

Key Points
HPA allele–specific HLA class I–negative MKs can be differentiated from CRISPR-edited human iPSCs.
Such cells can be stored frozen and thawed to use in whole-cell flow cytometric assays to detect anti-HPA-3a, -3b, and -9b alloantibodies.

Abstract
Human platelet membrane glycoprotein polymorphisms can be immunogenic in man and are frequently the cause of clinically important immune reactions responsible for disorders such as neonatal alloimmune thrombocytopenia. Platelets from individuals carrying rare polymorphisms are often difficult to obtain, making diagnostic testing and transfusion of matched platelets challenging. In addition, class I HLA antibodies frequently present in maternal sera interfere with the detection of platelet-reactive alloantibodies. Detection of alloantibodies to human platelet antigen 3 (HPA-3) and HPA-9 is especially challenging, in part because of the presence of cell type–specific glycans situated near the polymorphic amino acid that together form the alloepitope. To overcome these limitations, we generated a series of HLA class I–negative blood group O induced pluripotent stem cell (iPSC) lines that were gene edited to sequentially convert their endogenous HPA-3a alloantigenic epitope to HPA-3b, and HPA-9a to HPA-9b. Subjecting these cell lines, upon differentiation into CD41+/CD42b+ human megakaryocytes (MKs), to flow cytometric detection of suspected anti-HPA-3 and HPA-9 alloantisera revealed that the HPA-3a–positive MKs specifically reacted with HPA-3a patient sera, whereas the HPA-3b MKs lost reactivity with HPA-3a patient sera while acquiring reactivity to HPA-3b patient sera. Importantly, HPA-9b–expressing MKs specifically reacted with anti-HPA-9b–suspected patient samples that had been undetectable using conventional techniques. The provision of specialized iPSC-derived human MKs expressing intact homozygous glycoprotein alloantigens on the cell surface that carry the appropriate endogenous carbohydrate moieties should greatly enhance detection of clinically important and rare HPA-specific alloantibodies that, to date, have resisted detection using current methods.
Introduction
Human platelet alloantigens (HPAs) reside on functionally important platelet membrane glycoproteins. Currently, 6 biallelic HPA systems (HPA-1, -2, -3, -4, -5, and -15) as well as 26 single low-frequency antigens have been described on 6 platelet glycoproteins.1-4 Almost all the HPAs are caused by single amino acid substitutions encoded by single nucleotide polymorphisms on 1 of 6 different platelet glycoproteins. The only exception, HPA-14b, is caused by a single amino acid deletion resulting from an in-frame triplet deletion in the ITGB3 gene rather than a single nucleotide polymorphism.5
Antibodies that form against HPAs are responsible for several clinically important alloimmune bleeding disorders, including fetal and neonatal alloimmune thrombocytopenia (FNAIT, variously referred to in the literature as NATP, NAIT, and FNIT), posttransfusion purpura, and platelet transfusion refractoriness.6,7 In these conditions, serologic detection and characterization of anti-HPA alloantibodies are essential for proper diagnosis, treatment and, in FNAIT, the prevention of severe thrombocytopenia and its bleeding risks in subsequent pregnancies. Over the years, many creative assays have been developed for detecting alloantibodies against HPAs in patient sera, and they can be grouped into 2 categories: whole-platelet methods and glycoprotein-specific methods.8 That human platelets also express class I HLA antigens on their surface, coupled with the fact that patient HPA antisera often contain HLA class I antibodies (the binding of which can mask the presence of HPA-specific antibodies), creates a significant problem for whole-platelet antibody detection methods, thus limiting the HPA alloantibody detection to glycoprotein-specific assays for these patients. Currently, antigen capture assays based on the use of a monoclonal antibody such as modified antigen capture enzyme-linked immunosorbent assay (MACE)9 and monoclonal antibody immobilization of platelet antigens (MAIPA)10 are the most popular glycoprotein-specific assays in use because of their high specificity and sensitivity. However, these methods can be performed only by specialized laboratories, are tedious, and require solubilization of platelet glycoproteins with detergent, a procedure that can result in the loss of labile antigenic determinants.11,12 In addition, the monoclonal antibodies used to capture the glycoproteins sometimes compete with the binding of the alloantibodies present in patient sera, resulting in false-negative results.8
HPA-3, historically known as the Baka/Bakb alloantigen system, was first described in 1980 by von dem Borne et al13 as a new specificity present in a maternal alloantibody in a case of FNAIT. The antigen was localized to GPIIb in 198614 and was found to be the result of an Ile843Ser polymorphism near the carboxyl terminus (C terminus) of the GPIIb extracellular domain.15 Interestingly, amino acid 843 is only 6 amino acids away from a Val837Met polymorphism, identified as the HPA-9b antigen (also known as Maxa) in an FNAIT case.16 HPA-3 antibodies are relatively rare, but they can often induce severe FNAIT with intracranial hemorrhage.11,17-20 Several studies have identified anti-HPA-3 antibodies as the most problematic specificity to detect,11,12,17,20,21 likely because of the heterogeneity of HPA-3 epitope formation. In addition to the nearby Ile843Ser polymorphism, some HPA-3 epitopes depend upon the 3-dimensional conformation of GPIIb and are thus sensitive to fixation or detergents.11,12,17,20 Others require as part of their recognition epitope adjacent O-linked carbohydrate structures or terminal sialic acids that can be lost during platelet storage.21-23 One biochemical study identified Ser847 as a major O-glycosylation site required for anti-HPA-3a antibody binding.24
In contrast to the HPA-3 alloantigen system, HPA-9b is a low-frequency HPA located near the C terminus of the extracellular domain of GPIIb, with an estimated gene frequency of 0.002 to 0.003 in whites.16,25 Despite its low frequency, FNAIT caused by anti-HPA-9b alloantibodies is not uncommon, and nearly all cases are accompanied by severe thrombocytopenia and bleeding.25,26 Detection of anti-HPA-9b alloantibodies can be extremely difficult using existing serologic assays,25-27 probably because of the same complications encountered for detection of alloantibodies to the nearby HPA-3 polymorphism.
Induced pluripotent stem cells (iPSCs) have become an optimal source for large-scale in vitro megakaryocyte (MK) and platelet production because they offer the advantages of unlimited expansion in culture and are amenable to genetic manipulation.28,29 To overcome the limitations for current HPA alloantibody detection, we generated a series of genetically edited iPSC lines lacking HLA class I antigen expression and that display, upon differentiation into human MKs, homozygous forms of GPIIb that carry the HPA-3a, HPA-3b, or HPA-9b alloantigens. These iPSC-derived MKs are suitable for simple, 1-step flow cytometric detection of anti-HPA-3a, HPA-3b, and HPA-9b alloantibodies in patient sera without HLA class I antibody interference. Because these cell lines share an identical genetic background (except for the genetically targeted HPA-3a, HPA-3b, and HPA-9b isoforms of GPIIb), they offer extremely high specificity for diagnostic purposes compared with donor-derived platelets. Ready availability of MKs that express on the cell surface intact glycoproteins that carry carbohydrate moieties that likely mimic those found on human platelets should facilitate detection of HPA alloantibodies that are normally difficult or impossible to detect using existing techniques.
Materials and methods
Patient samples
Anti-HPA-3a (patient 1 [P1]-P3) and anti-HPA-9b (P1-P2) patient sera were from the Platelet and Neutrophil Immunology Laboratory (Versiti, Milwaukee, WI). All these samples, except anti-HPA-3a (P3), were positive in a MACE assay. The anti-HPA-3a (P3) sample tested negative in the MACE assay but was positive using the platelet suspension immune fluorescence test. Anti-HPA-3b (P1-P3) and anti-HPA-9b (P3) patient sera were from the Institute for Clinical Immunology and Transfusion Medicine (Giessen, Germany). These samples were all positive in an MAIPA assay. Anti-HPA-9b (P4-P6) suspected sera were provided by Richard Aster.
Guide RNA plasmid constructs
Guide RNAs (gRNAs) were designed using the CRISPR Design Tool (http://crispr.mit.edu/ and https://benchling.com/crispr) to minimize off-target effects, and they were selected to precede a 5′-NGG protospacer-adjacent motif (PAM). gRNA sequences are listed in supplemental Table 1 (available on the Blood Web site). Oligos were annealed and cloned into the BbsI site of the Cas9 expression plasmids PX459 V2.0 (Addgene, Cambridge, MA).
Design and generation of donor templates
To construct the donor plasmid for HPA-3b, a 1.6-kb ITGA2B gBlock Gene Fragment was synthesized by Integrated DNA Technologies (Coralville, IA). The sequence of the gBlock Gene Fragment is provided in supplemental Data. The fragment contains the T→G (HPA-3a to HPA-3b) substitution as well as silent mutations within the recognition sequence and the PAM sequence of 2 gRNAs to avoid repetitive digestions by Cas9 and also introduces an MfeI site into the genome for genotyping. The recognition sequence and the PAM sequence of guide 2 for generating HPA-3b were added to both ends of the gBlock Gene Fragment for linearizing the donor templates in the transfected cells. The gBlock Gene Fragment was cloned into the pMiniT 2.0 vector using an NEB polymerase chain reaction (PCR) cloning kit (New England Biolabs Inc., Ipswich, MA).
A single-stranded oligo-deoxynucleotide (ssODN) HPA-9b donor template was synthesized by Integrated DNA Technologies. The sequence of the oligo is provided in supplemental Data. This oligo corresponds to the antisense strand, contains a G→A (HPA-9a to HPA-9b) substitution, a silent mutation within the PAM sequence of gRNA to avoid repetitive digestions by Cas9, and introduces a PstI site into the genome for genotyping.
Cell culture and transfection
Human OT1-1 iPSCs30 were cultured on Matrigel (Corning, Corning, NY)-coated plates in mTeSR1 medium (STEMCELL Technologies Inc., Cambridge, MA) at 37°C in 4% O2 and 5% CO2. After incubation with 10 µM Rho kinase (ROCK) inhibitor Y27632 (StemRD Inc., Burlingame, CA), 2 × 105 cells were transfected with 0.5 or 1 μg of each guide plasmid in the presence or absence of 0.5 μg of the HPA-3b plasmid donor or 40 pmol of HPA-9b ssODN donor using the Amaxa P3 primary cell 4D Nucleofector Kit (Lonza, Allendale, NJ) and Nucleofector Program CB-150. The cells were then plated on Matrigel-coated plates with 10 μM Y27632. Puromycin was applied at 24 hours after transfection at a concentration of 1 μg/mL for 48 hours. Single clones were harvested at 12 to 14 days after puromycin selection and re-plated on Matrigel-coated plates. Karyotyping of the iPSC lines was performed every 15 passages by Wisconsin Diagnostic Laboratories (Milwaukee, WI) to verify continued diploidy of all iPSC lines.
Genotyping
Genomic DNA was extracted from each iPSC clone by using the QuickExtract DNA Extraction Solution (Epicenter, Madison, WI) following the manufacture’s protocol. The region surrounding the HPA-3 and HPA-9 polymorphisms was amplified by PCR using the pair of primers: ITGA2B for 5′-CGTGGAATTCAAGTGGAGCACACCTATGAG-3′ and ITGA2B rev 5′-GGCAAGC-TTACCTTGCTTTGGCATTGTTT-3′. PCR products were purified by using QiaQuick Spin Column, digested with MfeI or PstI (New England Biolabs Inc.), and analyzed on 2% agarose gels.
Differentiation of iPSCs
CRISPR-edited iPSC lines were differentiated to hematopoietic progenitor cells (HPCs) as previously described.31,32 Briefly, cells were plated on Matrigel for differentiation. Media and cytokine changes were observed as described except that the GSK-3β inhibitor CHIR99021 (1 μM) (Tocris Bioscience, Minneapolis, MN) was used instead of Wnt3a. Cells were cultured at 37°C with 4% O2 and 5% CO2 for 9 days, and loosely adherent HPCs were collected by carefully removing the supernatant. Cells were analyzed by flow cytometry to confirm surface expression of CD41a and CD235a. The HPCs were further differentiated to MKs in serum-free differentiation medium, which is composed of Iscove modified Dulbecco medium (Thermo Fisher Scientific, Waltham, MA) containing 25% Ham’s F12 (Corning), 0.5% N2 (Thermo Fisher Scientific), 1% B27 without vitamin A (Thermo Fisher Scientific), 0.05% bovine serum albumin (Sigma, St. Louis, MO), 2-mM l-glutamine and penicillin/streptomycin supplemented with 50 ng/mL stem cell factor (R&D Systems, Minneapolis, MN), and 50 ng/mL thrombopoietin (R&D Systems) at 37°C in 5% CO2 for 6 days. MKs were analyzed by flow cytometry to confirm the surface expression of CD41 and CD42b.
Flow cytometric analysis
3 × 105 iPSCs were incubated with fluorescein isothiocyanate–conjugated anti-human HLA-A, -B, -C and allophycocyanin-conjugated anti-human β2-microglobulin (β2M) antibodies (BioLegend, San Diego, CA) for 20 minutes at room temperature. 3 × 105 iPSC-derived MKs were incubated with 25 to 50 μL of normal human sera or patient sera for 30 minutes at room temperature. After washing, the cells were incubated with fluorescein isothiocyanate-conjugated anti-CD41, allophycocyanin-conjugated anti-CD42b (BioLegend), and phycoerythrin-conjugated donkey anti-human immunoglobulin G (Jackson ImmunoResearch Laboratories, West Grove, PA) at room temperature for 20 minutes. Flow cytometry was performed using a BD LSRII flow cytometer (BD Biosciences). Flow cytometry data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
Results
Generation of a blood type O HLA class I–negative iPSC founder line
In addition to platelet-specific antigens, platelets also express other non-HPA antigens, including blood group ABH antigens and HLA class I antigens. The ABO antibodies are formed naturally, and they are pre-existing in the serum of incompatible naive individuals. In contrast, anti-HLA class I antibodies are developed during incompatible transfusion or pregnancy, and about one-third of multiparous women are sensitized to HLA class I antigens.33 To prevent potential interference of anti-A, anti-B, and anti-HLA class I antibodies for anti-HPA alloantibody detection in patient sera, we aimed to generate a blood type O HLA class I–negative iPSC founder line. We recently reported the use of integration-free episomal vectors to generate a blood type O iPSC line (OT1-1) derived from human peripheral blood mononuclear cells obtained from a healthy donor.30 β2M, encoded by the B2M gene, is the light chain of HLA class I molecules and is required for trafficking of all class I heavy chains to the cell surface. Disrupting β2M expression using short hairpin RNA or transcription activator-like effector nucleases (TALEN) technology has been carried out for generating HLA class I knockdown/knockout iPSCs.34,35 Similarly, we designed a gRNA sequence to target exon 1 of the B2M gene that would introduce an insertion or deletion (indel) into the genome to eliminate expression of β2M, and consequently, the class I HLA heavy chain (Figure 1A). After transfection of these CRISPR/Cas9 constructs into the OT1-1 iPSCs, a puromycin-resistant β2M knockout clone (B2MKO) was selected. Flow cytometry analysis confirmed that surface expression of both β2M and the HLA class I heavy chain were abolished in the B2MKO iPSC line (Figure 1B). These blood group O HLA-negative cells were then used as the founder line into which all additional amino acid substitutions were introduced.
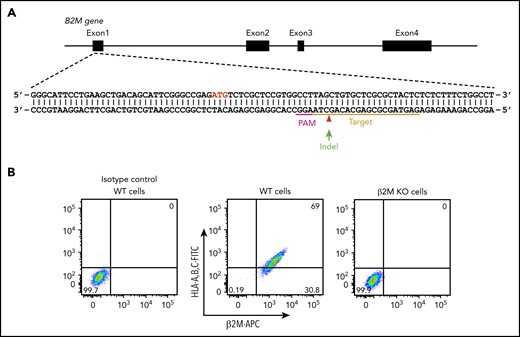
Generation of an HLA class I–negative iPSC founder line. (A) Schematic illustration of the B2M locus, showing the location of the gRNA binding site (orange bar) and the protospacer adjacent motifs sequence (magenta) necessary to guide Cas9 to its cleavage site (red arrow head). ATG start codon for β2M translation is highlighted in red. Green arrow indicates an insertion or deletion (indel) is expected to be introduced into the genome through non-homologous end joining DNA repair pathway to cause a frameshift mutation in the B2M gene. (B) Flow cytometry analysis demonstrating the loss of surface expression of both β2M and HLA in B2M knockout (KO) cells. APC, allophycocyanin; FITC, fluorescein isothiocyanate; WT, wild-type.
Generation of an HLA class I–negative iPSC founder line. (A) Schematic illustration of the B2M locus, showing the location of the gRNA binding site (orange bar) and the protospacer adjacent motifs sequence (magenta) necessary to guide Cas9 to its cleavage site (red arrow head). ATG start codon for β2M translation is highlighted in red. Green arrow indicates an insertion or deletion (indel) is expected to be introduced into the genome through non-homologous end joining DNA repair pathway to cause a frameshift mutation in the B2M gene. (B) Flow cytometry analysis demonstrating the loss of surface expression of both β2M and HLA in B2M knockout (KO) cells. APC, allophycocyanin; FITC, fluorescein isothiocyanate; WT, wild-type.
Detection of anti-HPA-3 alloantibodies using bioengineered iPSC-derived MKs
The HPA-3a and HPA-3b polymorphisms are caused by a single T13809G nucleotide substitution in the ITGA2B gene.15 DNA sequencing of the ITGA2B gene of OT1-1 cells (not shown) showed them to be homozygous for the HPA-3a and HPA-9a alleles. To convert HPA-3a to HPA-3b, a pair of gRNAs flanking exon 26 of ITGA2B were designed to remove the entire exon encoding the HPA-3a epitope (Figure 2A). The homology-directed repair (HDR) template contained the targeted T13809G point mutation for generating the HPA-3b epitope as well as silent mutations to introduce an MfeI site for genotyping. In addition, guide 2 sequences were added to both ends of the homology arms to linearize the HDR donor inside the cells upon Cas9 cleavage (Figure 2A), a feature that has previously been found to enhance HDR efficiency.36 After cotransfecting B2MKO (HPA-3a) cells with the CRISPR/Cas9 guide constructs that coexpress a puromycin resistance gene, together with HDR donor plasmids, clones from puromycin-resistant colonies were manually picked, expanded, and subjected to diagnostic MfeI restriction enzyme digestion to identify clones in which biallelic conversion of HPA-3a to HPA-3b had taken place. Figure 2B shows the MfeI digestion pattern of one such homozygous HPA-3b clone, the T>G 13809 genotype of which was verified by DNA sequencing (Figure 2C).
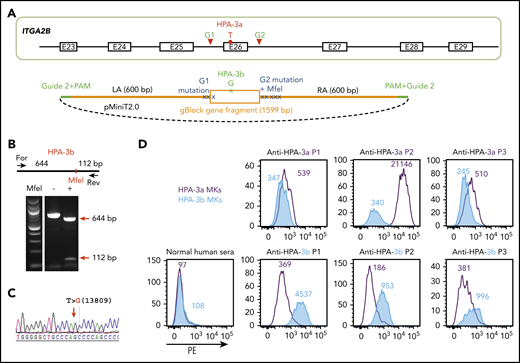
Detection of anti-HPA-3a and HPA-3b alloantibodies using genetically edited iPSC-derived MKs. (A) Schematic illustration of donor plasmid and targeting strategy for converting HPA-3a to HPA-3b in B2MKO iPSCs. Red triangles flanking exon 26 of the ITGA2B gene indicate the 2 gRNA binding sites that will guide Cas9 to remove the entire exon encoding HPA-3a. HDR donor plasmid contains the removed sequence by Cas9 cleavage (orange box) with targeted T>G mutation responsible for HPA-3b conversion, flanked by 600-bp homology arms on each side (orange line). The recognition sequence and the PAM sequence of guide 2 (green line) are added to both ends of the homology arms for linearizing the donor templates in the transfected cells. Donor plasmid also contains silent mutations (blue X) to prevent re-cleavage by Cas9 and to generate an MfeI site for genotyping. (B) Genomic DNA, isolated from puromycin-resistant iPSC clones was amplified by PCR and digested with MfeI, which differentiates the HPA-3b allelic isoform from WT HPA-3a. Red arrows indicate the expected fragment sizes of a typical clone that had been converted to HPA-3b. (C) Sequencing data confirmed the T>G 13809 point mutation in CRISPR-edited HPA-3b iPSCs. The red arrow indicates the target T>G mutation. (D) Reactions of anti-HPA-3a and anti-HPA-3b patient sera with allele-specific iPSC-derived MKs in flow cytometric analysis. Both HPA-3a (gray) and HPA-3b (blue) iPSC lines were differentiated into CD41+/CD42b+ MKs. The MKs were incubated with patient sera followed by phycoerythrin (PE)-conjugated donkey anti-human immunoglobulin G (IgG). Anti-HPA-3a P3 patient serum did not contain anti-HLA class I antibody and was detectable only by using a whole-platelet assay in a clinical diagnostic laboratory. Other anti-HPA-3a and anti-HPA-3b patient sera were all clinically confirmed with MACE or MAIPA assays. For, forward; Rev, reverse.
Detection of anti-HPA-3a and HPA-3b alloantibodies using genetically edited iPSC-derived MKs. (A) Schematic illustration of donor plasmid and targeting strategy for converting HPA-3a to HPA-3b in B2MKO iPSCs. Red triangles flanking exon 26 of the ITGA2B gene indicate the 2 gRNA binding sites that will guide Cas9 to remove the entire exon encoding HPA-3a. HDR donor plasmid contains the removed sequence by Cas9 cleavage (orange box) with targeted T>G mutation responsible for HPA-3b conversion, flanked by 600-bp homology arms on each side (orange line). The recognition sequence and the PAM sequence of guide 2 (green line) are added to both ends of the homology arms for linearizing the donor templates in the transfected cells. Donor plasmid also contains silent mutations (blue X) to prevent re-cleavage by Cas9 and to generate an MfeI site for genotyping. (B) Genomic DNA, isolated from puromycin-resistant iPSC clones was amplified by PCR and digested with MfeI, which differentiates the HPA-3b allelic isoform from WT HPA-3a. Red arrows indicate the expected fragment sizes of a typical clone that had been converted to HPA-3b. (C) Sequencing data confirmed the T>G 13809 point mutation in CRISPR-edited HPA-3b iPSCs. The red arrow indicates the target T>G mutation. (D) Reactions of anti-HPA-3a and anti-HPA-3b patient sera with allele-specific iPSC-derived MKs in flow cytometric analysis. Both HPA-3a (gray) and HPA-3b (blue) iPSC lines were differentiated into CD41+/CD42b+ MKs. The MKs were incubated with patient sera followed by phycoerythrin (PE)-conjugated donkey anti-human immunoglobulin G (IgG). Anti-HPA-3a P3 patient serum did not contain anti-HLA class I antibody and was detectable only by using a whole-platelet assay in a clinical diagnostic laboratory. Other anti-HPA-3a and anti-HPA-3b patient sera were all clinically confirmed with MACE or MAIPA assays. For, forward; Rev, reverse.
Both HPA-3a and HPA-3b iPSC lines were then differentiated into CD41+/CD42b+ MKs using a previously described serum-free, feeder-free, adherent differentiation system.31,32 The MKs derived from the 2 cell lines expressed similar levels of GPIIb (supplemental Figure 1) and showed similar levels of background binding to normal human sera in flow cytometry (Figure 2D). HPA-3a and HPA-3b patient samples, with the exception of anti-HPA-3a (P3 in Figure 2D), had tested positive in either the MACE or the MAIPA assay. All 3 HPA-3a patient serum samples reacted with MKs derived from an HPA-3a iPSC line, as expected, and reactivity was lost in MKs in which the HPA-3a allele had been converted to HPA-3b (Figure 2D). In contrast, HPA-3b–expressing MKs reacted strongly with HPA-3b– but not HPA-3a–specific patient sera. Notably, the anti-HPA-3a sample from P1 was tested because it had a known weak antibody in the MACE assay. As shown in Figure 2D, it also had the weakest binding of samples tested in our whole-cell flow cytometric detection system, demonstrating comparable sensitivity of this assay to MACE. We also examined the ability of the HPA-3a–expressing MKs to react positively with serum from a patient (P3) containing anti-HPA-3a, but that had been negative in MACE. Similar to previous reports describing certain HPA-3a–specific antibodies that are able to recognize their epitopes only in the context of an intact glycoprotein presented on the surface of intact cells,11,12,17,20 sample P3 was positive, although weakly so, against iPSC-derived HPA-3a– but not HPA-3b–expressing MKs. The absence of anti-HLA class I antibodies is a prerequisite for identifying HPA-specific alloantibodies that recognize labile epitopes and that have, to date, been detectable only by using whole-platelet assays. Accordingly, our HLA class I–negative whole-cell assay system seems to provide an important advantage for detecting these types of anti-HPA-3 alloantibodies that are missed or masked when HLA class I antibodies are also present in the sera.
Detection of anti-HPA-9b alloantibodies with bioengineered iPSC-derived MKs
The HPA-9a and HPA-9b polymorphisms are caused by a single G13790A nucleotide substitution in the ITGA2B gene. HPA-9b is genetically linked to HPA-3b because the mutation from HPA-9a to HPA-9b took place on an HPA-3b allele.16 Thus, we edited HPA-9a to HPA-9b in our newly generated homozygous HPA-3b iPSC line using a guide sequence that targets exon 26 of the ITGA2B gene (Figure 3A). The ssODN HDR donor contains the targeted G13790A mutation as well as silent mutations to introduce a PstI site for later genotyping. After cotransfection of the CRISPR/Cas9 guide construct and the HDR donor into HPA-3b iPSCs, puromycin-resistant clones were subjected to diagnostic PstI restriction enzyme digestion to identify homozygous HPA-9b clones (Figure 3B). DNA sequencing confirmed the G>A 13790 mutation (Figure 3C).

Detection of anti-HPA-9b alloantibodies using genetically edited iPSC-derived MKs. (A) Schematic illustration of HDR template and targeting strategy for converting HPA-9a to HPA-9b in HPA-3b iPSC clone. The gRNA binding site (orange bar) and the PAM sequence (magenta) will guide Cas9 to its cleavage site (red arrow head) next to the HPA-9 allele. A 199-bp HPA-9b HDR template was designed to introduce the Val→Met amino acid polymorphism. The G>A mutation responsible for the HPA-9a/HPA-9b polymorphism (highlighted in red) is flanked by 99 nucleotide homology arms. Silent mutations (highlighted in blue) were introduced to prevent re-cleavage by Cas9 and create a PstI site at the target locus that can be used for genotyping. (B) Genomic DNA, isolated from puromycin-resistant iPSC clones was amplified by PCR and digested with PstI, which differentiates the HPA-9b allelic isoform from HPA-9a. Red arrow indicates the expected fragment sizes of a typical clone that had been converted to HPA-9b. (C) Sequencing data confirmed the G>A 13790 point mutation in CRISPR-edited HPA-9b iPSCs. The red arrow indicates the target G>A mutation. (D) Reactions of anti-HPA-9b patient sera with allele-specific iPSC-derived MKs in flow cytometric analysis. All of the HPA-3a (gray), HPA-3b (blue) and HPA-9b (red) iPSC lines were differentiated into CD41+/CD42b+ MKs. The MKs were incubated with patient sera followed by PE-conjugated donkey anti-human IgG. Anti-HPA-9b P1-P3 sera were clinically confirmed with either an MACE or an MAIPA assay. Anti-HPA-9b P4-P6 were HPA-9b suspected patient samples from clinically unresolved FNAIT cases.
Detection of anti-HPA-9b alloantibodies using genetically edited iPSC-derived MKs. (A) Schematic illustration of HDR template and targeting strategy for converting HPA-9a to HPA-9b in HPA-3b iPSC clone. The gRNA binding site (orange bar) and the PAM sequence (magenta) will guide Cas9 to its cleavage site (red arrow head) next to the HPA-9 allele. A 199-bp HPA-9b HDR template was designed to introduce the Val→Met amino acid polymorphism. The G>A mutation responsible for the HPA-9a/HPA-9b polymorphism (highlighted in red) is flanked by 99 nucleotide homology arms. Silent mutations (highlighted in blue) were introduced to prevent re-cleavage by Cas9 and create a PstI site at the target locus that can be used for genotyping. (B) Genomic DNA, isolated from puromycin-resistant iPSC clones was amplified by PCR and digested with PstI, which differentiates the HPA-9b allelic isoform from HPA-9a. Red arrow indicates the expected fragment sizes of a typical clone that had been converted to HPA-9b. (C) Sequencing data confirmed the G>A 13790 point mutation in CRISPR-edited HPA-9b iPSCs. The red arrow indicates the target G>A mutation. (D) Reactions of anti-HPA-9b patient sera with allele-specific iPSC-derived MKs in flow cytometric analysis. All of the HPA-3a (gray), HPA-3b (blue) and HPA-9b (red) iPSC lines were differentiated into CD41+/CD42b+ MKs. The MKs were incubated with patient sera followed by PE-conjugated donkey anti-human IgG. Anti-HPA-9b P1-P3 sera were clinically confirmed with either an MACE or an MAIPA assay. Anti-HPA-9b P4-P6 were HPA-9b suspected patient samples from clinically unresolved FNAIT cases.
The newly generated HPA-3b/HPA-9b iPSC-derived cell line, when differentiated into CD41+/CD42b+ MKs, expressed levels of GPIIb similar to those of MKs derived from the HPA-3a and HPA-3b iPSC lines described above (supplemental Figure 1) and exhibited very low background binding to normal human sera in flow cytometry (Figure 3D). Three well-defined HPA-9b–specific alloantibodies (P1-P3) all reacted with HPA-3b/HPA-9b MKs in the flow cytometric test (Figure 3D). Two of them (P1 and P2) also bound weakly to HPA-3b/HPA-9a MKs (Figure 3D), but their reactivity was three- to four-fold stronger when the 9b polymorphism was present. Patient (P4-P6) sera were from unresolved cases of FNAIT suspected of containing anti-HPA-9b antibodies as a result of genetic incompatibility for HPA-9b in the parents, but in all 3 cases, the presence of anti-HPA-9b antibody or any other platelet-specific alloantibody could not be detected using standard techniques. As shown in Figure 3D, anti-HPA-9b alloantibodies in these maternal sera were easily detected with a simple 1-step flow cytometric test using intact class I HLA-negative blood group O iPSC-derived MKs as target cells.
Discussion
Although it became possible to genotype platelet-specific alloantigens beginning in the early 1990s, serological detection of HPA-specific alloantibodies remains critical for diagnosis, treatment, and prevention of platelet alloimmune disorders, including FNAIT, posttransfusion purpura, and platelet transfusion refractoriness. A wide range of techniques have been developed for HPA alloantibody detection over the last 40 years, but detection remains challenging in many cases. Maternal HPA alloantibodies can be identified in only 20% to 35% of apparent FNAIT cases referred for laboratory investigation.37 In particular, antibodies specific for HPA-3a, HPA-3b, and HPA-9b can be extremely difficult to detect using standard serologic tests.38,39
In this study, we generated blood group O HLA class I–negative, HPA-3a, HPA-3b, and HPA-9b allele-specific human iPSC lines using CRISPR gene editing technology. Upon differentiation, the iPSC-derived MKs expressed allele-specific HPAs on their surface that were easily adapted for flow cytometric detection of anti-HPA-3a, HPA-3b, and HPA-9b alloantibodies in patient sera. Importantly, patient sera that had previously been identified using standard clinical enzyme-linked immunosorbent assay-based MAIPA and MACE assays, as well as samples that had been negative using these methods could be typed using these intact, allele-specific bioengineered MKs. In contrast to MACE or MAIPA assays, which are time-consuming, labor-intensive, and difficult to standardize, we suggest that the flow cytometric assay described herein will be fast, simple to perform with either fresh or previously frozen cells (supplemental Figure 2), and require only 50 to 100 μL of patient serum.
Characterizing the precise specificity of anti-HPA alloantibodies present in maternal sera has historically required access to a well-characterized panel of platelets that have a wide variety of phenotypic specificities. This can be challenging, especially when platelets expressing rare low-frequency HPAs are needed. Previous attempts to circumvent this problem using transfected cell lines expressing allele-specific human platelet membrane glycoproteins have encountered technical issues that have precluded their wide-spread adoption. For example, COS cells (derived from fibroblasts of the African green monkey) expressing allelic isoforms of human GPIIb/IIIa are unable to detect a significant number of HPA-3a and HPA-3b alloantisera,40 likely because of their requirement for species-specific glycans that form part of their recognition epitope.21 Chinese hamster ovary (CHO) cells seem to be particularly poor in their ability to display human platelet alloantigenic epitopes, because a substantial number of human anti-HPA-3a,21 anti-HPA-9b,25 and anti-Lapa4 human alloantisera fail to react with human GPIIb expressed in this cell line. Transformed human cancer cell lines are not immune to this problem either. Hayashi et al41 discovered that only 4 of 6 anti-HPA-3a alloantisera are reactive with GPIIb derived from human erythroleukemia K562 cells, suggesting a limitation of using these cells in diagnostic HPA testing.
The inability to consistently detect anti-HPA-3a alloantibodies using cells of non-platelet origin can be at least partially attributed to the heterogeneous requirement21-23,40 for O-linked oligosaccharide chains that emanate from serine residues 845 and 847,42 both of which are proximal to the Ile843Ser polymorphic residue that defines the HPA-3 alloantigen system.15,40 In particular, although O-glycans attached to GPIIb Ser847 have been shown to participate in the formation of the HPA-3a epitope,24 the involvement of glycans in the formation of the HPA-3b epitope remains unclear, especially because it is formed as a result of replacing Ile843 (HPA-3a) with still another serine residue that has the potential to host a third glycan in the immediate vicinity of the polymorphic amino acid. The effect, if any, of these glycans on the HPA-9b alloantigenic epitope is completely unknown.
Mucin-type O-glycosylation is controlled by a large family of >20 genes that encode UDP-GalNAc:polypeptide GalNAc transferases (GalNAc-Ts) that are differentially expressed in cells and tissues, and marked changes in expression are also found in cancer cells.43 Because of the differential regulation of mucin-type O-glycosylation in cells and tissues, human GPIIb synthesized by non-human cells (COS, CHO) or human cancer cell lines (K562) are likely to express completely different glycan chains than would GPIIb produced in human platelets or MKs. Preliminary studies performed in our laboratory suggest that GPIIb expressed in iPSC-derived MKs contains the same sialylated T-antigens present in native O-glycosylated GPIIb from human platelets42 (supplemental Figure 3), making these cells uniquely suited for detecting HPA alloantibodies that recognize O-glycan-dependent epitopes.
Maternal sera containing platelet-specific alloantibodies often also harbor alloantibodies specific for 1 or more HLA class I polymorphisms. Thus, a major advantage of using HPA allele–specific MKs for detection of platelet-specific alloantibodies is that they can be engineered to not express the heavy and light chains of the HLA class I protein heterodimer complex, thereby lending themselves to widely available detection methods, like flow cytometry, that use intact cells. By knocking out β2M in our iPSC founder line (Figure 1), the resulting iPSCs could be easily modified using CRISPR/Cas9 gene editing technology to generate a series of cell lines that, upon differentiation, express homozygous allelic isoforms of GPIIb carrying the HPA-3a, HPA-3b, or HPA-9b alloantigenic determinants on their cell surface (Figures 2 and 3). Because they are all derived from a single clonal iPSC line, these cells should provide extremely high specificity for HPA detection because they all share an identical genetic background, and therefore phenotype, with the sole exception of the targeted polymorphic HPA allele. Because these HPA allele–specific MKs express GPIIb allele in homozygous form, the increase in alloantigen density on the cell surface should also result in increased sensitivity. This was demonstrated by the ability to identify and type anti-HPA alloantibody reactivity in several maternal sera that had previously shown weak or negative reactivity using currently existing methodology (Figure 3).
Recent advances in techniques, including the establishment of human iPSCs and CRISPR/Cas9-mediated gene editing have opened up new possibilities for stem cell–based therapies in a wide variety of disorders, especially for transfusion medicine. Solid groundwork has been laid for the ex vivo production of MKs and platelets, and the field is advancing rapidly. Platelet products are qualitatively and quantitatively approaching a clinically applicable level owing to advances in expandable MK lines, platelet-producing bioreactors, and novel reagents.28,29 Undoubtedly, the capacity to generate large-scale donor-independent MKs and platelets will promote their clinical translation and application sooner than had been anticipated. Incorporating our system into expandable MK cell lines holds great potential for production of designer platelets for diagnostic, investigative, and ultimately therapeutic use. We envision a future in which specialty units of in vitro–generated HPA-matched iPSC-derived platelets are used clinically to complement donor-derived platelets.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
For original data, please contact the corresponding author via e-mail at pjnewman@versiti.org.
The online version of this article contains a data supplement.
Acknowledgment
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01 HL130054 and R35 HL139937).
Authorship
Contribution: N.Z. conducted experiments and analyzed data; S.S. provided intellectual input, participated in discussions, and contributed vital reagents; R.H.A. and B.R.C. provided rare human anti-HPA-3a and HPA-9b alloantisera; and N.Z. and P.J.N. designed experiments and wrote the manuscript.
Conflict-of-interest disclosure: B.R.C. has served as a consultant for Prophylix Pharma regarding platelet antigen and antibody detection. The remaining authors declare no competing financial interests.
Correspondence: Peter J. Newman, Blood Research Institute, Versiti, 8727 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: pjnewman@versiti.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal