

Key Points
Conjugation of GSK3 inhibitor–loaded nanoparticles with donor cells enhances their proliferation kinetics post-IUHCT.
Resulting therapeutic levels of engraftment could allow single-step in utero treatment of congenital hematological disorders.

Abstract
Host cell competition is a major barrier to engraftment after in utero hematopoietic cell transplantation (IUHCT). Here we describe a cell-engineering strategy using glycogen synthase kinase-3 (GSK3) inhibitor–loaded nanoparticles conjugated to the surface of donor hematopoietic cells to enhance their proliferation kinetics and ability to compete against their fetal host equivalents. With this approach, we achieved remarkable levels of stable, long-term hematopoietic engraftment for up to 24 weeks post-IUHCT. We also show that the salutary effects of the nanoparticle-released GSK3 inhibitor are specific to donor progenitor/stem cells and achieved by a pseudoautocrine mechanism. These results establish that IUHCT of hematopoietic cells decorated with GSK3 inhibitor–loaded nanoparticles can produce therapeutic levels of long-term engraftment and could therefore allow single-step prenatal treatment of congenital hematological disorders.
Introduction
In utero hematopoietic cell transplantation (IUHCT) has the potential to cure any congenital disease that can be diagnosed prenatally and treated postnatally by hematopoietic stem cell (HSC) transplantation (HSCT).1 IUHCT is based on the preimmune window of fetal development, resulting in donor-specific tolerance and allogeneic donor-cell engraftment, without the need for myeloablation or immunosuppression.2-4 Although stem cells from various sources might be used for IUHCT, the most likely candidates for broad clinical application to date are bone marrow (BM)–derived adult HSCs.1
Despite the potential advantages of IUHCT over postnatal transplantation, host cell competition remains a major barrier to overcome before IUHCT becomes a therapeutic option.1,3,4 In the normal fetus, stem cell and progenitor populations in the liver and other hematopoietic niches have a competitive advantage over their postnatal equivalents as a result of favorable cell cycling and expansion kinetics.1 Because of this advantage, IUHCT of adult HSCs yields subtherapeutic levels of long-term chimerism in many animal models with robust fetal hematopoietic compartments, including mouse5 and dog models,6 requiring postnatal boosting with HSCs from the same donor in conjunction with nonmyeloablative conditioning.7
Glycogen synthase kinase-3 (GSK3) is a constitutively active serine-threonine kinase8 involved in the regulation of several signaling pathways (including Wnt,9 Hedgehog,10 and Notch11 ) associated with modulation of HSC homeostasis.12-16 The importance of GSK3 has been long suggested by the clinical finding that lithium increases circulating HSCs (eg, CD34+ cells)17 and peripheral blood counts18-20 in >90% of patients. In addition, there are laboratory findings that lithium increases transplantable HSCs in mice.21 Because lithium directly inhibits GSK3,22 these clinical and laboratory observations implicate GSK3 as an important regulator of HSC homeostasis.9-11,23-25
The use of GSK3 inhibitors in experimental postnatal HSCT results in enhanced repopulating capacity in vivo.26 Although the regulatory effect of GSK3 inhibitor treatment preferentially affects the primitive hematopoietic compartment, it has been shown that this augmented reconstitution is dependent upon sustained GSK3 inhibitor administration.26 To increase adult HSC competitiveness in IUHCT, the cells will need to be continuously treated with the GSK3 inhibitor. Unfortunately, fetuses are inaccessible for continuous dosing. Even if sustained fetal treatment were possible, systemic GSK3 inhibition would affect both donor and host cells alike and may have adverse effects on fetal development.
Cell engineering with surface-conjugated synthetic nanoparticles can be a strategy for adjuvant drug delivery in cell therapies. Chemically conjugating submicron-sized drug-loaded nanoparticles directly onto the plasma membrane of donor cells enables continuous and selective pseudoautocrine stimulation of transferred cells in vivo.27-29 Nanoparticles have been shown to increase in vivo repopulation rates of donor HSCs after HSCT in adult mice, when compared with systemic dosing.27
We hypothesized that treatment of adult HSCs with GSK3 inhibitor–loaded nanoparticles before IUHCT would enhance the competitive capacity of donor cells in the fetal hematopoietic environment. Here we demonstrate a remarkable increase in allogeneic donor cell engraftment after IUHCT using this strategy.
Methods
Cell isolation and animal procedures
Balb/c (H2Kd+) and B6.SJL-Ptprca-Pep3b/BoyJ (H2Kb+, CD45.1+; B6-CD45.1) mice were purchased from Jackson Laboratory. C57BL/6TgN(act-EGFP)OsbY01 (H2Kb+, GFP+; B6-GFP) mice were kindly provided by Dr Okabe (Osaka University). All protocols and procedures were approved by the Institutional Animal Care and Use Committee at the Children’s Hospital of Philadelphia and followed guidelines set forth in the National Institutes of Health Guide for the NC3Rs and ARRIVE guidelines.
For the isolation of donor cells, BM was harvested from 6- to 8-week-old donors (B6-GFP and B6-CD45.1), and mononuclear cells (BM-MNCs) were obtained as previously described.30 The protocol for isolation of hematopoietic stem cells (lineage negative [Lin−/Sca1+/cKit+ [LSK] cells or BM-HSCs) is summarized in supplemental Methods (available on the Blood Web site). Intravenous IUHCT of allogeneic BM-MNCs (B6-GFP or B6-CD45.1; 107 cells) was performed at embryonic day 14 of Balb/c fetuses as previously described (supplemental Methods).3,30
GSK3 inhibitor–loaded nanoparticle synthesis and conjugation on cell surface
The nanoparticles used in this study are referred to as multilamellar lipid vesicles (MLVs). To synthesize MLVs, we adapted the protocol described by Stephan et al27 (supplemental Methods; supplemental Figure 1A). They were loaded with CHIR99021-HCl (MLV-CHIR99021s), a hydrochloride of CHIR99021 that is a GSK3α/β inhibitor, with half maximal inhibitory concentrations of 10 nM/6.7 nM in cell-free assays (Selleck Chemicals, Houston, TX). For MLV conjugation on the surface of hematopoietic cells, we mixed MLVs with freshly isolated BM-MNCs or BM-HSCs in serum-free media to achieve a nanoparticle concentration of 300 to 900 MLVs per cell and cell concentrations of 6 × 106 to 1.2 × 107/mL. Protocols for MLV characterization are summarized in supplemental Methods.
Experimental protocols
Protocol 1
After preliminary studies that assessed potential effects of MLV conjugation on the ability of BM-MNCs to home to and engraft in fetal hematopoietic tissues post–allogeneic IUHCT (Figure 1; supplemental Methods), we performed a proof-of-principle in vivo study aiming to establish whether gradual release of CHIR99021 from MLVs conjugated on the surface of donor (B6-GFP) BM-MNCs could enhance engraftment post-IUHCT. There were 3 experimental groups (Figure 2A): group 1, 107 BM-MNCs without nanoparticles; group 2, BM-MNCs with a bolus dose of CHIR99021 similar to that loaded on MLVs (BM-MNC-CHIR99021s); and group 3, BM-MNC cell surface engineered with MLV-CHIR99021s (BM-MNC-MLV-CHIR99021s).
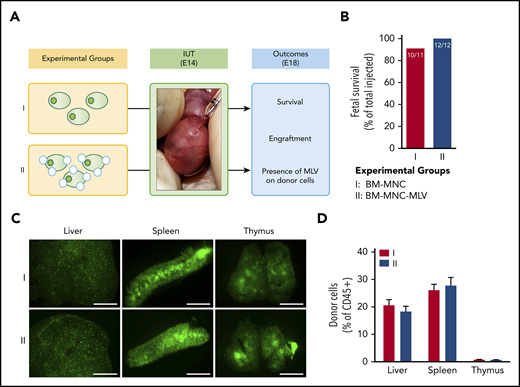
IUHCT of B6-GFP BM-MNCs conjugated with empty MLVs in Balb/c fetuses. (A) Experimental design: effect of empty MLV conjugation on ability of BM-MNCs to home and engraft post-IUHCT; group 1, unconjugated BM-MNC and group 2, DiD-labeled empty MLV-conjugated BM-MNCs. (B) Fetal survival 4 days post-IUHCT in both groups. Nanoparticle conjugation had no effect on fetal survival (group 1, 91% vs group 2, 100%). (C) Qualitative assessment of donor cell homing to liver, spleen, and thymus by fluorescent stereotactic microscopy (4 days post-IUHCT) demonstrated no differences between experimental groups. (D) Quantitative confirmation of donor cell engraftment (4 days post-IUHCT) using flow cytometry (liver: group 1, 20.6% ± 2.1% vs group 2, 18.3% ± 1.9%; spleen: group 1, 26.1% ± 2.1% vs group 2, 27.7% ± 2.9%; thymus: group 1, 0.8% ± 0.1% vs group 2, 0.7% ± 0.1%). E18, embryonic day 18; IUT, in utero transplantation.
IUHCT of B6-GFP BM-MNCs conjugated with empty MLVs in Balb/c fetuses. (A) Experimental design: effect of empty MLV conjugation on ability of BM-MNCs to home and engraft post-IUHCT; group 1, unconjugated BM-MNC and group 2, DiD-labeled empty MLV-conjugated BM-MNCs. (B) Fetal survival 4 days post-IUHCT in both groups. Nanoparticle conjugation had no effect on fetal survival (group 1, 91% vs group 2, 100%). (C) Qualitative assessment of donor cell homing to liver, spleen, and thymus by fluorescent stereotactic microscopy (4 days post-IUHCT) demonstrated no differences between experimental groups. (D) Quantitative confirmation of donor cell engraftment (4 days post-IUHCT) using flow cytometry (liver: group 1, 20.6% ± 2.1% vs group 2, 18.3% ± 1.9%; spleen: group 1, 26.1% ± 2.1% vs group 2, 27.7% ± 2.9%; thymus: group 1, 0.8% ± 0.1% vs group 2, 0.7% ± 0.1%). E18, embryonic day 18; IUT, in utero transplantation.
![Conjugation of GSK3 inhibitor–loaded MLVs on B6-GFP BM-MNCs enhances long-term hematopoietic engraftment after IUHCT in Balb/c fetuses. (A) Experimental design: IUHCT of BM-MNCs alone (group 1), BM-MNCs associated with a bolus of GSK3 inhibitor (group 2), and BM-MNCs conjugated with GSK3 inhibitor MLVs (group 3). (B) Fetal survival after 4 days post-IUHCT. There were no subsequent deaths. (C) Flow cytometry gating strategy for identifying CD45+ and GFP+ cells at P28 (light blue contour plots: CD45+, GFP− controls). (D) Donor cell levels in blood at P28, P84, and P168 after IUHCT in 3 groups. At 4 weeks, chimerism was similar between animals in group 1 and survivors in group 2 (group 1, 15.5% ± 1.4% vs group 2, 10.5% ± 2.3%) but was more than 3 times higher in group 3 animals that received donor BM-MNCs carrying MLV-CHIR99021s (group 3, 52.9% ± 2.8%). (C-D) Engraftment levels in group 1 and 2 offspring were significantly reduced at 12 and 24 weeks of age compared with 4 weeks (group 1 at 12 weeks, 5.9% ± 0.7%; group 1 at 24 weeks, 3.4% ± 0.7%; group 2 at 12 weeks, 4.5% ± 1.0%; group 2 at 24 weeks, 2.9% ± 0.9%) but were maintained in group 3 animals (group 3 at 12 weeks, 48.3% ± 2.2%; group 3 at 24 weeks, 48.5% ± 3.1%). (E) Histograms for hematopoietic multilineage analysis using CD3, B220, Cd11b, and Gr1 antibodies in blood at 6 months (light blue histograms: CD45+, GFP+ [donor] or GFP− [host] lineage-specific antibody-negative controls). (F) Multilineage reconstitution of all 3 groups at 6 months in blood compared with donor lineages (GFP control, normal nontransplanted donor). (B) *P < .05 group 2 vs groups 1 and 3; (D) *P < .05 group 1 at 4 weeks vs group 1 at 12 and 24 weeks; **P < .001 group 2 at 4 weeks vs group 2 at 12 and 24 weeks; ***P < .0001 vs groups 1 and 2 (all timepoints).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/22/10.1182_blood.2019001037/3/m_bloodbld2019001037f2.png?Expires=1769428588&Signature=IApwqGxh6HLGdhCa403cbaNmh8iSo2P~Hi9grmMEx2hFL8c1aN2iqM6kmGQbFtIecDEgjxHeLr5kUM8MBvRQLS6eqE7hdYSvMXyrz7vaikGqxXNcsq7bjjo9JrH0mq07vO5-wByec2L7cUv2im6fPTkDlZDcNKhHdQyBVuYMDUiBLeNlWer7jmEPngo97OFOZewzDCCSwgHW7Jq51nXC8AGgbxjB3sE1lYWMvSjO0TEzgL~b-g8STn4bvSnGlervsH22UieWxHUS2oS5ZEYvFXEZz2Ah~SNqiiRakQNLjtzKS0HFJQRJ3EDH7iVO6D0S0r2rjQFmjFlkRi0K9uSqRg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Conjugation of GSK3 inhibitor–loaded MLVs on B6-GFP BM-MNCs enhances long-term hematopoietic engraftment after IUHCT in Balb/c fetuses. (A) Experimental design: IUHCT of BM-MNCs alone (group 1), BM-MNCs associated with a bolus of GSK3 inhibitor (group 2), and BM-MNCs conjugated with GSK3 inhibitor MLVs (group 3). (B) Fetal survival after 4 days post-IUHCT. There were no subsequent deaths. (C) Flow cytometry gating strategy for identifying CD45+ and GFP+ cells at P28 (light blue contour plots: CD45+, GFP− controls). (D) Donor cell levels in blood at P28, P84, and P168 after IUHCT in 3 groups. At 4 weeks, chimerism was similar between animals in group 1 and survivors in group 2 (group 1, 15.5% ± 1.4% vs group 2, 10.5% ± 2.3%) but was more than 3 times higher in group 3 animals that received donor BM-MNCs carrying MLV-CHIR99021s (group 3, 52.9% ± 2.8%). (C-D) Engraftment levels in group 1 and 2 offspring were significantly reduced at 12 and 24 weeks of age compared with 4 weeks (group 1 at 12 weeks, 5.9% ± 0.7%; group 1 at 24 weeks, 3.4% ± 0.7%; group 2 at 12 weeks, 4.5% ± 1.0%; group 2 at 24 weeks, 2.9% ± 0.9%) but were maintained in group 3 animals (group 3 at 12 weeks, 48.3% ± 2.2%; group 3 at 24 weeks, 48.5% ± 3.1%). (E) Histograms for hematopoietic multilineage analysis using CD3, B220, Cd11b, and Gr1 antibodies in blood at 6 months (light blue histograms: CD45+, GFP+ [donor] or GFP− [host] lineage-specific antibody-negative controls). (F) Multilineage reconstitution of all 3 groups at 6 months in blood compared with donor lineages (GFP control, normal nontransplanted donor). (B) *P < .05 group 2 vs groups 1 and 3; (D) *P < .05 group 1 at 4 weeks vs group 1 at 12 and 24 weeks; **P < .001 group 2 at 4 weeks vs group 2 at 12 and 24 weeks; ***P < .0001 vs groups 1 and 2 (all timepoints).
Conjugation of GSK3 inhibitor–loaded MLVs on B6-GFP BM-MNCs enhances long-term hematopoietic engraftment after IUHCT in Balb/c fetuses. (A) Experimental design: IUHCT of BM-MNCs alone (group 1), BM-MNCs associated with a bolus of GSK3 inhibitor (group 2), and BM-MNCs conjugated with GSK3 inhibitor MLVs (group 3). (B) Fetal survival after 4 days post-IUHCT. There were no subsequent deaths. (C) Flow cytometry gating strategy for identifying CD45+ and GFP+ cells at P28 (light blue contour plots: CD45+, GFP− controls). (D) Donor cell levels in blood at P28, P84, and P168 after IUHCT in 3 groups. At 4 weeks, chimerism was similar between animals in group 1 and survivors in group 2 (group 1, 15.5% ± 1.4% vs group 2, 10.5% ± 2.3%) but was more than 3 times higher in group 3 animals that received donor BM-MNCs carrying MLV-CHIR99021s (group 3, 52.9% ± 2.8%). (C-D) Engraftment levels in group 1 and 2 offspring were significantly reduced at 12 and 24 weeks of age compared with 4 weeks (group 1 at 12 weeks, 5.9% ± 0.7%; group 1 at 24 weeks, 3.4% ± 0.7%; group 2 at 12 weeks, 4.5% ± 1.0%; group 2 at 24 weeks, 2.9% ± 0.9%) but were maintained in group 3 animals (group 3 at 12 weeks, 48.3% ± 2.2%; group 3 at 24 weeks, 48.5% ± 3.1%). (E) Histograms for hematopoietic multilineage analysis using CD3, B220, Cd11b, and Gr1 antibodies in blood at 6 months (light blue histograms: CD45+, GFP+ [donor] or GFP− [host] lineage-specific antibody-negative controls). (F) Multilineage reconstitution of all 3 groups at 6 months in blood compared with donor lineages (GFP control, normal nontransplanted donor). (B) *P < .05 group 2 vs groups 1 and 3; (D) *P < .05 group 1 at 4 weeks vs group 1 at 12 and 24 weeks; **P < .001 group 2 at 4 weeks vs group 2 at 12 and 24 weeks; ***P < .0001 vs groups 1 and 2 (all timepoints).
Protocol 2
In our next round of in vivo experiments, we sought to determine whether the GSK3 inhibitor released by MLVs affects engraftment by acting on donor BM-HSCs. To achieve this, we isolated LSK cells from B6-GFP BM-MNCs and decorated their cell membranes with either empty MLVs or MLV-CHIR99021s. We also cultured BM-HSCs over a 7-day period with (or without) an amount of CHI99021 similar to that carried by MLVs (per cell) added to culture media (supplemental Methods). Some of the cultured cells were used for in vitro hematopoietic colony-forming unit assays and real-time polymerase chain reaction (Wnt, Notch, and Hedgehog signaling pathways; supplemental Methods). Before IUHCT, decorated or cultured BM-HSCs were mixed with fresh non-LSK BM-MNCs to achieve similar proportions of BM-HSCs in the 107 B6-GFP cells transplanted into each fetus (∼0.1%, or 10 000 HSCs). There were 5 experimental groups in this study (Figure 4F): group 1, BM-MNCs without nanoparticles; group 2, BM-MNCs in which fresh BM-HSCs were replaced by a similar number of BM-HSCs cultured without CHIR99021 (cultured HSCs without CHIR99021); group 3, BM-MNCs in which fresh BM-HSCs were replaced by similar numbers of BM-HSCs cultured with CHIR99021 (cultured HSCs with CHIR99021); group 4, BM-MNCs in which fresh BM-HSCs were decorated with empty MLVs (fresh HSC-MLVs); and group 5, BM-MNCs in which MLV-CHIR99021s were conjugated on the cell surface of fresh BM-HSCs (fresh HSC-MLV-CHIR99021s).
![Long-term engraftment in BM after IUHCT of B6-GFP BM-MNCs conjugated with GSK3 inhibitor–loaded MLVs in Balb/c fetuses. (A) Flow cytometry gating strategy for identifying CD45+ and GFP+ cells in BM in 3 groups: BM-MNCs alone (group 1), BM-MNCs associated with a bolus of GSK3 inhibitor (group 2), and BM-MNCs conjugated with GSK3 inhibitor MLVs (group 3) (light blue contour plots: CD45+, GFP− controls). (B) Percentage of CD45+ GFP+ donor cells on host BM at 6 months in the 3 groups. Group 1 and 2 animals had similar levels of donor cell engraftment (group 1, 1.6% ± 0.4% vs group 2, 2.1% ± 0.9%), whereas levels in group 3 animals were almost 25 times higher (group 3, 50.4% ± 2.6%). (C) Flow cytometry gating strategy for hematopoietic multilineage analysis using CD3, B220, Cd11b, and Gr1 antibodies in BM at 6 months (light blue histograms: lineage-specific antibody-negative controls). (D) Multilineage reconstitution of all 3 groups at 6 months in BM compared with donor lineages demonstrated no differences between groups. (E) BM histology with immunohistochemistry for GFP (brown) at 6 months in all 3 groups and Balb/c and GFP control mice (magnification: ×40; scale bar 100 μm; stain: diaminobenzidine [brown] and hematoxylin [blue]). (B) ***P < .0001 vs groups 1 and 2.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/22/10.1182_blood.2019001037/3/m_bloodbld2019001037f3.png?Expires=1769428588&Signature=Q7~UZfi2t70Xn2fzE7LmyMjObjdDw9Fnoio89oH~rmymluxD~2NPIQ0ocpO3EKhgWF7DErtF~XozrlnAhzg~4WGsblYJQWaW1-Zxvop3XmmPEQ4pAmolsG5Xgzas5K79qt5nSKImzHHP96ABex-3hQar-ioGZt3Hmgz9WlFwXdRMoMzYTK9b3Gy9LQfatYUgNdMV1mQAaYqrBKE-GNC3gLVjXRQJ40mvOp9oDtJmxwIDbhEZTg-l7c6PnQjJxE~6f-AtG8IliLDhRiICYIDCkvZcrYeO3defNdITTvs07mRlZmu3v~HX-OZtAv7RPq3WZ7vQGisyhklRcuD93o1xYw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Long-term engraftment in BM after IUHCT of B6-GFP BM-MNCs conjugated with GSK3 inhibitor–loaded MLVs in Balb/c fetuses. (A) Flow cytometry gating strategy for identifying CD45+ and GFP+ cells in BM in 3 groups: BM-MNCs alone (group 1), BM-MNCs associated with a bolus of GSK3 inhibitor (group 2), and BM-MNCs conjugated with GSK3 inhibitor MLVs (group 3) (light blue contour plots: CD45+, GFP− controls). (B) Percentage of CD45+ GFP+ donor cells on host BM at 6 months in the 3 groups. Group 1 and 2 animals had similar levels of donor cell engraftment (group 1, 1.6% ± 0.4% vs group 2, 2.1% ± 0.9%), whereas levels in group 3 animals were almost 25 times higher (group 3, 50.4% ± 2.6%). (C) Flow cytometry gating strategy for hematopoietic multilineage analysis using CD3, B220, Cd11b, and Gr1 antibodies in BM at 6 months (light blue histograms: lineage-specific antibody-negative controls). (D) Multilineage reconstitution of all 3 groups at 6 months in BM compared with donor lineages demonstrated no differences between groups. (E) BM histology with immunohistochemistry for GFP (brown) at 6 months in all 3 groups and Balb/c and GFP control mice (magnification: ×40; scale bar 100 μm; stain: diaminobenzidine [brown] and hematoxylin [blue]). (B) ***P < .0001 vs groups 1 and 2.
Long-term engraftment in BM after IUHCT of B6-GFP BM-MNCs conjugated with GSK3 inhibitor–loaded MLVs in Balb/c fetuses. (A) Flow cytometry gating strategy for identifying CD45+ and GFP+ cells in BM in 3 groups: BM-MNCs alone (group 1), BM-MNCs associated with a bolus of GSK3 inhibitor (group 2), and BM-MNCs conjugated with GSK3 inhibitor MLVs (group 3) (light blue contour plots: CD45+, GFP− controls). (B) Percentage of CD45+ GFP+ donor cells on host BM at 6 months in the 3 groups. Group 1 and 2 animals had similar levels of donor cell engraftment (group 1, 1.6% ± 0.4% vs group 2, 2.1% ± 0.9%), whereas levels in group 3 animals were almost 25 times higher (group 3, 50.4% ± 2.6%). (C) Flow cytometry gating strategy for hematopoietic multilineage analysis using CD3, B220, Cd11b, and Gr1 antibodies in BM at 6 months (light blue histograms: lineage-specific antibody-negative controls). (D) Multilineage reconstitution of all 3 groups at 6 months in BM compared with donor lineages demonstrated no differences between groups. (E) BM histology with immunohistochemistry for GFP (brown) at 6 months in all 3 groups and Balb/c and GFP control mice (magnification: ×40; scale bar 100 μm; stain: diaminobenzidine [brown] and hematoxylin [blue]). (B) ***P < .0001 vs groups 1 and 2.
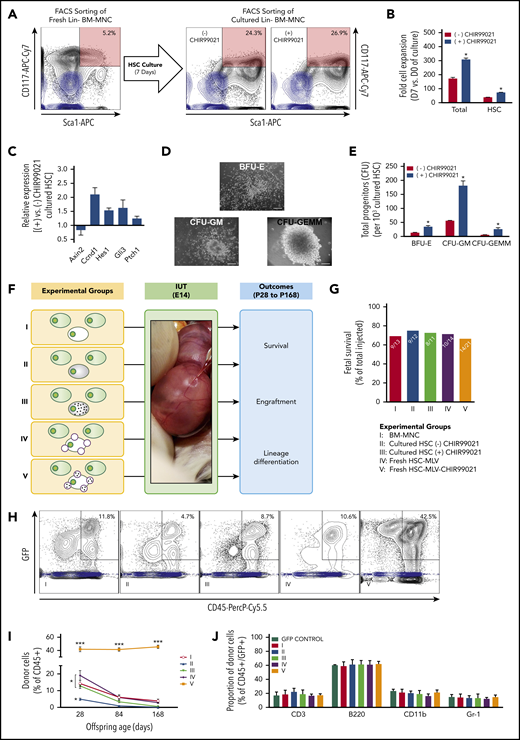
GSK3 inhibitor released by MLVs increases engraftment after IUHCT by acting on HSCs/HPCs. (A) BM-HSCs were isolated and cultured over a 7-day period with (or without) an amount of CHI99021 similar to that carried by MLVs (per cell) added to the serum-free media. At the end of the culture period, ∼25% of the harvested cells were LSK, and this was not affected by the presence of CHIR99021 in the media. Flow cytometry gating strategy for sorting B6-GFP BM-HSCs (CD117+ and Sca1+ double-positive cells shown; light blue contour plots: CD117− and Sca1− double-negative controls). (B) Fold cell expansion after 7 days in culture in both groups (cultured without or with CHIR99021); total cells (left) and those only in the HSC compartment (right). Exposure of BM-HSCs to CHIR99021 over the 7-day culture period resulted in significant increase in the total number of both BM-MNCs and BM-HSCs. (C) Quantitative real-time polymerase chain reaction on the HSC group cultured with CHIR99021 demonstrated modulation of the Wnt (downregulation of Axin2 expression, upregulation of Ccnd1 expression), Notch (upregulation of Hes1 expression), and Hedgehog (upregulation Gli3 and Ptch1 expression) pathways, which have been shown to affect HSC function, proliferation kinetics, and repopulating capacity. (D) Hematopoietic colony-forming unit (CFU) assay; record of colony growth. (E) Hematopoietic CFU assays comparing CFU numbers in the different lineages in both cultured groups (without or with CHIR99021). BM-HSCs cultured in serum-free media with CHIR99021 had enhanced clonogenic potential evident in CFU (semisolid media) assays compared with untreated controls. (F) Experimental design: group 1, untreated BM-MNCs; group 2, BM-MNCs with cultured HSCs; group 3, BM-MNCs with HSCs cultured with GSK3 inhibitor; group 4, BM-MNCs in which fresh HSCs were conjugated with empty MLVs; and group 5, BM-MNCs in which fresh HSCs were conjugated with GSK3 inhibitor–loaded MLVs. Similar number of HSCs contained on the isolated fresh BM-MNCs was replaced by cultured HSCs. The HSC population is usually 0.1% of all BM-MNCs. Because the fetus received 107 BM-MNCs, this included 10 000 HSC, fresh, cultured, or MLV decorated. (G) Fetal survival after 4 days post-IUHCT in Balb/c fetuses in the 5 groups; there was no difference between groups in fetal survival (group 1, 69% vs group 2, 75% vs group 3, 72% vs group 4, 71% vs group 5, 67%). (H) Density plots of a sample of each group demonstrating the gating strategy for the identification of the CD45+ GFP+ population (light blue contour plots: CD45+, GFP− controls). (I) Donor cell (B6-GFP) levels in blood at P28, P84, and P168 after IUHCT in 5 groups. Levels of engraftment gradually decreased during the follow-up period (group 1 at 4 weeks, 14.3% ± 1.9%; group 1 at 12 weeks, 6.1% ± 0.8%; group 1 at 24 weeks, 3.9% ± 1.1%). Replacement of BM-HSCs from freshly isolated BM-MNCs with expanded BM-HSCs resulted in reduced blood engraftment at 4 weeks (group 4 at 4 weeks, 4.9% ± 0.6%), and only microchimerism was detected thereafter. Addition of CHIR99021 to the culture media of BM-HSCs prevented the latter drop in chimerism at 4 weeks (4 weeks: group 3, 12.9% ± 1.6%) but did not enhance it any further, resulting in an engraftment profile in blood similar to that observed in animals that received IUHCT of unprocessed/freshly isolated BM-MNCs (group 1). Conjugation of MLV-CHIR99021s on donor BM-HSCs resulted in an impressive increase in baseline engraftment levels post-IUHCT (4 weeks: group 5, 41.8% ± 3.7%), and this remained unchanged up to 24 weeks of recipient age (12 weeks: group 5, 41.4% ± 2.6%; 24 weeks: group 5, 45.4% ± 2.3%). The effect of MLV-CHIR99021s on engraftment was due to the gradual release and activity of the inhibitor, because conjugation of donor cells with empty particles did not reproduce the chimerism rise seen in group 5 animals (4 weeks: group 4, 19.2% ± 2.9%). (J) Multilineage reconstitution of all 5 groups at 6 months in blood compared with donor lineages did not demonstrate any differences between groups. (B,E) *P < .05 vs (-) CHIR99021; (I) *P < .05 groups 1-4: 4 weeks vs groups 1-4: 12 and 24 weeks and group 2: 4 weeks vs groups 1, 3, 4: 4 weeks; ***P < .0001 vs groups 1-4 (all timepoints).
GSK3 inhibitor released by MLVs increases engraftment after IUHCT by acting on HSCs/HPCs. (A) BM-HSCs were isolated and cultured over a 7-day period with (or without) an amount of CHI99021 similar to that carried by MLVs (per cell) added to the serum-free media. At the end of the culture period, ∼25% of the harvested cells were LSK, and this was not affected by the presence of CHIR99021 in the media. Flow cytometry gating strategy for sorting B6-GFP BM-HSCs (CD117+ and Sca1+ double-positive cells shown; light blue contour plots: CD117− and Sca1− double-negative controls). (B) Fold cell expansion after 7 days in culture in both groups (cultured without or with CHIR99021); total cells (left) and those only in the HSC compartment (right). Exposure of BM-HSCs to CHIR99021 over the 7-day culture period resulted in significant increase in the total number of both BM-MNCs and BM-HSCs. (C) Quantitative real-time polymerase chain reaction on the HSC group cultured with CHIR99021 demonstrated modulation of the Wnt (downregulation of Axin2 expression, upregulation of Ccnd1 expression), Notch (upregulation of Hes1 expression), and Hedgehog (upregulation Gli3 and Ptch1 expression) pathways, which have been shown to affect HSC function, proliferation kinetics, and repopulating capacity. (D) Hematopoietic colony-forming unit (CFU) assay; record of colony growth. (E) Hematopoietic CFU assays comparing CFU numbers in the different lineages in both cultured groups (without or with CHIR99021). BM-HSCs cultured in serum-free media with CHIR99021 had enhanced clonogenic potential evident in CFU (semisolid media) assays compared with untreated controls. (F) Experimental design: group 1, untreated BM-MNCs; group 2, BM-MNCs with cultured HSCs; group 3, BM-MNCs with HSCs cultured with GSK3 inhibitor; group 4, BM-MNCs in which fresh HSCs were conjugated with empty MLVs; and group 5, BM-MNCs in which fresh HSCs were conjugated with GSK3 inhibitor–loaded MLVs. Similar number of HSCs contained on the isolated fresh BM-MNCs was replaced by cultured HSCs. The HSC population is usually 0.1% of all BM-MNCs. Because the fetus received 107 BM-MNCs, this included 10 000 HSC, fresh, cultured, or MLV decorated. (G) Fetal survival after 4 days post-IUHCT in Balb/c fetuses in the 5 groups; there was no difference between groups in fetal survival (group 1, 69% vs group 2, 75% vs group 3, 72% vs group 4, 71% vs group 5, 67%). (H) Density plots of a sample of each group demonstrating the gating strategy for the identification of the CD45+ GFP+ population (light blue contour plots: CD45+, GFP− controls). (I) Donor cell (B6-GFP) levels in blood at P28, P84, and P168 after IUHCT in 5 groups. Levels of engraftment gradually decreased during the follow-up period (group 1 at 4 weeks, 14.3% ± 1.9%; group 1 at 12 weeks, 6.1% ± 0.8%; group 1 at 24 weeks, 3.9% ± 1.1%). Replacement of BM-HSCs from freshly isolated BM-MNCs with expanded BM-HSCs resulted in reduced blood engraftment at 4 weeks (group 4 at 4 weeks, 4.9% ± 0.6%), and only microchimerism was detected thereafter. Addition of CHIR99021 to the culture media of BM-HSCs prevented the latter drop in chimerism at 4 weeks (4 weeks: group 3, 12.9% ± 1.6%) but did not enhance it any further, resulting in an engraftment profile in blood similar to that observed in animals that received IUHCT of unprocessed/freshly isolated BM-MNCs (group 1). Conjugation of MLV-CHIR99021s on donor BM-HSCs resulted in an impressive increase in baseline engraftment levels post-IUHCT (4 weeks: group 5, 41.8% ± 3.7%), and this remained unchanged up to 24 weeks of recipient age (12 weeks: group 5, 41.4% ± 2.6%; 24 weeks: group 5, 45.4% ± 2.3%). The effect of MLV-CHIR99021s on engraftment was due to the gradual release and activity of the inhibitor, because conjugation of donor cells with empty particles did not reproduce the chimerism rise seen in group 5 animals (4 weeks: group 4, 19.2% ± 2.9%). (J) Multilineage reconstitution of all 5 groups at 6 months in blood compared with donor lineages did not demonstrate any differences between groups. (B,E) *P < .05 vs (-) CHIR99021; (I) *P < .05 groups 1-4: 4 weeks vs groups 1-4: 12 and 24 weeks and group 2: 4 weeks vs groups 1, 3, 4: 4 weeks; ***P < .0001 vs groups 1-4 (all timepoints).
Protocol 3
In our final in vivo experimental series, we attempted to provide more definitive evidence of targeted/pseudoautocrine bioactivity by adapting Harrison’s competitive repopulation assay31 and using it in the setting of IUHCT. Our donor cell inoculum consisted of a 1:1 mixture of B6-GFP (5 × 106) and B6-CD45.1 (5 × 106) BM-MNCs. MLV-CHIR99021s were conjugated either on GFP+ or CD45.1+ donor cells before IUHCT. There were 3 experimental groups in our in vivo competitive experiments (Figure 5A): group 1, mixture of unprocessed B6-GFP and B6-CD45.1 BM-MNCs (GFP plus CD45.1); group 2, B6-GFP BM-MNCs decorated with MLV-CHIR99021s mixed with unprocessed B6-CD45.1 BM-MNCs (GFP-MLV-CHIR99021s plus CD45.1); and group 3, unprocessed B6-GFP BM-MNCs mixed with B6-CD45.1 BM-MNCs decorated with MLV-CHIR99021s (GFP plus CD45.1-MLV-CHIR99021s).
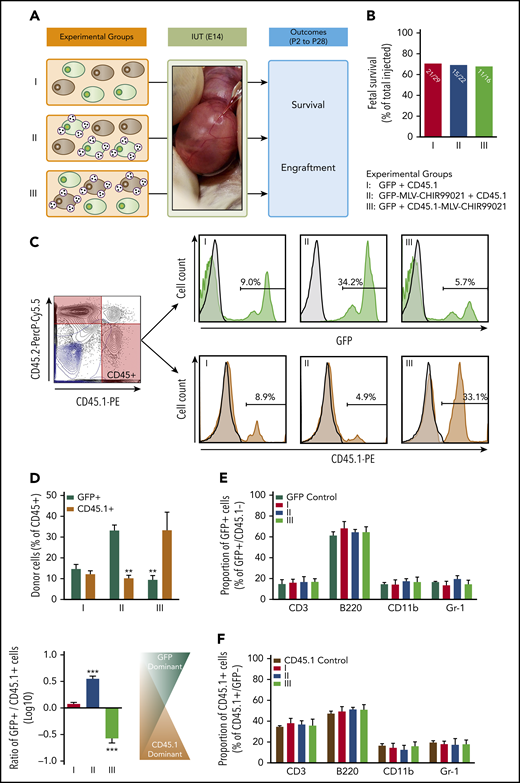
Pseudoautocrine bioactivity of GSK3 inhibitor released by MLVs results in targeted augmentation of donor cell repopulating function. (A) Experimental design: IUHCT in Balb/c fetuses of a 1:1 mixture of B6GFP and B6-CD45.1 BM-MNCs with both populations unconjugated (group 1), with only B6GFP cells conjugated to GSK3 inhibitor–loaded MLVs (group 2), and with only B6-CD45.1 cells conjugated (group 3). (B) Fetal survival after 4 days post-IUHCT in 3 groups; there was no difference between groups (group 1, 71% vs group 2, 68% vs group 3, 69%). (C) Flow cytometry gating strategy for determining the percentage of both populations in blood; CD45.1+ (B6-CD45.1 cells) and GFP+ (B6-GFP cells) within the CD45 population; samples from the 3 groups (light blue contour plots: 45.1− and 45.2− double-negative controls; light blue histograms: GFP− or CD45.1− negative controls). (D) Mean percentage of GFP+ and CD45.1+ donor cells within groups 1, 2, and 3 in blood. Group 1 showed similar levels of engraftment in blood of recipients at 4 weeks of age (group 1: GFP+, 14.7% ± 2.2% vs CD45.1+, 12.1% ± 1.7%; GFP+/CD45.1+ ratio, 1.2 ± 1.7). Conjugation of MLV-CHIR99021s on either B6-GFP or B6-CD45.1 cells resulted in targeted enhancement of blood chimerism of the donor cell subpopulation carrying the inhibitor-loaded particles. In group 2, the percentage of GFP+ cells were significantly higher than CD45.1+ cells (group 2: GFP+, 33.2% ± 2.6% vs CD45.1+, 10.2% ± 1.4%; GFP+/CD45.1+ ratio, 3.3 ± 1.3). In group 3, the opposite was found (group 3: GFP+, 9.23% ± 2.1% vs CD45.1+, 33.3% ± 8.7%; GFP+/CD45.1+ ratio, 0.3 ± 1.1). (E-F) Multilineage analysis in GFP+ (E) and CD45.1+ (F) donor cells; no differences between groups in the proportion of lymphoid (CD3+, B220+) or myeloid (CD11b+, Gr-1+) cells in blood. (D) **P < .001 groups 2, 3: GFP+ vs CD45.1+; ***P < .0001 vs groups 1 and 2 or 1 and 3.
Pseudoautocrine bioactivity of GSK3 inhibitor released by MLVs results in targeted augmentation of donor cell repopulating function. (A) Experimental design: IUHCT in Balb/c fetuses of a 1:1 mixture of B6GFP and B6-CD45.1 BM-MNCs with both populations unconjugated (group 1), with only B6GFP cells conjugated to GSK3 inhibitor–loaded MLVs (group 2), and with only B6-CD45.1 cells conjugated (group 3). (B) Fetal survival after 4 days post-IUHCT in 3 groups; there was no difference between groups (group 1, 71% vs group 2, 68% vs group 3, 69%). (C) Flow cytometry gating strategy for determining the percentage of both populations in blood; CD45.1+ (B6-CD45.1 cells) and GFP+ (B6-GFP cells) within the CD45 population; samples from the 3 groups (light blue contour plots: 45.1− and 45.2− double-negative controls; light blue histograms: GFP− or CD45.1− negative controls). (D) Mean percentage of GFP+ and CD45.1+ donor cells within groups 1, 2, and 3 in blood. Group 1 showed similar levels of engraftment in blood of recipients at 4 weeks of age (group 1: GFP+, 14.7% ± 2.2% vs CD45.1+, 12.1% ± 1.7%; GFP+/CD45.1+ ratio, 1.2 ± 1.7). Conjugation of MLV-CHIR99021s on either B6-GFP or B6-CD45.1 cells resulted in targeted enhancement of blood chimerism of the donor cell subpopulation carrying the inhibitor-loaded particles. In group 2, the percentage of GFP+ cells were significantly higher than CD45.1+ cells (group 2: GFP+, 33.2% ± 2.6% vs CD45.1+, 10.2% ± 1.4%; GFP+/CD45.1+ ratio, 3.3 ± 1.3). In group 3, the opposite was found (group 3: GFP+, 9.23% ± 2.1% vs CD45.1+, 33.3% ± 8.7%; GFP+/CD45.1+ ratio, 0.3 ± 1.1). (E-F) Multilineage analysis in GFP+ (E) and CD45.1+ (F) donor cells; no differences between groups in the proportion of lymphoid (CD3+, B220+) or myeloid (CD11b+, Gr-1+) cells in blood. (D) **P < .001 groups 2, 3: GFP+ vs CD45.1+; ***P < .0001 vs groups 1 and 2 or 1 and 3.
Outcomes
In protocols 1 and 2, fetal and postnatal survival (up to 6 months) was documented. Donor cell (GFP+) chimerism and multilineage differentiation were assessed in blood (4, 12, and 24 weeks of recipient age), as well as in BM and spleen (24 weeks; flow cytometry and fluorescence-activated cell sorting [FACS]; supplemental Methods). Qualitative confirmation of engraftment results obtained by flow cytometry was achieved using immunohistochemistry (BM only; 6 months; supplemental Methods).
In protocol 3, fetal and postnatal survival (up to 4 weeks) was documented. Donor cell (GFP+, CD45.1+) chimerism and multilineage differentiation were assessed in blood, BM, and spleen at 4 weeks of recipient age by FACS. In addition, we examined the hematopoietic progenitor cells (HPCs)/HSCs (Lin−) and HSCs (LSK) containing compartments of donor cells found in BM (4 weeks; FACS; supplemental Methods).
Statistical analyses
Results are expressed as mean plus or minus SEM, and statistical analyses were performed using 1- or 2-way analysis of variance with Bonferroni post hoc tests. P < .05 was considered significant. Statistical analyses were performed with Prism 6 statistical software (GraphPad Software, Inc., La Jolla, CA).
Results
Nanoparticle characterization
Our protocol resulted in polydisperse MLV-CHIR99021 solutions, with a nanoparticle diameter of 496 ± 5 nm, a loaded CHIR99021 mass of 27 ± 1 μg per 1010 particles, and the ability to release the inhibitor over 7 days (supplemental Results; supplemental Figures 1 and 2). Using a nanoparticle/cell ratio of 600:1 for conjugation, we achieved a mean number of 55 ± 8 MLV-CHIR99021s per BM-MNC or BM-HSC (supplemental Results; supplemental Figure 3). On the basis of this, we estimated that the mean CHIR99021 dose used was 0.15 ± 0.02 pg per cell. Preliminary studies demonstrated that conjugation of MLVs to the cell surface of BM-MNCs did not affect their ability to home to or engraft in fetal hematopoietic tissues post-IUHCT (Figure 1; supplemental Results; supplemental Figure 4).
Conjugation of GSK3 inhibitor–loaded MLVs on BM-MNCs enhances long-term hematopoietic engraftment after IUHCT
Conjugation of MLV-CHIR99021s on BM-MNCs and gradual release of the inhibitor (1.5 μg per fetus) did not affect fetal survival, which contrasted with what we observed when the same amount of CHIR99021 was administered as a bolus at the time of IUHCT (P < .05; Figure 2B). Donor cell engraftment in blood at 4 weeks of recipient age was similar between animals in group 1 and survivors in group 2 (group 1, 15.5% ± 1.4% vs group 2, 10.5% ± 2.3%) but was >3 times higher in group 3 animals (group 3, 52.9% ± 2.8%; P < .0001 vs groups 1 and 2; Figure 2C-D). In contrast to groups 1 and 2, engraftment in group 3 animals remained stable at 12 and 24 weeks (12 weeks, 48.3% ± 2.2%; 24 weeks, 48.5% ± 3.1%; P < .0001 vs groups 1 and 2; Figure 2D). There were no differences in white blood cell count between groups (group 1, 4.5 × 109 ± 0.4 × 109/L; group 2, 5.3 × 109 ± 0.5 × 109/L; group 3, 4.9 × 109 ± 0.3 × 109/L); multilineage analysis of donor and host cells did not demonstrate any differences in the relative levels of different lineages in the hematopoietic compartments of experimental offspring (Figure 2E-F; supplemental Figures 5 and 6A), and there was no evidence of graft-versus-host disease or malignancy. Similar results were observed in BM and spleen at 24 weeks (Figure 3; supplemental Figures 6B and 7).
GSK3 inhibitor released by MLVs increases engraftment after IUHCT by acting on HSCs/HPCs
BM-HSCs were isolated and cultured over a 7-day period with (or without) an amount of CHI99021 similar to that carried by MLVs (per cell) added to the media. At the end of the culture period, ∼25% of the harvested cells were LSK (Figure 4A); this was not affected by the presence of CHIR99021. In agreement with what has been published previously, exposure of BM-HSCs to CHIR99021 over the 7-day culture period resulted in a significant increase in the total number of both BM-MNCs and BM-HSCs (Figure 4B) and was associated with modulation of the Wnt (downregulation of Axin2 expression, upregulation of Ccnd1 expression), Notch (upregulation of Hes1 expression), and Hedgehog (upregulation Gli3 and Ptch1 expression) pathways, which have been shown to affect HSC function, proliferation kinetics, and repopulating capacity (Figure 4C).12-17 Moreover, we found that BM-HSCs cultured with CHIR99021 had enhanced clonogenic potential evident in colony-forming unit assays compared with untreated controls (Figure 4D-E). At the end of the culture and before IUHCT, BM-HSCs from freshly isolated BM-MNCs were replaced 1:1 with expanded BM-HSCs (cultured with or without CHIR99021).
In agreement with our previous in vivo experiments, donor cell chimerism in blood after IUHCT of 107 BM-MNCs was 14.3% ± 1.9% at 4 weeks (group 1; Figure 4H-I), and levels of engraftment gradually decreased during the follow-up period (Figure 4I). Replacement of BM-HSCs from freshly isolated BM-MNCs with expanded BM-HSCs led to reduced blood engraftment at 4 weeks (group 2, 4.9% ± 0.6%; P < .05 vs group 1; Figure 4H-I) and only microchimerism thereafter (Figure 4I). Addition of CHIR99021 to the culture media of BM-HSCs prevented the latter drop in chimerism at 4 weeks (group 3, 12.9% ± 1.6%; P < .05 vs group 2; Figure 4H-I) but did not enhance it any further (Figure 4I). Conjugation of MLV-CHIR99021s on donor BM-HSCs (dose of CHIR99021, 1.5 ng per fetus) resulted in an impressive increase in baseline engraftment levels post-IUHCT (4 weeks; group 5, 41.8% ± 3.7%; P < .0001 vs groups 1-4; Figure 4H, I), and this remained unchanged up to 24 weeks of recipient age (Figure 4I). Conjugation of donor cells with empty particles (group 4) did not reproduce the chimerism rise seen in group 5 animals (Figure 4H-I). More importantly, the levels of engraftment achieved by conjugating MLV-CHIR99021s only on BM-HSCs were similar to those seen when all donor BM-MNCs were decorated with MLV-CHIR99021s. There were no differences in white blood cell count (group 1, 4.7 × 109 ± 0.2 × 103/µL; group 2, 4.3 × 109 ± 0.6 × 109/L; group 3, 5.2 × 109 ± 0.4 × 103/L; group 4, 4.4 × 109 ± 0.4 × 109/L; group 5, 5.4 × 109 ± 0.7 × 109/µL) or multilineage analysis of donor and host cells between groups (Figure 4J; supplemental Figure 8), and there was no evidence of graft-versus-host disease or malignancy. Similar results were seen in the BM and spleen at 24 weeks (supplemental Figure 9).
Pseudoautocrine bioactivity of GSK3 inhibitor released by MLVs results in targeted augmentation of donor cell repopulating function
IUHCT of a 1:1 mixture of unmodified B6-GFP and B6-CD45.1 (CD45.1+) cells resulted in similar levels of engraftment in blood of recipients at 4 weeks of age (group 1 (GFP+), 14.7% ± 2.2% vs group 1 (CD45.1+), 12.1% ± 1.7%; group 1 (GFP+/CD45.1+ ratio), 1.2 ± 1.7; Figure 5C-D). Conjugation of MLV-CHIR99021s on either B6-GFP or B6-CD45.1 cells resulted in targeted enhancement of blood chimerism of the donor cell subpopulation carrying the inhibitor-loaded particles (group 2 (GFP+), 33.2% ± 2.6% vs group 2 (CD45.1+), 10.2% ± 1.4%; P < .001; group 2 (GFP+/CD45.1+ ratio), 3.3 ± 1.3; P < .0001 vs group 1; group 3 (GFP+), 9.23% ± 2.1% vs group 3 (CD45.1+), 33.3% ± 8.7%; P < .001; group 3 (GFP+/CD45.1+ ratio), 0.3 ± 1.1; P < .0001 vs groups 1 and 2; Figure 5C-D). Multilineage analysis did not demonstrate any differences between groups (Figure 5E-F). Results in BM and spleen mirrored those in blood (Figure 5A; supplemental Figures 10 and 11).
The proportion of donor Lin− cells (percentage of the sum of CD45.1+ and CD45.2+) was 2.8% ± 0.2% in group 1, with no differences among experimental groups (Figure 6B). However, when we looked at the absolute number of donor Lin− cells (per hind limb long bone), we saw a significant increase in the hematopoietic progenitor number only in the donor cell subpopulation subjected to targeted GSK3 inhibition (Figure 6C-D). Similar effects were observed in the donor BM-MNC HSC-containing compartment (LSK cells). The proportion of donor primitive LSK cells (percentage of Lin− within the sum of CD45.1+ and CD45.2+ BM-MNCs) was 11.2% ± 0.6% in group 1, and no differences were observed among experimental groups (Figure 6E). However, when we estimated the absolute number of donor LSK cells, we saw a significant rise in the HSC number only in the donor cell subpopulation carrying MLV-CHIR99021s (Figure 6F-G).
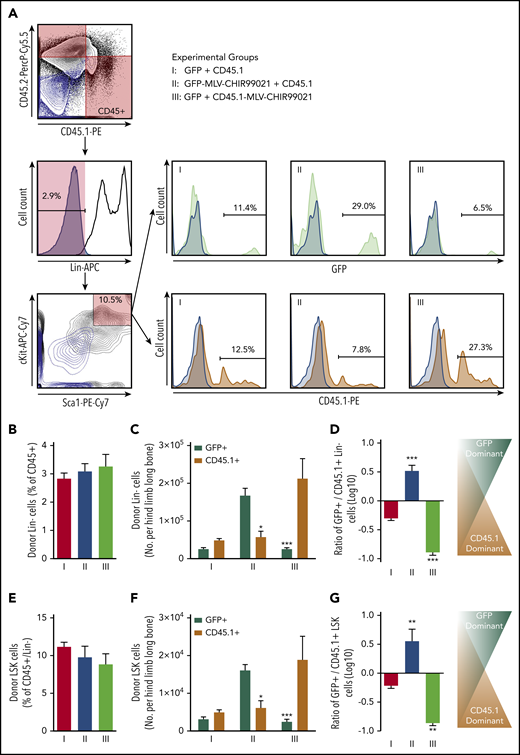
Effect of targeted pseudoautocrine bioactivity of GSK3 inhibitor released by MLVs on donor HSCs/HPCs. (A) Flow cytometry gating strategy for determining the HPC/HSC (Lin−) and HSC-containing compartments of donor cells found in BM of IUHCT recipients (Balb/c fetuses); samples from the 3 groups (light blue contour plots: 45.1− and 45.2− double-negative controls or CD117− and Sca1− double-negative controls; light blue histograms: Lin−, GFP−, or CD45.1− negative controls). (B) Proportion of donor Lin− cells (expressed as a percentage of the sum of CD45.1+ and CD45.2+ BM-MNCs) was 2.8% ± 0.2% in group 1, and no differences were observed among experimental groups (Lin−: group 2, 3.1% ± 0.3% vs group 3, 3.3% ± 0.4%). (C) Absolute number of donor Lin− cells (per hind limb long bone); significant increase in hematopoietic progenitor number only in the donor cell subpopulation subjected to targeted GSK3 inhibition (group 1: Lin−, GFP+, 2.5 × 103 ± 0.4 × 103 vs Lin−, CD45.1+, 4.8 × 103 ± 0.5 × 103; group 2: Lin−, GFP+, 16.7 × 103 ± 1.9 × 103 vs Lin−, CD45.1+, 5.7 × 103 ± 1.5 × 103; group 3: Lin−, GFP+, 2.5 × 103 ± 0.3 × 103 vs Lin−, CD45.1+, 21.2 × 103 ± 5.2 × 103). (D) Ratio of GFP+/CD45.1+ Lin− cells. (E) Proportion of donor primitive LSK cells (expressed as a percentage of the Lin− cells in the sum of CD45.1+ and CD45.2+ BM-MNCs); no differences were observed among experimental groups (LSK: group 1, 11.2% ± 0.6% vs group 2, 9.8% ± 1.5% vs group 3, 8.8% ± 1.4%). (F) Absolute number of LSK cells (per hind limb long bone); significant increase only in the donor cell subpopulation carrying MLV-CHIR99021s (group 1: LSK, GFP+, 3.1 × 102 ± 0.6 × 102 vs LSK, CD45.1+, 4.9 × 102 ± 0.7 × 102 CD45.1+ cells; group 2: LSK, GFP+, 16.1 × 102 ± 1.6 × 102 vs LSK, CD45.1+, 6.1 × 102 ± 1.9 × 102; group 3: LSK, GFP+, 2.4 × 102 vs LSK, CD45.1+, 18.8 × 102 ± 6.2 × 102. (G) Ratio of GFP+/CD45.1+ LSK cells. (C) *P < .05 group 2: Lin−, GFP+ vs Lin−, CD45.1+, **P < .001 group 3: Lin−, GFP+ vs Lin−, CD45.1+; (D) ***P < .0001 vs groups 1 and 2 or 1 and 3; (F) *P < .05 group 2: GFP+ vs CD45.1+, **P < .001 group 3: GFP+ vs CD45.1+; (G) **P < .001 vs groups 1 and 2 or 1 and 3.
Effect of targeted pseudoautocrine bioactivity of GSK3 inhibitor released by MLVs on donor HSCs/HPCs. (A) Flow cytometry gating strategy for determining the HPC/HSC (Lin−) and HSC-containing compartments of donor cells found in BM of IUHCT recipients (Balb/c fetuses); samples from the 3 groups (light blue contour plots: 45.1− and 45.2− double-negative controls or CD117− and Sca1− double-negative controls; light blue histograms: Lin−, GFP−, or CD45.1− negative controls). (B) Proportion of donor Lin− cells (expressed as a percentage of the sum of CD45.1+ and CD45.2+ BM-MNCs) was 2.8% ± 0.2% in group 1, and no differences were observed among experimental groups (Lin−: group 2, 3.1% ± 0.3% vs group 3, 3.3% ± 0.4%). (C) Absolute number of donor Lin− cells (per hind limb long bone); significant increase in hematopoietic progenitor number only in the donor cell subpopulation subjected to targeted GSK3 inhibition (group 1: Lin−, GFP+, 2.5 × 103 ± 0.4 × 103 vs Lin−, CD45.1+, 4.8 × 103 ± 0.5 × 103; group 2: Lin−, GFP+, 16.7 × 103 ± 1.9 × 103 vs Lin−, CD45.1+, 5.7 × 103 ± 1.5 × 103; group 3: Lin−, GFP+, 2.5 × 103 ± 0.3 × 103 vs Lin−, CD45.1+, 21.2 × 103 ± 5.2 × 103). (D) Ratio of GFP+/CD45.1+ Lin− cells. (E) Proportion of donor primitive LSK cells (expressed as a percentage of the Lin− cells in the sum of CD45.1+ and CD45.2+ BM-MNCs); no differences were observed among experimental groups (LSK: group 1, 11.2% ± 0.6% vs group 2, 9.8% ± 1.5% vs group 3, 8.8% ± 1.4%). (F) Absolute number of LSK cells (per hind limb long bone); significant increase only in the donor cell subpopulation carrying MLV-CHIR99021s (group 1: LSK, GFP+, 3.1 × 102 ± 0.6 × 102 vs LSK, CD45.1+, 4.9 × 102 ± 0.7 × 102 CD45.1+ cells; group 2: LSK, GFP+, 16.1 × 102 ± 1.6 × 102 vs LSK, CD45.1+, 6.1 × 102 ± 1.9 × 102; group 3: LSK, GFP+, 2.4 × 102 vs LSK, CD45.1+, 18.8 × 102 ± 6.2 × 102. (G) Ratio of GFP+/CD45.1+ LSK cells. (C) *P < .05 group 2: Lin−, GFP+ vs Lin−, CD45.1+, **P < .001 group 3: Lin−, GFP+ vs Lin−, CD45.1+; (D) ***P < .0001 vs groups 1 and 2 or 1 and 3; (F) *P < .05 group 2: GFP+ vs CD45.1+, **P < .001 group 3: GFP+ vs CD45.1+; (G) **P < .001 vs groups 1 and 2 or 1 and 3.
Discussion
The data presented here demonstrate that temporally and spatially targeted GSK3 inhibition of BM-HSCs results in remarkable levels of stable hematopoietic engraftment after experimental IUHCT. The gradual release of the inhibitor from liposome-based, synthetic nanoparticles conjugated on the surface of donor cells resulted in inhibition of GSK3 activity via a pseudoautocrine mechanism. The targeted and sustained in vivo GSK3 inhibition led to significant increases in overall cellularity of the donor cell pool post-IUHCT, with an associated expansion of the donor HPC and HSC populations (LSK). The enhanced proliferation kinetics of cell-engineered BM-HSCs mitigated the competitive advantage of fetal equivalents, with no evidence of increased differentiation or exhaustion of BM-HSCs with long-term repopulating capacity. Our study has significant translational potential, because the levels of engraftment achieved after IUHCT of nanoparticle-conjugated donor cells in our murine model would be therapeutic for common congenital hematological disorders, including thalassemia and sickle cell disease,32-35 allowing for single-step prenatal treatment.36
The most compelling rationale for the use of IUHCT for the treatment of congenital diseases of hematopoiesis is based on unique opportunities related to normal developmental events that may facilitate engraftment and avoid complications associated with postnatal transplantation.1 The most important of these is the development of the immune system, which allows the induction of actively acquired tolerance and hematopoietic engraftment of allogeneic fetal (fetal liver)37 and adult (BM-derived) cells2 across immune barriers. Despite the potential advantages of IUHCT over postnatal transplantation, IUHCT in many animal models with intact fetal hematopoiesis has been limited by low levels of engraftment, which are enough for tolerance induction but inadequate for therapeutic effect for many target disorders.38-41
The main cause of the latter is host cell competition. The first successful animal studies of IUHCT were performed in stem cell–defective murine models42 and immunodeficient mouse strains.43 In a fetus with intact hematopoiesis during in utero development (eg, inherited hemoglobinopathies), fetal stem cell and progenitor populations have a competitive advantage over their postnatal (adult) equivalents. As a result, IUHCT of large doses of donor cells (2 × 1011 adult BM-MNCs per kg intravenously) results in rather modest levels of long-term chimerism of <10%.3,38 Such engraftment would not be therapeutic for most congenital hematological disorders targeted by IUHCT, which would necessitate chimerism levels of ∼20%.32-35,44 Although current allogeneic IUHCT protocols allow the induction of donor-specific tolerance that could be combined with nonmyeloablative postnatal HSCT to achieve clinically relevant engraftment,7,45 the development of a single-step prenatal therapeutic strategy would be clinically, financially, and ethically desirable. In previous murine studies, we and others attempted to address this by enhancing donor BM-HSC homing to fetal hematopoietic niches46 or mobilizing host HSCs from fetal hematopoietic niches, thus generating more space for donor cells to engraft.47,48 An additional approach that could be used in parallel with the latter strategies would be to attempt to make adult BM-HSCs more competitive by enhancing the proliferation kinetics of donor cells. An appealing cellular target for such an approach would be GSK3.
In the seminal study by Trowbridge et al,26 a 30-mg/kg dose of the GSK3 inhibitor CHIR99021 was administered twice per week to sublethally irradiated recipients of mouse or human BM-HSCs over a period of 5 weeks, resulting in improved proliferation kinetics of donor cells and enhanced hematopoietic repopulation. The latter would not be feasible in IUHCT because of limited access to the fetus and potential fetal toxicity of systemic GSK3 inhibition. An elegant solution to this problem was introduced by Stephan et al,27,28 using novel nanoparticle technology.29 They described a strategy to enhance the efficacy of cell therapy via the conjugation of adjuvant drug–loaded nanoparticles to the cell surface. Multilayered, liposome-based nanoparticles (MLVs) were formed with a hydrophilic core that allowed loading and slow release of various bioactive chemicals, providing targeted and sustained pseudoautocrine stimulation of donor cells. The results of the present experimental series extend these observations to the fetal hematopoietic environment and establish a strong salutary effect of sustained and targeted donor cell GSK3 inhibition in hematopoietic engraftment after IUHCT. Our experiments demonstrated that continuous but time-limited in vivo inhibition of GSK3 in donor cells resulted in remarkable enhancement in engraftment levels after allogeneic IUHCT. The near 50% chimerism achieved is reproducible and, more importantly, stable for up to 6 months. The latter contrasts with control observations and would be consistent with a GSK3 inhibition–mediated increase in the overall number of donor HSCs with long-term repopulating capacity. Single, systemic administration of the same small amount of CHIR99021 to the fetus at the time of IUHCT resulted in increased fetal mortality and failed to reproduce the effects achieved by pseudoautocrine release from MLV-CHIR99021s. The reduction in fetal survival can be explained by well-recognized fetotoxic effects of global GSK3 inhibition,49 and the lack of benefit can be explained by the requirement for sustained GSK3 inhibition for therapeutically relevant effects to become manifest.21,26,50 Our findings also confirm previous observations26 that the biological effect of GSK3 inhibition is specific to the primitive, HSC/HPC-containing subpopulation of LSK cells; cell surface decoration of donor LSK cells with MLV-CHIR99021s resulted in levels of stable hematopoietic engraftment after IUHCT similar to those observed when nanoparticles are conjugated to the whole BM-MNC inoculum. This occurred despite the even smaller total dose of CHIR99021 administered and suggests that it is the total dose of CHIR99021 per cell (0.15 pg per cell) that is important in this setting.27
In our competitive in utero repopulation experiments, we adapted the protocols introduced by Harrison31 in 1980 and applied them in the setting of the competitively robust fetal hematopoietic environment. We demonstrated that targeted GSK3 inhibition, achieved by pseudoautocrine activity of the inhibitor released from MLV-CHIR99021s, offered a distinct competitive advantage to the donor cell population carrying the particles. This competitive boost was both against the nonengineered donor BM-MNC population, as well as against fetal hematopoietic cells. In addition, we showed that the CHIR99021-induced increase in engraftment was associated with a proportional increase in the absolute number of donor HSCs/HPCs, consistent with augmented expansion of the HPC and HSC compartments of donor MNCs (in number but not in proportion because of improved cycling kinetics) as a possible explanation of the enhanced engraftment of cells decorated with MLV-CHIR99021s. The stable levels of long-term hematopoietic engraftment observed in our 6-month follow-up studies suggest that the sustained but transient inhibition of GSK3 expands both donor HSC and HPC pools without affecting the long-term repopulating capacity of HSCs, although the latter would need to be confirmed by transplanting donor HSCs in serial recipients. This contrasts with what has been found in assays of long-term HSC function, in which disruption of GSK3 progressively depleted HSCs through activation of mammalian target of rapamycin.21,50 Our findings also differ from the original observations by Trowbridge et al,26 who showed that GSK3 inhibition does not induce expansion of the long-term repopulating HSC pool. These differences can be attributed to the transient and brief GSK3 inhibition used in the present experimental series, as well as to the very small dose of the inhibitor used. Potential human application may require larger CHIR99021 doses (per cell) and inhibitor release over a more prolonged period of time, which could result in mammalian target of rapamycin activation–related effects on HSCs. Such issues could be addressed by decorating the cell surface with nanoparticles containing both GSK3b inhibitors and rapamycin. Identification of the optimal dose/concentration of candidate growth factors/small-molecule agents as well as the optimal duration of stimulation in human HSCs (to achieve maximal in vivo donor HSCs without affecting long-term repopulating capacity) would be essential before clinical translation.
Although we did not investigate the direct molecular effects of GSK3 inhibition on BM-derived cells used in IUHCT experiments, 7-day culture of BM-derived LSK cells in the presence of an amount of CHIR99021 similar to that carried per cell in MLV-CHIR99021–conjugated LSK cells resulted in an increase in the number of primitive HSCs (but not their proportion compared with lineage-committed hematopoietic cells). The observed enhanced proliferation kinetics were associated with modulation of the Wnt, Notch, and Hedgehog pathways, similar to those observed by Trowbridge et al,26 as well as an augmented ability compared with controls to form hematopoietic progenitor colonies in semisolid media. It was not possible to use MLV-CHIR99021s in our culture experiments (serum-free culture conditions do not allow gradual MLV degradation or inhibitor release). As a result, our in vitro observations provided only indirect evidence of a mechanism for the GSK3 inhibition–associated enhancement in HSC/HPC repopulating capacity via the modulation of cellular pathways known to regulate their homeostasis and proliferation kinetics.21,26,50 More importantly, accurate determination of the exact molecular pathways that mediate the salutary effect of in vivo GSK3 inhibition in the setting of IUHCT would require transcriptomic analysis of isolated donor MLV-CHIR99021–carrying HSCs soon after transplantation, as well as analysis of isolated long-term repopulating donor HSCs (derived from the original cell-engineered donor HSCs) at later time points. Such analyses were not performed in the present study but will be the focus of future work in our laboratory.
When 7-day cultured, CHIR99021-pretreated BM-HSCs were used for IUHCT, they (as expected) outperformed control/untreated cultured equivalents but failed to generate engraftment levels similar to those achieved when MLV-CHIR99021–decorated BM-HSCs were used. Although these findings could be partly attributed to the well-recognized effects of culture on the long-term repopulating ability of HSCs,51-53 they nonetheless support the requirement for sustained in vivo GSK3 inhibition of donor cells to achieve the remarkable improvement in engraftment. The MLV-CHIR99021 nano-delivery platform has unique properties that allow such a targeted release of the inhibitor in the setting of IUHCT and resulted in levels of long-term engraftment that would be therapeutic for many inherited diseases of hematopoiesis.32-35,44 The scalability and adaptability (inhibitor loading mass and release profile) of the MLV-CHIR99021 platform, as well as the low associated costs (US$7 per 107 engineered cells), add further translational value to our findings.
We did not perform dedicated dose-determining, efficacy, or toxicity studies in this experimental series, and the choice of CHIR99021 dose per cell was made based on previously published data.27 Although the total dose of CHIR99021 administered per fetus varied from 6 μg/kg to 6 mg/kg, we saw no MLV-CHIR99021–associated toxic effects on the fetus at any inhibitor dose. Even though the maximum dose of the inhibitor used in our experiments was much lower than that used in previously published work in postnatal transplantation,26 a bolus 6-mg/kg dose of CHIR99021 at the time of IUHCT resulted in increased fetal mortality. A similar CHIR99021 dose carried in MLVs had no adverse effects on fetal survival. This, along with the lack of effect of the nanoparticles on homing of donor cells to fetal hematopoietic tissues or on induction of central (thymic) immune tolerance, highlights the favorable safety characteristics of the nano-delivery platform in the setting of IUHCT. Although we did not include secondary transplantation experiments in our experimental design and did not perform specific analysis for long-term repopulating HSCs (CD150+, CD48+) within donor LSK cells, the minimal change in engraftment levels over the 24-week follow-up period observed when LSK cells decorated with MLV-CHIR99021s were used for IUHCT supports a salutary effect of the inhibitor on HSCs, with long-term repopulating capacity. However, serial transplantation experiments will be crucial to establish unequivocally the effect of our therapeutic platform on long-term repopulating HSCs before clinical application to the human fetus. Finally, the observation of improved competitive capacity of MLV-CHIR99021–treated donor cells observed in this model of a nonmyeloablated, highly competitive host hematopoietic system has obvious implications beyond IUHCT for minimally myeloablative strategies in postnatal HSC transplantation.
For original data, please contact sloukogeorgakis@gmail.com.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the staff at the Metabolomic Core Facility, Children's Hospital of Philadelphia for measuring the CHIR99021 level in various experiments.
This work was supported by grants from the Wellcome Trust postdoctoral fellowship for MB/PhD graduates (098539/Z/12/Z) (S.P.L.), the National Institute for Health Research (P.D.C.), and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) agency, Brazil (99999.002599/2014-07) (C.G.F.).
Authorship
Contribution: S.P.L., C.G.F., A.I.B.S.D., H.L., L.T., D.J.I., and A.W.F. designed the work; S.P.L., C.G.F., A.I.B.S.D., H.L., A.G.K., J.D.V., J.D.S., and N.J.A. performed the experiments and contributed to data collection; Ilana Nissim and Izhtak Nissim developed the method and performed the measurements of CHIR99021; S.P.L., C.G.F., A.I.B.S.D., H.L., Ilana Nissim, Izhtak Nissim, W.H.P., P.D.C., D.J.I., and A.W.F. contributed to data analysis and interpretation; S.P.L., C.G.F., J.D.S., and N.J.A. prepared the figures; S.P.L., C.G.F., and A.W.F. wrote the manuscript; and S.P.L., C.G.F., A.I.B.S.D., H.L., J.D.S., N.J.A., A.F.M., J.L.M., W.H.P., P.D.C., D.J.I., and A.W.F. contributed to the revision of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stavros P. Loukogeorgakis, Stem Cells and Regenerative Medicine, UCL Great Ormond Street Institute of Child Health, 30 Guilford St, London WC1N 1EH, United Kingdom; e-mail: sloukogeorgakis@gmail.com.
