based immunotherapy approaches.38,40,41 An extensive meta-analysis of our HM immunopeptidome data revealed only a small set of entity-spanning antigens that was predominantly characterized by low presentation frequencies within the different patient cohorts,42 indicating that T-cell–based immunotherapies for HMs should be designed in an entity-specific manner. For CML, very few nonmutated tumor-associated antigens43-46 or peptides derived from the BCR-ABL fusion region47-49 have been described and validated as immunogenic targets of anticancer T-cell responses.50-52 Here, we comprehensively mapped the landscape of naturally presented HLA class I and II peptides in primary CML samples to identify novel CML-associated antigens covering a broad range of HLA allotypes. These antigens were further validated for their potential to induce T-cell responses, particularly in the context of immunomodulatory effects induced by TKI treatment in CML patients.53-56
Methods
A detailed description of the methods used can be found in supplemental Methods (available on the Blood Web site).
Patients and blood samples
Peripheral blood mononuclear cells (PBMCs) from CML patients were collected at the Departments of Hematology and Oncology in Tübingen, Leipzig, and Aachen, Germany. Informed consent was obtained in accordance with the Declaration of Helsinki protocol. The study was performed according to the guidelines of the local ethics committees. Patient characteristics are provided in supplemental Table 1.
HLA surface molecule quantification
HLA surface expression was determined using a QIFIKIT quantification flow cytometric assay (Dako).40,57 Cells were stained with the pan-HLA class I–specific W6/32, HLA-DR–specific L243 monoclonal antibodies (mAbs), or isotype control. Surface marker staining was performed with fluorescence-conjugated antibodies against CD33, CD13, CD117, and CD34.
Isolation of HLA ligands
Analysis of HLA ligands by liquid chromatography–tandem MS
HLA ligand extracts were analyzed as described previously.38 Peptides were separated by nanoflow high-performance liquid chromatography. Eluted peptides were analyzed in an online-coupled LTQ Orbitrap XL mass spectrometer. Furthermore, parallel reaction monitoring targeting BCR-ABL– and ABL-BCR–derived peptides (supplemental Table 2) was performed on an Orbitrap Fusion Lumos mass spectrometer.
Data processing
Data processing was performed as described previously.38,57 Proteome Discoverer (v1.3, Thermo Fisher Scientific) was used to integrate the search results of the Mascot search engine (v2.2.04, Matrix Science) against the human proteome (Swiss-Prot database). For the search of BCR-ABL– and ABL-BCR–derived neoantigens, the human proteome was extended by BCR-ABL sequences from the TrEMBL database and by published ABL-BCR sequences.59,60
The false discovery rate (FDR; estimated by the Percolator algorithm 2.0461 ) was limited to 5% for HLA class I and 1% for HLA class II. HLA class I annotation was performed using SYFPEITHI 1.062 and NetMHCpan 3.0.63,64 The lists of HLA class I and II peptides identified on CML, CMLMR, and hematological benign tissue samples are provided in supplemental Data Set 1.
Peptide synthesis
Peptides were produced with the Liberty Blue Automated Peptide Synthesizer (CEM) using the 9-fluorenylmethyl-oxycarbonyl/tert-butyl strategy.65
Amplification of peptide-specific T cells and IFN-γ ELISPOT assay
aAPC priming of naive CD8+ T cells
Cytokine and tetramer staining
The functionality of peptide-specific CD8+ T cells was analyzed by intracellular cytokine staining (ICS).66,68 Cells were pulsed with peptide, brefeldin A, and GolgiStop. Staining was performed using mAbs against CD8, tumor necrosis factor (TNF), IFN-γ, and CD107a. The frequency of peptide-specific CD8+ T cells was determined by anti-CD8 and tetramer staining.69
Cytotoxicity assay
The cytolytic capacity of peptide-specific CD8+ T cells was analyzed using the flow cytometry–based VITAL assay.70,71 Autologous target cells were loaded with test peptides or irrelevant control peptides and labeled with CFSE or FarRed, respectively. Effector cells were added at the indicated effector-to-target ratios. Specific lysis of peptide-loaded target cells was calculated relative to control targets.
Data availability
The MS data have been submitted to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE72 partner repository (dataset identifier PXD010450).
Results
Myeloid and precursor cells of primary CML samples express high levels of HLA molecules
T-cell–based immunotherapy requires sufficient HLA expression on target cells, which, in the case of CML, consist of myeloid cells and myeloid precursor cells. Thus, as a first step, we quantified HLA surface expression on CD33+ and CD13+ myeloid cells, as well as on CD117+CD34+ precursor cells, using PBMCs from CML patients (n = 7; supplemental Table 1). HLA class I surface levels showed substantial heterogeneity, with molecule counts per cell of 78 600 to 202 100 (mean 153 600) for CD33+ cells and 69 500 to 322 000 (mean 184 600) for CD13+ cells (Figure 1A). HLA class II expression ranged from 37 700 to 230 000 (mean 101 200) molecules per cell for DR+CD33+ cells and from 39 000 to 286 300 (mean 158 900) molecules per cell for DR+CD13+ cells (Figure 1B). Notably, the highest HLA surface levels were detected on precursor cells, with 112 000 to 316 500 (mean 221 100) and 99 700 to 487 200 (mean 228 300) molecules per cell for HLA class I and II, respectively (Figure 1).
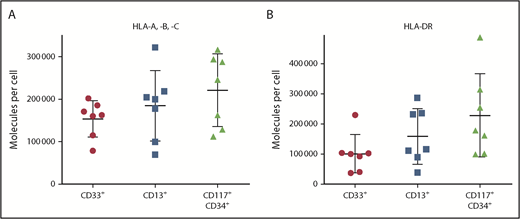
HLA surface expression of primary CML cells. HLA class I (A) and HLA-DR (B) expression was determined by flow cytometry for CD33+ and CD13+ myeloid cells, as well as for CD117+CD34+ precursor cells, from the peripheral blood of CML patients (n = 7) at the time of diagnosis. Data points represent individual samples. Horizontal lines indicate mean values ± standard deviation.
HLA surface expression of primary CML cells. HLA class I (A) and HLA-DR (B) expression was determined by flow cytometry for CD33+ and CD13+ myeloid cells, as well as for CD117+CD34+ precursor cells, from the peripheral blood of CML patients (n = 7) at the time of diagnosis. Data points represent individual samples. Horizontal lines indicate mean values ± standard deviation.
MS identifies naturally presented CML-associated HLA class I ligands in CML patient samples
MS analysis of 21 primary CML samples revealed a total of 11 945 unique HLA class I ligands (range 535-2107; mean 1080 per sample) from 5478 source proteins (supplemental Figure 3A; supplemental Data Set 1), obtaining 82% of the estimated maximum attainable coverage in HLA ligand source proteins (Figure 2A). For the identification of CML-associated antigens, we established a comparative cohort of hematological benign tissues (n = 108), including PBMCs (n = 63), granulocytes (n = 14), CD19+ B cells (n = 5), bone marrow (n = 18), and CD34+ hematopoietic progenitor cells (HPCs; n = 8). A total of 51 232 naturally presented HLA class I ligands (range 101-7587; mean 1404 per sample) from 11 437 source proteins (supplemental Data Set 1), obtaining 95% of maximum attainable coverage (supplemental Figure 4A), were identified. Furthermore, we created an additional comparative benign ligandome dataset of PBMCs from CML patients in deep molecular remission (CMLMR, n = 15) comprising a total of 5907 unique HLA class I ligands (range 311-1145; mean 655 per sample; supplemental Data Set 1).
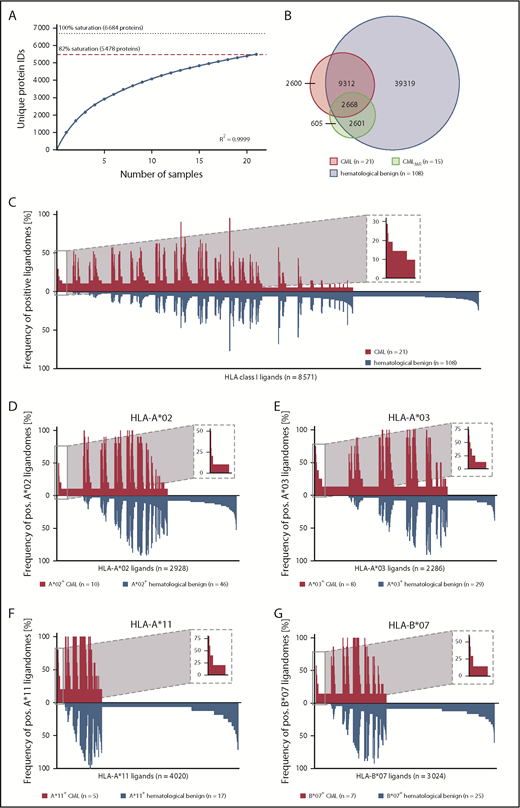
Comparative HLA class I ligandome profiling and identification of CML-associated antigens. (A) Saturation analysis of HLA class I ligand source proteins of the CML patient cohort. Number of unique HLA ligand source protein identifications are shown as a function of cumulative HLA ligandome analysis of CML samples (n = 21). Exponential regression allowed for the robust calculation (R2 = 0.9999) of the maximum attainable number of different source protein identifications (dotted line). The dashed red line depicts the source proteome coverage achieved in our CML patient cohort. (B) Overlap analysis of HLA class I ligand identifications of primary CML samples (n = 21), CMLMR samples (n = 15), and hematological benign samples (n = 108), including PBMCs (n = 63), granulocytes (n = 14), CD19+ B cells (n = 5), bone marrow (n = 18), and CD34+ HPCs (n = 8). (C) Comparative profiling of HLA class I ligands based on the frequency of HLA-restricted presentation in CML and hematological benign ligandomes. Frequencies of positive immunopeptidomes for the respective HLA ligands (x-axis) are indicated on the y-axis. To allow for better readability, HLA ligands identified on <5% of the samples within the respective cohort were not depicted in this plot. The box on the left and its magnification highlight the subset of CML-associated antigens showing CML-exclusive high frequent presentation. Allotype-specific comparative profiling of HLA-A*02–positive (D), HLA-A*03–positive (E), HLA-A*11–positive (F), and HLA-B*07–positive (G) samples, as described above. ID, identifications; pos., positive.
Comparative HLA class I ligandome profiling and identification of CML-associated antigens. (A) Saturation analysis of HLA class I ligand source proteins of the CML patient cohort. Number of unique HLA ligand source protein identifications are shown as a function of cumulative HLA ligandome analysis of CML samples (n = 21). Exponential regression allowed for the robust calculation (R2 = 0.9999) of the maximum attainable number of different source protein identifications (dotted line). The dashed red line depicts the source proteome coverage achieved in our CML patient cohort. (B) Overlap analysis of HLA class I ligand identifications of primary CML samples (n = 21), CMLMR samples (n = 15), and hematological benign samples (n = 108), including PBMCs (n = 63), granulocytes (n = 14), CD19+ B cells (n = 5), bone marrow (n = 18), and CD34+ HPCs (n = 8). (C) Comparative profiling of HLA class I ligands based on the frequency of HLA-restricted presentation in CML and hematological benign ligandomes. Frequencies of positive immunopeptidomes for the respective HLA ligands (x-axis) are indicated on the y-axis. To allow for better readability, HLA ligands identified on <5% of the samples within the respective cohort were not depicted in this plot. The box on the left and its magnification highlight the subset of CML-associated antigens showing CML-exclusive high frequent presentation. Allotype-specific comparative profiling of HLA-A*02–positive (D), HLA-A*03–positive (E), HLA-A*11–positive (F), and HLA-B*07–positive (G) samples, as described above. ID, identifications; pos., positive.
The CML cohort included a total of 31 different HLA class I allotypes, with the most frequent being HLA-C*07 (n = 11), HLA-A*02 (n = 10), HLA-A*03 (n = 8), HLA-B*07 (n = 7), and HLA-B*35 (n = 6; supplemental Figure 5A). Among the world’s population, 99.3% of individuals carry ≥1 HLA class I allotype that is represented within this cohort73,74 (supplemental Figure 6A). The comparative hematological benign cohort showed an HLA allotype population coverage of 99.9% (supplemental Figure 6B) and matched 89% of HLA-A allotypes, 100% of HLA-B allotypes, and 88% of HLA-C allotypes within the CML cohort (supplemental Figure 5B).
To identify CML-associated antigens, we performed comparative HLA class I ligandome profiling of the CML cohort with the hematological benign and CMLMR cohorts. Overlap analysis revealed that 2600 HLA class I ligands were presented exclusively on CML samples (Figure 2B) and never detected on hematological benign or CMLMR samples. For the identification of broadly applicable CML-associated antigens, we aimed for the selection of target antigens that not only fulfill the criterion of CML exclusivity, but also exhibit high prevalence within the CML cohort. At a target-definition FDR <5% (<1%) a total of 23 (5) HLA class I ligands with a representation frequency ≥19% (≥24%) were identified (Figure 2C; supplemental Figure 7A; supplemental Table 4). The most common HLA allotype restrictions of these HLA ligands included HLA-A*02, HLA-A*03, HLA-A*11, and HLA-B*07. To identify CML-associated targets with even higher representation frequencies, we subsequently performed HLA allotype-specific immunopeptidome profiling. Setting the target FDR <5% (<1%), we identified 4 (1) HLA-A*02-, 35 (15) HLA-A*03-, 3 (0) HLA-A*11-, and 8 (2) HLA-B*07-restricted ligands with representation frequencies of ≥40% (≥50%), ≥38% (≥50%), ≥80% (≥80%), and ≥43% (≥57%), respectively (Figure 2D-G; supplemental Figure 7B-E; supplemental Table 4). To further validate these CML-associated targets, we compared them with an additional benign dataset comprising 28 different nonhematological tissue entities (n = 166; eg, liver, lung, brain, skin) with a total of 128 590 unique HLA class I peptides from 16 405 source proteins. Thus, we selected a panel of 8 CML-exclusive target antigens, including 2 HLA-A*02–restricted, 3 HLA-A*03–restricted, 1 HLA-A*11–restricted, and 2 HLA-B*07–restricted ligands, for further immunological characterization.
HLA class II ligandome profiling delineates 3 novel groups of CML-associated antigens
Mapping the HLA class II ligandomes of 20 primary CML samples, we identified 5991 different HLA class II–restricted peptides (range 172-1162; mean 641 per sample) derived from 1302 source proteins (supplemental Figure 3B; supplemental Data Set 1), achieving 72% of maximum attainable coverage (Figure 3A). Our HLA class II hematological benign tissue cohort (n = 88; PBMCs, n = 38; granulocytes, n = 18; CD19+ B cells, n = 9; bone marrow, n = 15; CD34+ HPCs, n = 8) contained 42 753 unique peptides (range 111-6267; mean 1197 per sample) from 4877 source proteins (supplemental Data Set 1), obtaining 84% of maximum attainable coverage (supplemental Figure 4B). The benign CMLMR ligandome dataset (n = 15) included a total of 1529 HLA class II peptides (range 74-281; mean 164 per sample; supplemental Data Set 1).
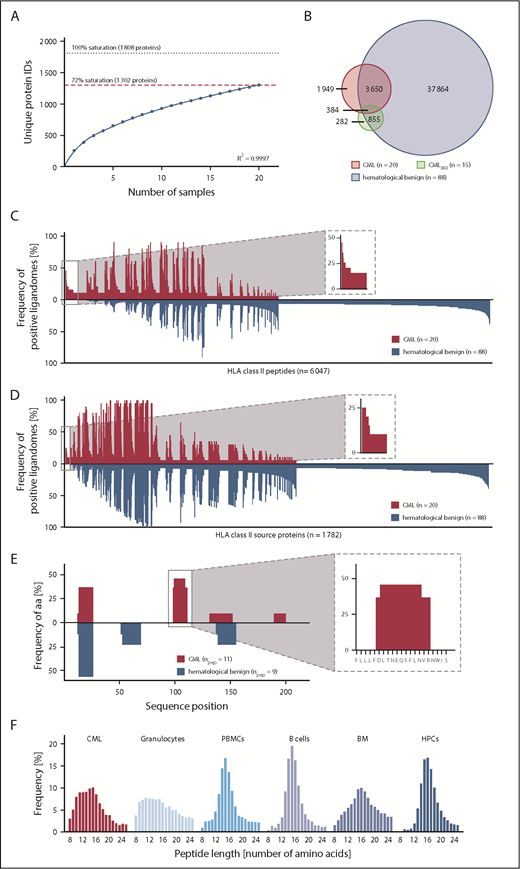
Comparative HLA class II ligandome profiling and identification of CML-associated antigens. (A) Saturation analysis of HLA class II peptide source proteins of the CML patient cohort. Number of unique HLA peptide source protein identifications as a function of cumulative HLA ligandome analysis of CML samples (n = 20). Exponential regression allowed for the robust calculation (R2 = 0.9997) of the maximum attainable number of different source protein identifications (dotted line). The dashed red line depicts the source proteome coverage achieved in our CML patient cohort. (B) Overlap analysis of HLA class II peptides of primary CML samples (n = 20), CMLMR samples (n = 15), and hematological benign samples (n = 88), including PBMCs (n = 38), granulocytes (n = 18), CD19+ B cells (n = 9), bone marrow (n = 15), and CD34+ HPCs (n = 8). Comparative profiling of HLA class II peptides (C) and HLA class II source proteins (D) based on the frequency of HLA-restricted presentation in CML and hematological benign ligandomes. The frequencies of positive immunopeptidomes for the respective HLA peptides or source proteins (x-axis) are indicated on the y-axis. To allow for better readability, HLA peptides or source proteins identified on <5% of the samples within the respective cohort are not depicted. The boxes on the left and their magnifications highlight the subset of CML-associated antigens showing CML-exclusive high frequent presentation in CML samples. (E) Hotspot analysis of the protein RB27A by peptide clustering. Identified peptides were mapped to their amino acid positions within the source protein. Representation frequencies of amino acid counts within each cohort for the respective amino acid position (x-axis) were calculated and are indicated on the y-axis. The box on the left and its magnification highlight the identified hotspot with the respective amino acids on the x-axis. (F) Tissue-specific HLA class II peptide length distribution (number of amino acids) of all identified peptides on primary CML samples (n = 20), granulocytes (n = 18), PBMCs (n = 38), CD19+ B cells (n = 9), bone marrow (n = 15), and CD34+ HPCs (n = 8). aa, amino acids; IDs, identifications; npep, number of peptides.
Comparative HLA class II ligandome profiling and identification of CML-associated antigens. (A) Saturation analysis of HLA class II peptide source proteins of the CML patient cohort. Number of unique HLA peptide source protein identifications as a function of cumulative HLA ligandome analysis of CML samples (n = 20). Exponential regression allowed for the robust calculation (R2 = 0.9997) of the maximum attainable number of different source protein identifications (dotted line). The dashed red line depicts the source proteome coverage achieved in our CML patient cohort. (B) Overlap analysis of HLA class II peptides of primary CML samples (n = 20), CMLMR samples (n = 15), and hematological benign samples (n = 88), including PBMCs (n = 38), granulocytes (n = 18), CD19+ B cells (n = 9), bone marrow (n = 15), and CD34+ HPCs (n = 8). Comparative profiling of HLA class II peptides (C) and HLA class II source proteins (D) based on the frequency of HLA-restricted presentation in CML and hematological benign ligandomes. The frequencies of positive immunopeptidomes for the respective HLA peptides or source proteins (x-axis) are indicated on the y-axis. To allow for better readability, HLA peptides or source proteins identified on <5% of the samples within the respective cohort are not depicted. The boxes on the left and their magnifications highlight the subset of CML-associated antigens showing CML-exclusive high frequent presentation in CML samples. (E) Hotspot analysis of the protein RB27A by peptide clustering. Identified peptides were mapped to their amino acid positions within the source protein. Representation frequencies of amino acid counts within each cohort for the respective amino acid position (x-axis) were calculated and are indicated on the y-axis. The box on the left and its magnification highlight the identified hotspot with the respective amino acids on the x-axis. (F) Tissue-specific HLA class II peptide length distribution (number of amino acids) of all identified peptides on primary CML samples (n = 20), granulocytes (n = 18), PBMCs (n = 38), CD19+ B cells (n = 9), bone marrow (n = 15), and CD34+ HPCs (n = 8). aa, amino acids; IDs, identifications; npep, number of peptides.
For the identification of HLA class II–restricted CML-associated antigens, we established an innovative HLA class II ligandome–profiling platform that delineated 3 groups of antigens: peptide targets, protein targets, and hotspot targets. First, we performed comparative ligandome profiling at the peptide level. Overlap analysis revealed that 1949 peptides were exclusively presented on CML (Figure 3B) and were never detected on hematological benign or CMLMR samples. Of these, 36 peptides were identified with a representation frequency ≥20% based on an FDR <1%; however, 30 of 36 peptide targets showed length variants (>50% overlap) presented on benign hematological samples and, therefore, were excluded (peptide targets; Figure 3C; supplemental Figure 8A; supplemental Table 5). Further ligandome profiling was performed at the HLA class II source protein level. Based on an FDR <5% (<1%), a total of 4 (2) source proteins were identified with a frequency ≥20% (≥25%) representing 10 (4) unique HLA class II peptides (protein targets; Figure 3D; supplemental Figure 8B; supplemental Table 5). As a third group of CML-associated antigens, we analyzed CML-exclusive hotspots by peptide clustering, which validated the previously described targets and identified 1 additional CML-associated hotspot with a representation frequency of 20% comprising 3 unique HLA class II peptides (hotspot targets; Figure 3E; supplemental Table 5). Subsequent validation of these targets using our nonhematological benign tissue dataset (n = 166, 28 tissues, 143 652 HLA class II peptides, 13 410 source proteins) delineated a panel of 6 strongly CML-associated target antigens for immunological characterization.
Notably, most of the identified targets showed unusual short peptide lengths for HLA class II–restricted peptides (mean 12 amino acids), which are reflected by a general length distribution shift in myeloid cell–containing samples representing shorter HLA class II–restricted peptides (Figure 3F; supplemental Figure 9).
The roles of CTAs, LAAs, and BCR-ABL–derived neoantigens in the immunopeptidome of CML
In addition to the definition of novel CML-associated antigens, we focused on the identification and ranking of established cancer/testis antigens (CTAs)75,76 and leukemia-associated antigens (LAAs)43,77 in our dataset of naturally presented HLA peptides. We identified 170 different HLA class I peptides and 382 HLA class II peptides from 39 and 18 CTAs, respectively, as well as 1429 HLA class I peptides and 3428 HLA class II peptides from 131 and 77 LAAs, respectively (supplemental Tables 6-9). Notably, these antigens were represented in CML immunopeptidomes, as well as on hematological benign samples (Figure 4). Hence, this analysis delineated only a small panel of 7 (4% of total) CML-exclusive, but infrequent, CTAs and LAAs that represent suitable candidates for T-cell–based immunotherapy in selected CML patients.
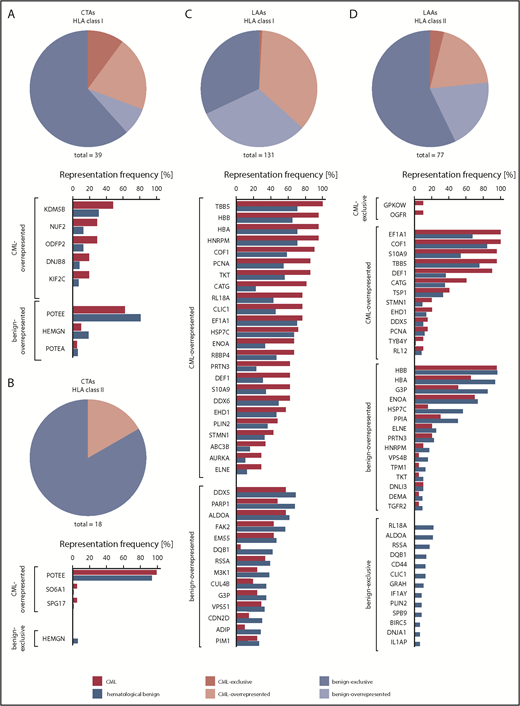
Representation of published CTAs and LAAs in CML and hematological benign HLA ligandomes. Representation frequencies of published CTAs in HLA class I (A) and class II (B) ligandomes, as well as published LAAs in HLA class I (C) and class II (D) ligandomes in CML patient and hematological benign samples. Pie charts represent the total amount of identified CTAs and LAAs assigned to their degree of CML association (ie, CML-exclusive, CML-overrepresented, benign-overrepresented, benign-exclusive). Bar diagrams depict the relative representation (%) of the respective antigens on CML and hematological benign samples allocated to their CML association. Only antigens with representation frequencies >5% (A-B,D) or >25% (C) in the respective cohort are shown.
Representation of published CTAs and LAAs in CML and hematological benign HLA ligandomes. Representation frequencies of published CTAs in HLA class I (A) and class II (B) ligandomes, as well as published LAAs in HLA class I (C) and class II (D) ligandomes in CML patient and hematological benign samples. Pie charts represent the total amount of identified CTAs and LAAs assigned to their degree of CML association (ie, CML-exclusive, CML-overrepresented, benign-overrepresented, benign-exclusive). Bar diagrams depict the relative representation (%) of the respective antigens on CML and hematological benign samples allocated to their CML association. Only antigens with representation frequencies >5% (A-B,D) or >25% (C) in the respective cohort are shown.
Because the characteristic BCR-ABL translocation may result in the presentation of BCR-ABL or ABL-BCR neoepitopes, we further screened our CML cohort for naturally presented BCR-ABL–derived and ABL-BCR–derived peptides by data-dependent acquisition of all CML samples, as well as by targeted parallel reaction monitoring of 4 CML samples (supplemental Table 2). Despite the fact that the BCR-ABL and ABL-BCR fusion sites potentially provide HLA-binding motifs for several HLA allotypes, no naturally presented HLA peptides were identified.
HLA class I–restricted CML-associated antigens induce functional peptide-specific T cells in samples from HVs and CML patients
To confirm immunogenicity and detect preexisting memory T-cell responses against the identified CML-associated antigens (Figure 5A), we performed IFN-γ ELISPOT and tetramer staining assays using HLA-matched PBMCs from CMLTKI patients and HVs. We observed IFN-γ secretion for 1 of 8 CML-associated ligands in 2 of 17 (12%) analyzed CMLTKI patients (Figure 5B), as well as for 2 peptides in 1 HV, respectively (supplemental Figure 10). It appears to be unlikely that cross-reacting microorganism-specific or virus-specific T cells are the reason for the observed T-cell responses in single HVs, because no sequence similarity was found between the CML-associated antigens and the proteins from microorganisms and viruses. In addition, low frequent peptide-specific CD8+ T cells were detected by tetramer staining for 4 of 8 peptides in 3 of 18 CMLTKI patient samples after a 12-day stimulation without any detectable preexisting peptide-specific T cells ex vivo prior to stimulation (supplemental Figure 11). To assess the immunogenicity of the remaining HLA class I–restricted ligands, we performed in vitro aAPC-based priming experiments using CD8+ T cells from HVs and CML patients. Effective priming and expansion of antigen-specific T cells were observed for all 8 CML-associated peptides in ≥70% of analyzed HVs, with frequencies of peptide-specific T cells ranging from 0.1% to 33.9% (mean 2.2%) within the CD8+ T-cell population (Figure 5A,C; supplemental Figure 12). Furthermore, all analyzed CML-associated peptides induced peptide-specific T cells using CML patient samples with frequencies of 0.1% to 2.2% (mean 0.4%) within the CD8+ T-cell population (Figure 5A,D). Notably, peptide-specific immune responses were even induced in CMLTKI patient samples that had not shown preexisting immune responses. Priming experiments with control peptides frequently presented by HLA-A*02 and HLA-A*03 on tumor and benign tissues (peptide presentation >90% in HLA-matched sources) confirmed the CML specificity of the induced T-cell responses (Figure 5E). Furthermore, multifunctionality of peptide-specific T cells was shown for 6 of 8 CML-associated peptides by IFN-γ and TNF production and upregulation of the degranulation marker CD107a (Figure 6A-B). Finally, cytotoxicity assays with polyclonal peptide–specific effector T cells revealed the capacity to induce antigen-specific lysis for 3 of 4 analyzed peptides (Figure 6A,C-E; supplemental Figure 13).
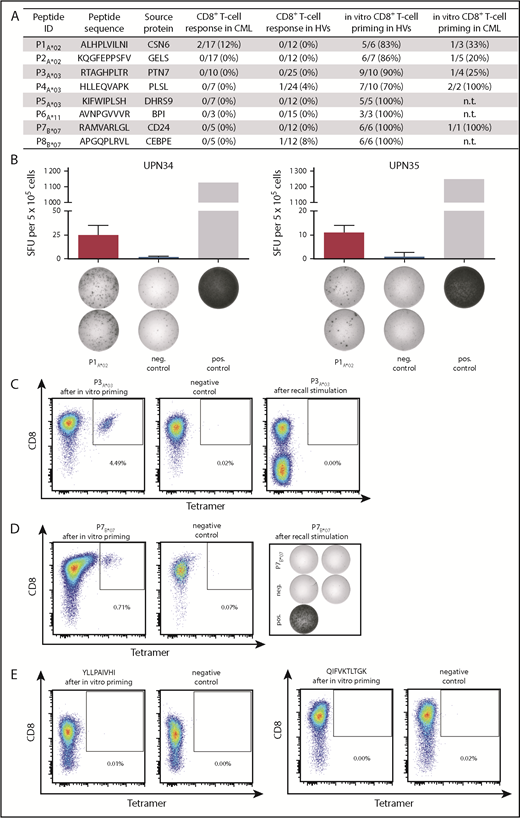
Immunogenicity of HLA class I–restricted CML-associated antigens. (A) Immunogenicity analysis results for the 8 HLA class I–restricted CML-associated peptides with their respective frequencies of preexisting immune recognition by PBMCs from CML patients or HVs in IFN-γ ELISPOT assays (CD8+ T-cell response in CML/HVs), as well as the frequencies of peptide-specific CD8+ T cells detected after in vitro aAPC-based priming experiments with naive CD8+ T cells from HVs and CML patients. (B) Examples of CML-associated ligands evaluated in IFN-γ ELISPOT assays after a 12-day stimulation using PBMCs from CML patients. Results are shown for immunoreactive peptides only. Phytohemagglutinin was used as positive control and the HLA-A*02–restricted DDX5_HUMAN148-156 peptide YLLPAIVHI served as negative control. Data are expressed as mean ± standard deviation of 2 independent replicates. Naive CD8+ T cells from HVs (C) and CML patients (D) were primed in vitro using aAPCs. Graphs show single viable cells stained for CD8 and PE-conjugated multimers of indicated specificity. Tetramer staining was performed after 4 stimulation cycles with peptide-loaded aAPCs. The left panels show P3A*03-tetramer (C) or P7B*07-tetramer (D) staining. The middle panels (negative control) depict P3A*03-tetramer (C) or P7B*07-tetramer (D) staining of respective T cells primed with an irrelevant peptide. The right panels show T cells from the same donor that were tested for the absence of preexisting memory T cells after a 12-day recall stimulation by tetramer staining (C) or IFN-γ ELISPOT assay (D). (E) Tetramer staining after 4 stimulation cycles with negative control peptide-loaded aAPCs (HLA-A*02, YLLPAIVHI, DDX5_HUMAN148-156 and HLA-A*03, QIFVKTLTGK, UBC_HUMAN2-11). ID, identification; neg., negative; n.t., not tested; pos., positive; SFU, spot-forming unit; UPN, uniform patient number.
Immunogenicity of HLA class I–restricted CML-associated antigens. (A) Immunogenicity analysis results for the 8 HLA class I–restricted CML-associated peptides with their respective frequencies of preexisting immune recognition by PBMCs from CML patients or HVs in IFN-γ ELISPOT assays (CD8+ T-cell response in CML/HVs), as well as the frequencies of peptide-specific CD8+ T cells detected after in vitro aAPC-based priming experiments with naive CD8+ T cells from HVs and CML patients. (B) Examples of CML-associated ligands evaluated in IFN-γ ELISPOT assays after a 12-day stimulation using PBMCs from CML patients. Results are shown for immunoreactive peptides only. Phytohemagglutinin was used as positive control and the HLA-A*02–restricted DDX5_HUMAN148-156 peptide YLLPAIVHI served as negative control. Data are expressed as mean ± standard deviation of 2 independent replicates. Naive CD8+ T cells from HVs (C) and CML patients (D) were primed in vitro using aAPCs. Graphs show single viable cells stained for CD8 and PE-conjugated multimers of indicated specificity. Tetramer staining was performed after 4 stimulation cycles with peptide-loaded aAPCs. The left panels show P3A*03-tetramer (C) or P7B*07-tetramer (D) staining. The middle panels (negative control) depict P3A*03-tetramer (C) or P7B*07-tetramer (D) staining of respective T cells primed with an irrelevant peptide. The right panels show T cells from the same donor that were tested for the absence of preexisting memory T cells after a 12-day recall stimulation by tetramer staining (C) or IFN-γ ELISPOT assay (D). (E) Tetramer staining after 4 stimulation cycles with negative control peptide-loaded aAPCs (HLA-A*02, YLLPAIVHI, DDX5_HUMAN148-156 and HLA-A*03, QIFVKTLTGK, UBC_HUMAN2-11). ID, identification; neg., negative; n.t., not tested; pos., positive; SFU, spot-forming unit; UPN, uniform patient number.

Functional characterization of CML-associated antigen-specific CD8+T cells. (A) Functional characterization of CML-associated antigen-specific CD8+ T cells, including their CD107a and cytokine expression profile detected by ICS following aAPC-based priming experiments and their cytotoxic capability (VITAL assay). (B) Representative example of increased IFN-γ and TNF production, as well as CD107a expression, after stimulation with the respective P7B*07-peptide used for the stimulation with aAPCs compared with the corresponding negative control peptide (HLA-B*07, TPGPGVRYPL, NEF_HV1BR128-137). Phorbol myristate acetate (PMA) and ionomycin served as positive control. The P7B*07-specific CD8+ T-cell population showed a frequency of 1.01%, as detected by tetramer staining (far left panel). (C-E) Selective cytotoxicity of P5A*03-specific effector T cells analyzed in a VITAL cytotoxicity assay with in vitro primed CD8+ T cells from an HV. Tetramer staining of polyclonal effector cells before performance of the VITAL assay determined the amount of P5A*03-specific effector cells in the population of successfully P5A*03-primed CD8+ T cells (C) and in the population of control cells (D) from the same donor primed with an HLA-matched irrelevant peptide. (E) At an effector-to-target ratio of 2.5:1, P5A*03-specific effectors (red) exerted 20.9% (± 0.4%) P5A*03-specific and significant higher lysis of P5A*03-loaded autologous target cells in comparison with control peptide-loaded target cells (HLA-A*03, RLRPGGKKK, GAG_HV1BR20-28). P5A*03-unspecific effectors (blue) only showed 2.0% (± 0.4%) unspecific lysis of the same targets. Results are shown as mean ± standard error of the mean for 3 independent replicates. ***P < .001. FSC, forward scatter; ID, identification; n.s., not significant; n.t., not tested.
Functional characterization of CML-associated antigen-specific CD8+T cells. (A) Functional characterization of CML-associated antigen-specific CD8+ T cells, including their CD107a and cytokine expression profile detected by ICS following aAPC-based priming experiments and their cytotoxic capability (VITAL assay). (B) Representative example of increased IFN-γ and TNF production, as well as CD107a expression, after stimulation with the respective P7B*07-peptide used for the stimulation with aAPCs compared with the corresponding negative control peptide (HLA-B*07, TPGPGVRYPL, NEF_HV1BR128-137). Phorbol myristate acetate (PMA) and ionomycin served as positive control. The P7B*07-specific CD8+ T-cell population showed a frequency of 1.01%, as detected by tetramer staining (far left panel). (C-E) Selective cytotoxicity of P5A*03-specific effector T cells analyzed in a VITAL cytotoxicity assay with in vitro primed CD8+ T cells from an HV. Tetramer staining of polyclonal effector cells before performance of the VITAL assay determined the amount of P5A*03-specific effector cells in the population of successfully P5A*03-primed CD8+ T cells (C) and in the population of control cells (D) from the same donor primed with an HLA-matched irrelevant peptide. (E) At an effector-to-target ratio of 2.5:1, P5A*03-specific effectors (red) exerted 20.9% (± 0.4%) P5A*03-specific and significant higher lysis of P5A*03-loaded autologous target cells in comparison with control peptide-loaded target cells (HLA-A*03, RLRPGGKKK, GAG_HV1BR20-28). P5A*03-unspecific effectors (blue) only showed 2.0% (± 0.4%) unspecific lysis of the same targets. Results are shown as mean ± standard error of the mean for 3 independent replicates. ***P < .001. FSC, forward scatter; ID, identification; n.s., not significant; n.t., not tested.
Reduced functionality of CD8+ T cells in CMLTKI patients
Subsequently, we reasoned that weak preexisting immune responses against the CML-associated HLA class I–restricted peptides in our IFN-γ ELISPOT assays could have been caused by an impairment of CD8+ T-cell functionality that reportedly occurs upon TKI treatment.53-56 Therefore, we compared T-cell responses against viral epitopes of CMLTKI patients, HVs, and CLL patients38 in IFN-γ ELISPOT assays. Although CD8+ T-cell counts themselves were not reduced in CMLTKI patients (Figure 7A), we observed significantly reduced IFN-γ release by T cells compared with HVs and CLL patients (P < .001, Figure 7B). In contrast, no significantly reduced IFN-γ production was observed upon stimulation with HLA class II–restricted viral epitopes (Figure 7C). These results were confirmed by the functional characterization of 6 HLA class II–restricted CML-associated peptides in IFN-γ ELISPOT assays (Figure 7D-E). Frequencies of CD4+ T-cell responses reached up to 24% (4/17) of analyzed CML patient samples; however, some peptides were only analyzed in pooled read-outs because of low cell numbers.
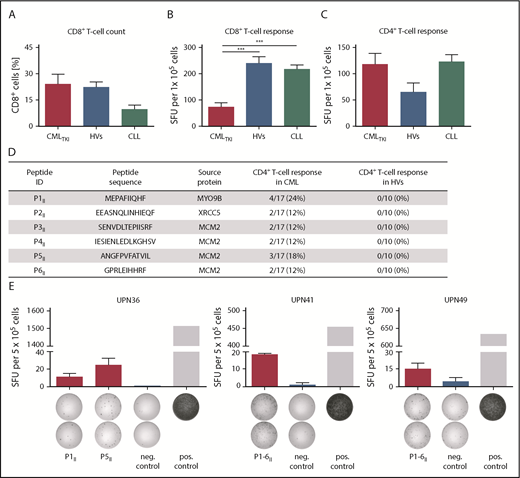
General functionality of T cells in CMLTKIpatients and immunogenicity of HLA class II–restricted CML–associated antigens. (A) CD8+ T-cell counts for CML patients under TKI treatment (CMLTKI patients, n = 7) compared with HVs (n = 10) and CLL patients (n = 10). Retrospective analysis of preexisting immune responses directed against HLA class I–restricted (B) and HLA class II–restricted (C) viral T-cell epitopes (supplemental Table 3) analyzed in IFN-γ ELISPOT assays after a 12-day recall stimulation of PBMCs from CMLTKI patients (HLA class I, n = 10; HLA class II, n = 12), HVs (HLA class I, n = 14; HLA class II, n = 6), and CLL patients (HLA class I, n = 31; HLA class II, n = 24). (D) HLA class II–restricted CML-associated peptides with their corresponding source proteins and frequencies of preexisting immune recognition by CD4+ T cells from CML patients or HVs in IFN-γ ELISPOT assays after a 12-day stimulation. (E) Examples of CML-associated HLA class II–restricted peptides evaluated in IFN-γ ELISPOT assays using PBMCs from CML patients. Results are shown for immunoreactive peptides only. Phytohemagglutinin was used as positive control and the HLA class II–restricted FLNA_HUMAN1669-1683 peptide ETVITVDTKAAGKGK served as negative control. Because of low cell numbers, the results for UPN41 and UPN49 are shown as pool read-outs of all 6 HLA class II–restricted CML-associated peptides. Data are expressed as mean ± standard deviation of 2 independent replicates. ***P < .001. ID, identification; neg., negative; pos., positive; SFU, spot-forming unit; UPN, uniform patient number.
General functionality of T cells in CMLTKIpatients and immunogenicity of HLA class II–restricted CML–associated antigens. (A) CD8+ T-cell counts for CML patients under TKI treatment (CMLTKI patients, n = 7) compared with HVs (n = 10) and CLL patients (n = 10). Retrospective analysis of preexisting immune responses directed against HLA class I–restricted (B) and HLA class II–restricted (C) viral T-cell epitopes (supplemental Table 3) analyzed in IFN-γ ELISPOT assays after a 12-day recall stimulation of PBMCs from CMLTKI patients (HLA class I, n = 10; HLA class II, n = 12), HVs (HLA class I, n = 14; HLA class II, n = 6), and CLL patients (HLA class I, n = 31; HLA class II, n = 24). (D) HLA class II–restricted CML-associated peptides with their corresponding source proteins and frequencies of preexisting immune recognition by CD4+ T cells from CML patients or HVs in IFN-γ ELISPOT assays after a 12-day stimulation. (E) Examples of CML-associated HLA class II–restricted peptides evaluated in IFN-γ ELISPOT assays using PBMCs from CML patients. Results are shown for immunoreactive peptides only. Phytohemagglutinin was used as positive control and the HLA class II–restricted FLNA_HUMAN1669-1683 peptide ETVITVDTKAAGKGK served as negative control. Because of low cell numbers, the results for UPN41 and UPN49 are shown as pool read-outs of all 6 HLA class II–restricted CML-associated peptides. Data are expressed as mean ± standard deviation of 2 independent replicates. ***P < .001. ID, identification; neg., negative; pos., positive; SFU, spot-forming unit; UPN, uniform patient number.
Taken together, we characterized a panel of novel CML-associated HLA class I and II antigens that, even in the context of the immunosuppressive effects induced by TKI treatment, was able to induce multifunctional T-cell responses and, therefore, could serve as prime targets for the development of antigen-specific immunotherapies in CML.
Discussion
Several studies have shown that immunological control plays a major role in the course of disease and for treatment success in CML.12,14,16 Therefore, various immunotherapeutic approaches are currently being evaluated,17-20,24,29 with the main goal to achieve deep remissions that enable long-term TKI-free survival or even cure of CML patients. An attractive approach is the further development of tailored peptide-based immunotherapy, which enables specific targeting of CML cells with minor side effects. Therefore, the identification of novel naturally presented and highly frequent CML-associated target antigens is required. In this study, we present a large-scale immunopeptidomics-based approach to identify and functionally characterize such CML-associated HLA class I– and class II–restricted peptides.
We confirmed strong HLA surface expression on myeloid cells, as well as on hematopoietic precursor cells, from CML patients in a range that is comparable to different healthy hematological cell types,40,41 other HMs,38,40,41 and solid tumors37 ; this constitutes a major prerequisite for immunotherapeutic approaches. The comprehensive comparison of HLA ligandomes from CML samples with benign tissues and PBMC samples from CMLMR revealed a total of 50 CML-associated HLA class I ligands for 4 of the most common HLA allotypes.78 The allele-specific prevalence of these CML-associated targets reached up to 80%. This enables the creation of personalized multipeptide vaccine cocktails, as well as the broadly applicable off-the-shelf development of single-peptide–based immunotherapeutic approaches, such as adoptive T-cell transfer or T-cell receptor therapies.
In addition to cytotoxic CD8+ T cells, CD4+ T cells play important direct and indirect roles in anticancer immunity.79-85 Thus, we expanded our profiling approach to the HLA class II peptidome identifying 19 additional CML-associated peptides. Interestingly, length distribution of HLA class II–restricted peptides could be correlated with specific cell types and lineages, because, in general, myeloid cell–derived peptides are represented by shorter peptide sequences. This is in line with the previous observation that the immunopeptidome directly mirrors cell type biology and specificity, which is reflected by the general peptide composition,42 as well as by the length distribution of HLA-presented peptides, as demonstrated by our data.
Because spontaneous pathophysiologically relevant T-cell responses against nonmutated LAAs were described for other HMs,38,86,87 we analyzed our CML patient cohort for preexisting T-cell responses against our newly defined targets. Of note, although preexisting T-cell responses against HLA class II peptides were identified with comparable frequencies as previously described for CLL,38 acute myeloid leukemia,40 and multiple myeloma,41 functional T cells targeting HLA class I antigens were only of low frequency in CMLTKI patient samples. In line with previous studies reporting a negative53-56 or dysregulating88 impact of TKI treatment on immune responses, CD8+ T-cell functionality in our CMLTKI patient cohort was impaired, potentially explaining the reduced frequencies of preexisting memory T-cell responses to CML-associated HLA class I ligands. Of note, because no CML patients without TKI treatment were included in the immunogenicity analyses, the reduced T-cell functionality could not be directly correlated with TKI treatment; it might also be linked to a general immunosuppressive state in CML disease caused, for example, by HLA-G,89 elevated myeloid-derived suppressor cells,12 and regulatory T cells,12,90 as well as by increased PD-1 expression on immune cells.12 However, the immunogenicity of all of our CML-associated HLA class I antigens was proven by in vitro induction of multifunctional and cytotoxic T cells from HVs. Strikingly, CML-specific T cells could also be induced de novo using PBMCs from CMLTKI patients, which qualifies the identified targets as promising candidates for peptide-based immunotherapy approaches in CML patients after termination of TKI therapy, as well as for tailored combinations with TKI treatment. Furthermore, several studies showed the pathophysiological relevance of preexisting peptide-specific T-cell responses to clinical outcomes in cancer patients,38,86,87 suggesting that such a T-cell response, induced or boosted by peptide-based immunotherapies, might result in clinical effectiveness.
Mutated neoantigens have been described as the main specificities of anticancer T-cell responses induced by immune checkpoint inhibitors in solid tumors with high mutational burden.91 However, only a very small fraction of mutations at the DNA sequence level results in peptides naturally presented in the HLA ligandome.92-94 This raises the question about the relevance of mutated neoepitopes for T-cell-based immunotherapy, in particular for malignancies with low mutational burden, including CML. Despite an extensive search for naturally presented BCR-ABL– and ABL-BCR–derived peptides, none could be validated in our CML cohort by MS. However, we have to emphasize that the absence of evidence does not mean that there is evidence of absence; the sensitivity of shotgun mass spectrometric discovery approaches, even in the context of immense technical improvements in the last decades,95 is limited because the HLA immunopeptidome is a highly dynamic, rich, and complex assembly of peptides. Therefore, we cannot exclude low-level presentation of mutation-derived peptides in our CML patient cohort. Nevertheless, MS-based immunopeptidomics is the only unbiased methodology that can identify all naturally processed and presented HLA peptides in primary tissue samples96 ; this enables us to identify and characterize target antigens in low mutational–burden cancer entities that are nonmutated, naturally presented, highly frequent, and tumor-specific.
This is further emphasized, because the extensive screening of our CML and hematological benign cohorts for HLA-presented peptides derived from previously described CTAs75,76 and LAAs43,77 did not reveal any highly frequent tumor-exclusive presentation. Together with previous findings showing a distorted correlation between gene expression and HLA-restricted antigen presentation meaning that the immunopeptidome that does not mirror neither the transcriptome nor the proteome,40,93,97-100 this precludes, in our view, these antigens as optimal candidates for T-cell–based immunotherapy. Nevertheless, tumor exclusivity can be determined at the level of HLA ligands or at the level of entire antigens. In this study, CTA and LAA analyses were performed at the level of entire antigens and do not consider presentation of CTA- and LAA-derived single HLA ligands, as they might potentially be tumor-exclusive as a result of differential antigen processing in cancer cells.
In conclusion, the cell biology–specific character of the immunopeptidome42 calls for entity-centered identification of tumor-associated targets. Therefore, our study provides profound insights into the naturally presented immunopeptidome of CML, delineating a panel of novel, immunogenic, nonmutated, and CML-associated T-cell epitopes. These antigens aid in the development of different antigen-specific therapeutic approaches that may provide options to enable achievement of deep remission, long-term TKI-free survival, or even cure for CML patients.
The MS data reported in this article have been deposited in the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (dataset identifier PXD010450).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Ulrike Schmidt, Claudia Falkenburger, Patricia Hrstić, Nicole Bauer, Beate Pömmerl, and Ulrich Wulle for excellent technical support.
This work was supported by the German Cancer Consortium, the Deutsche Forschungsgemeinschaft (STI 704/1-1 and SFB 685), the Wilhelm Sander Stiftung (2016.177.1), the Bundesministerium für Bildung und Forschung (031A535A), and the European Union (ERC AdG339842 MUTAEDITING).
Authorship
Contribution: T.B., A.N., H.-G.R., S.S., and J.S.W. designed the study; A.N., D.J.K., H.S., A.M., J.B., and M.M. performed HLA ligandome experiments; T.B., M.L., J.R., and J.K.P. conducted in vitro T-cell experiments; L.B., C.-C.T., M.C.N., M.S., T.H.B., V.V., D.N., O.K., and J.S.W. provided new reagents/analytic tools/samples; H.R.S., M.C.N., M.S., T.H.B., V.V., D.N., M.R., R.K., L.K., and J.S.W. collected patient data and performed medical evaluations; T.B., A.N., L.B., D.J.K., H.S., S.S., and J.S.W. analyzed data; T.B., A.N., H.R.S., L.K., H.-G.R., S.S., and J.S.W. drafted the manuscript; and H.-G.R., S.S., and J.S.W. supervised the study.
Conflict-of-interest disclosure: D.J.K. and H.S. are employees of Immatics Biotechnologies. C.-C.T. is an employee of Immatics US. H.-G.R. is a shareholder of Immatics Biotechnologies and Curevac. The remaining authors declare no competing financial interests.
Correspondence: Juliane S. Walz, Otfried-Müller-Str. 10, 72076 Tübingen, Germany; e-mail: juliane.walz@med.uni-tuebingen.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal