

Key Points
Synthetic C-miR146a mimic inhibits inflammatory and tumorigenic NF-κB activity in macrophages and myeloid leukemia in vitro and in vivo.
C-miR146a alleviates monocyte-mediated cytokine storm without affecting CD19-specific CAR T-cell activity against B-cell lymphoma.

Abstract
NF-κB is a key regulator of inflammation and cancer progression, with an important role in leukemogenesis. Despite its therapeutic potential, targeting NF-κB using pharmacologic inhibitors has proven challenging. Here, we describe a myeloid cell–selective NF-κB inhibitor using an miR-146a mimic oligonucleotide conjugated to a scavenger receptor/Toll-like receptor 9 agonist (C-miR146a). Unlike an unconjugated miR146a, C-miR146a was rapidly internalized and delivered to the cytoplasm of target myeloid cells and leukemic cells. C-miR146a reduced expression of classic miR-146a targets (IRAK1 and TRAF6), thereby blocking activation of NF-κB in target cells. IV injections of C-miR146a mimic to miR-146a–deficient mice prevented excessive NF-κB activation in myeloid cells, and thus alleviated myeloproliferation and mice hypersensitivity to bacterial challenge. Importantly, C-miR146a showed efficacy in dampening severe inflammation in clinically relevant models of chimeric antigen receptor (CAR) T-cell–induced cytokine release syndrome. Systemic administration of C-miR146a oligonucleotide alleviated human monocyte-dependent release of IL-1 and IL-6 in a xenotransplanted B-cell lymphoma model without affecting CD19-specific CAR T-cell antitumor activity. Beyond anti-inflammatory functions, miR-146a is a known tumor suppressor commonly deleted or expressed at reduced levels in human myeloid leukemia. Using The Cancer Genome Atlas acute myeloid leukemia data set, we found an inverse correlation of miR-146a levels with NF-κB–related genes and with patient survival. Correspondingly, C-miR146a induced cytotoxic effects in human MDSL, HL-60, and MV4-11 leukemia cells in vitro. The repeated IV administration of C-miR146a inhibited expression of NF-κB target genes and thereby thwarted progression of disseminated HL-60 leukemia. Our results show the potential of using myeloid cell–targeted miR-146a mimics for the treatment of inflammatory and myeloproliferative disorders.
Introduction
MicroRNAs (miRNAs) are small noncoding RNAs that control expression of a broad set of target genes based on sequence complementarity. By binding to the 3′ untranslated (3′UTR) regions of the target messenger RNA (mRNA), miRNAs regulate gene expression and enable control of multiple gene targets within the same or distinct signaling pathways.1,2 Many miRNAs are dysregulated in cancer and cardiovascular and autoimmune diseases.3 Genomic mutations, deletions, or changes in the key enzymes in miRNA biogenesis may all lead to alterations in miRNA levels.4,5 Genome-wide miRNA screening of leukemia-associated loci identified miR-146a as a major mediator of the chromosome-5q deletion myelodysplastic syndrome [del(5q) MDS] and acute myeloid leukemia (AML).6-8 The reduced miR-146a expression contributes to the development of del(5q) MDS and progression to AML through IRAK1- and TRAF6-dependent activation of NF-κB.9,10 In nonmalignant myeloid cells (eg, monocytes), decreased miR-146a levels result in expression of IL-6 and other proinflammatory mediators implicated in the pathogenesis of autoimmune diseases and cancers.11-13 The miR-146a dysregulation and IL-6 elevation in hematopoietic stem/progenitor and myeloid cells is also associated with many autoinflammatory diseases such as rheumatoid arthritis, systemic lupus erythematosus, type 2 diabetes, Sjögren’s syndrome, and endotoxemia-related cytokine storm.14
Although the role of miR-146a remains unclear, the NF-κB–mediated release of IL-6 from monocytes was also shown to be responsible for cytokine release syndrome (CRS), a serious adverse effect of CAR T-cell therapies.12,15 Due to the lack of pharmacologic NF-κB inhibitors, synthetic miR-146a mimics are an attractive opportunity for immunomodulation or elimination of tumorigenic signaling. However, the effective delivery of miRNA therapeutic agents is challenging, complicated by safety concerns and potential off-target effects.2,16 Several types of miRNA delivery vehicles, including liposomes, lipid nanoparticles, dendrimers, or hydrogels, had been tested previously.17-19 Only a few of the synthetic miRNA mimics, including antifibrotic and transforming growth factor β–targeting miR-29/remlarsen, progressed to initial clinical testing.19 The current article describes an original approach for the targeted delivery of a chemically modified miR-146a mimic to myeloid cells and verifies miR-146a mimic activity in models of inflammatory and myeloproliferative diseases.
Methods
Mice and in vivo studies
All animal experiments were conducted following institutional guidance and approved protocols from the Institutional Animal Care and Use Committee. C57BL/6, BALB/c mice 6 to 8 weeks of age were purchased from the National Cancer Institute; female C.B-Igh-1b/GbmsTac-Prkdcscid-LystbgN7 (SCID-Beige) mice were from Taconic, and NOD/SCID/IL-2RgKO (NSG) and NSG Tg(IL3,CSF2,KITLG)1Eav/MloySzJ (SGM3) mice were from The Jackson Laboratory. miR-146a−/− and LyzM-Cre/miR-146afl/fl mice9,20 were bred and housed in the Laboratory Animal Resources facility at the California Institute of Technology.
C-miR146a synthesis
The C-miR146a conjugates were synthesized in the DNA/RNA Synthesis Core (City of Hope) by linking CpG-D19 to miR-146a passenger strands as previously described.21 These were hybridized with complementary guide strands of mature miR-146a, creating chimeric C-miR146a mimic. The single-stranded sequences are listed in the following text (x = C3-unit; asterisks = phosphorothioation; underline = 2′-O-methylation):
C-miR146a passenger:
5′ G*G*TGCATCGATGCAGG*G*G*G*G-xxxxx-CCCAUGGAAUUCAGUUCUCAAA 3′.
miR-146a guide:
5′ UGAGAACUGAAUUCCAUGGGUU 3′.
control scrambled RNA (C-scrRNA) passenger:
5′ G*G*TGCATCGATGCAGG*G*G*G*G -xxxxx-AUUUAGCCUUAAUACACGCCAA 3′
scrRNA guide:
5′ GGCGUGUAUUAAGGCUAAAUCU 3′
For internalization studies, the miR-146a guide strand was labeled on the 3′ end by using Cy3 fluorochrome.
RNA-binding protein immunoprecipitation
RNA-binding protein immunoprecipitation was performed by using the Magna RNA Immunoprecipitation Kit (MilliporeSigma) according to the manufacturer’s instructions. Details are described in the supplemental Methods (available on the Blood Web site).
In vivo biodistribution
miR-146a−/− mice or syngeneic Cbfb/Myh11/Mpl mouse leukemia–bearing C57BL/6 mice were injected retro-orbitally with 2.5 to 20 mg/kg of C-miR146a, C-miR146aCy3, or miR146aCy3 as previously described.22 At indicated times, mice were euthanized to collect organs for flow cytometry, quantitative polymerase chain reaction (qPCR), or western blot analysis. EasySep PE-positive selection kits (Stemcell Technologies) were used for cell subset enrichment.
Studies of Listeria monocytogenes
A total of 105 colony-forming units of Listeria monocytogenes (strain-10 403/serotype-1) were injected to each wild-type (WT) C57BL/6 or LyzM-Cre/miR-146afl/fl mouse using retro-orbital injections. Mice were treated every day using 5 mg/kg of C-miR146a or C-scrRNA (negative control) for 3 days before and 3 days after infection. The mice were euthanized, and various organs were collected for further analysis.
CRS models
For in vitro studies, mock or CD19 CAR T cells from 4 donors were cultured with target Nalm6 leukemia with or without CD14+ monocytes from the same donor. Healthy donors’ peripheral blood mononuclear cells were collected in accordance with the Declaration of Helsinki. The cocultured cells were treated with C-miR146a or C-scrRNA (500 nM) for 2 days. After 48 hours, supernatants were collected, and IL-1 and IL-6 levels were analyzed by using an enzyme-linked immunosorbent assay (ELISA) (Thermo Fisher Scientific). In vivo CRS studies used female SCID-Beige mice injected using 3 × 106 RajiGFP/Luc cells, followed by intraperitoneal injections of C-miR146a (5 mg/kg) or phosphate-buffered saline (PBS) daily for 3 days before transfer of 12.5 × 106 mock-transfected or CD19-specific CAR T cells, as previously described.15,23,24
Human AML xenotransplants
NSG-SGM3 mice were injected intravenously with 2 × 106 of HL-60LUC cells, and tumor progression was monitored by using BLI/AmiX (Spectral Instruments). Mice were injected retro-orbitally with 10 mg/kg of C-miR146a or C-scrRNA every other day. Total RNA was isolated from bone marrow cells and analyzed by using RT2 Profiler PCR Arrays (Qiagen).
Gene expression analysis
The overall survival analysis was conducted by using R package survival (version 2.44.1.1). Log-rank statistics were applied to identify the optimal cut-point for transforming the continuous variable of gene expression into categorical high and low expression groups in a survfit model, using the cutp function of the R package survMisc (version 0.5.5).
Statistics
An unpaired Student t test was used to calculate 2-tailed P values to estimate statistical significance between 2 treatment groups. One- or 2-way analysis of variance plus the Bonferroni posttest assessed differences between multiple groups or in tumor growth kinetics, respectively. Statistically significant P values were indicated in figures compared with untreated or PBS groups. Data were analyzed by using Prism software version 7 (GraphPad Software).
Results
Targeted delivery of functional miR-146a mimic into myeloid cells and B cells
To overcome challenges in the efficient and cell-selective delivery of miR-146a, we modified the scavenger receptor (SR)/Toll-like receptor 9 (TLR9)–targeting platform originally developed for transfer of 25/27-mer Dicer-substrate small interfering RNA.21 The double-stranded miR-146a was conjugated through the 5′ end of the passenger strand to the 3′ end of a single-stranded, partly phosphorothioated oligodeoxynucleotide (CpG ODN) using a synthetic carbon linker. The specific type A CpG ODN sequence of the conjugate was selected for monocytes/myeloid cell specificity25 and poor ability to activate NF-κB and its downstream IL-6 and IL-10 targets.26 To ensure the maximum activity and target specificity, the miR-146a was chemically modified for nuclease resistance by a 2′-O-methyl-modification in the 3′ end of the passenger strand (Figure 1A). This design significantly improved serum stability of the C-miR146a conjugate without affecting activity. Compared with miR-146a (half-life, ∼1 hour), the conjugated C-miR146a had a 34-hour half-life in human serum (Figure 1B; supplemental Figure 1).
![Targeted delivery of miR-146a mimic to RISC/Ago2 complexes in myeloid cells. (A) The design of C-miRNA146a mimic conjugate; underlined, phosphorothioated nucleotides; “o”, C3 units of the carbon linker; green, 2′-O-methyl-modified nucleotide. (B) Serum stability of miR146a (left) and C-miR146a (right). Oligonucleotides were incubated in 50% human serum at 37°C for the indicated times and then resolved on 7.5 M urea/15% polyacrylamide gel electrophoresis. Shown is a representative result from 1 of 3 independent experiments; the band intensities were quantified, and the estimated oligonucleotide half-lives (T1/2) are indicated. (C) The intracellular uptake of C-miR146aCy3 compared with miR146aCy3 alone by primary human immune cells (monocytes, CD14+; myeloid dendritic cells [mDCs], CD1c+; plasmacytoid dendritic cells [pDCs], CD303+; B cells, CD19+; and T cells, CD3+), mouse RAW264.7 macrophages, human MDSL and HL-60 leukemia, human Raji lymphoma, and TAIL7 T-cell leukemia cells. Cells were incubated for 1 hour with 100 nM of C-miR146aCy3 or miR146aCy3 without any transfection reagents, and the uptake was measured by using flow cytometry. (D) The intracellular localization of C-miR146aCy3 or miR146aCy3 oligonucleotides (100 nM/red) as visualized by using confocal microscopy in RAW264.7 cells after 1 hour of incubation. Hoechst33342 (blue) was used for nuclear counterstain. (E) The successful loading of miR146a into RISC/Ago2 by C-miRNA conjugate. RAW264.7 or splenocytes isolated from miR-146a−/− mice were incubated for 1 hour with 1 μM of C-miR146a or miR146a alone. The RNA-protein complexes were immunoprecipitated by using anti-Ago2 or control IgG antibodies, and the miR-146a levels were quantified by using qPCR. The equal protein loading was confirmed with western blotting. The data shown represent results from 3 independent experiments; shown are means ± SEM. ***P < .001 compared to untreated.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/3/10.1182_blood.2019002045/2/m_bloodbld2019002045f1.png?Expires=1769087287&Signature=D0jHobeUi3VAXpJmDKvafQhrv2j4lxHdqTno28jujIRaj4ZvFqPl4Xt5akbrOgPve08MefeXeXUbKY7PPpaANSs8IddCnfUb4hqyin-0X05Xgko~33shLVaYAjc2Ou~qw0TLr~Z~Af~eOza0hTLjdyJunqtNb6P7PxNIjAlRM7-~uO4q2lQE4y-0vOHbQGGs9AfNzSRm~fe663rLH4nvSOgdUqeNpMacepYoT4ZpciPLYAGHJIrg~vFlVABtuG-Y6OPzt9LsQrdTxV350pDSdMr9-xefvOa8LrjxEMKVF2-kdIeCtdsbXB8MypABRzzJscDWoG1Df1-xw3qBD~l9gw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Targeted delivery of miR-146a mimic to RISC/Ago2 complexes in myeloid cells. (A) The design of C-miRNA146a mimic conjugate; underlined, phosphorothioated nucleotides; “o”, C3 units of the carbon linker; green, 2′-O-methyl-modified nucleotide. (B) Serum stability of miR146a (left) and C-miR146a (right). Oligonucleotides were incubated in 50% human serum at 37°C for the indicated times and then resolved on 7.5 M urea/15% polyacrylamide gel electrophoresis. Shown is a representative result from 1 of 3 independent experiments; the band intensities were quantified, and the estimated oligonucleotide half-lives (T1/2) are indicated. (C) The intracellular uptake of C-miR146aCy3 compared with miR146aCy3 alone by primary human immune cells (monocytes, CD14+; myeloid dendritic cells [mDCs], CD1c+; plasmacytoid dendritic cells [pDCs], CD303+; B cells, CD19+; and T cells, CD3+), mouse RAW264.7 macrophages, human MDSL and HL-60 leukemia, human Raji lymphoma, and TAIL7 T-cell leukemia cells. Cells were incubated for 1 hour with 100 nM of C-miR146aCy3 or miR146aCy3 without any transfection reagents, and the uptake was measured by using flow cytometry. (D) The intracellular localization of C-miR146aCy3 or miR146aCy3 oligonucleotides (100 nM/red) as visualized by using confocal microscopy in RAW264.7 cells after 1 hour of incubation. Hoechst33342 (blue) was used for nuclear counterstain. (E) The successful loading of miR146a into RISC/Ago2 by C-miRNA conjugate. RAW264.7 or splenocytes isolated from miR-146a−/− mice were incubated for 1 hour with 1 μM of C-miR146a or miR146a alone. The RNA-protein complexes were immunoprecipitated by using anti-Ago2 or control IgG antibodies, and the miR-146a levels were quantified by using qPCR. The equal protein loading was confirmed with western blotting. The data shown represent results from 3 independent experiments; shown are means ± SEM. ***P < .001 compared to untreated.
Targeted delivery of miR-146a mimic to RISC/Ago2 complexes in myeloid cells. (A) The design of C-miRNA146a mimic conjugate; underlined, phosphorothioated nucleotides; “o”, C3 units of the carbon linker; green, 2′-O-methyl-modified nucleotide. (B) Serum stability of miR146a (left) and C-miR146a (right). Oligonucleotides were incubated in 50% human serum at 37°C for the indicated times and then resolved on 7.5 M urea/15% polyacrylamide gel electrophoresis. Shown is a representative result from 1 of 3 independent experiments; the band intensities were quantified, and the estimated oligonucleotide half-lives (T1/2) are indicated. (C) The intracellular uptake of C-miR146aCy3 compared with miR146aCy3 alone by primary human immune cells (monocytes, CD14+; myeloid dendritic cells [mDCs], CD1c+; plasmacytoid dendritic cells [pDCs], CD303+; B cells, CD19+; and T cells, CD3+), mouse RAW264.7 macrophages, human MDSL and HL-60 leukemia, human Raji lymphoma, and TAIL7 T-cell leukemia cells. Cells were incubated for 1 hour with 100 nM of C-miR146aCy3 or miR146aCy3 without any transfection reagents, and the uptake was measured by using flow cytometry. (D) The intracellular localization of C-miR146aCy3 or miR146aCy3 oligonucleotides (100 nM/red) as visualized by using confocal microscopy in RAW264.7 cells after 1 hour of incubation. Hoechst33342 (blue) was used for nuclear counterstain. (E) The successful loading of miR146a into RISC/Ago2 by C-miRNA conjugate. RAW264.7 or splenocytes isolated from miR-146a−/− mice were incubated for 1 hour with 1 μM of C-miR146a or miR146a alone. The RNA-protein complexes were immunoprecipitated by using anti-Ago2 or control IgG antibodies, and the miR-146a levels were quantified by using qPCR. The equal protein loading was confirmed with western blotting. The data shown represent results from 3 independent experiments; shown are means ± SEM. ***P < .001 compared to untreated.
The C-miR146aCy3 was rapidly internalized by primary human immune cells and mouse RAW264.7 macrophages (Figure 1C) or splenocytes (supplemental Figure 2) within 1 hour of incubation, without any transfection reagents. Compared with the unconjugated miR146aCy3, C-miR146aCy3 was also efficiently taken up by human MDSL or HL60 myeloid leukemia cells and Raji Burkitt’s lymphoma cells but not human T-cell leukemia TAIL7 cells. The uptake of C-miR146a depended on the scavenger receptor A– and clathrin-mediated endocytosis, as verified by using internalization inhibitors (dextran sulfate, fucoidan, and cadaverine) (supplemental Figure 3). TLR9 is commonly expressed by myeloid and B-cell malignancies, including tested myeloid leukemia and B-cell lymphoma cells (supplemental Figure 4).26 Confocal microscopy showed that C-miR146a localized to the cytosol of target cells within 1 hour, whereas miR146a alone was undetectable (Figure 1D). To inhibit target mRNAs, the miRNA guide strand must bind Argonaute-2 (Ago2) protein within the RNA-induced silencing complex (RISC). To assess whether the guide strand of C-miR146a was successfully loaded onto the RISC, we performed RNA-binding protein immunoprecipitation using Ago2-specific antibodies (Figure 1E). In fact, the level of Ago2-bound miR-146a guide strand increased several fold in mouse RAW264.7 macrophages incubated with the C-miR146a but not with miR146a. This effect was even more prominent in primary mouse splenocytes derived from miR-146a−/− mice. These results indicate that the C-miR146a allows for the delivery of functional miR-146a to human and mouse myeloid target cells.
C-miR146a mimic targets upstream regulators of NF-κB signaling
miR-146a targets IRAK1 and TRAF6, the critical upstream regulators of NF-κB signaling.10 We used 3′-UTR luciferase reporter assays to verify whether C-miR146a can alter IRAK1 and TRAF6 expression. As shown in Figure 2A, C-miR146a conjugate reduced IRAK1 and TRAF6 3′-UTR luciferase reporter activities in a sequence-specific manner, verified by mutating 3′-UTR sequences. Neither miR-146a alone nor negative C-scrRNA affected these 3′-UTR luciferase reporters. Moreover, C-miR146a, but not the control oligonucleotides, reduced IRAK1 and TRAF6 protein levels in mouse RAW264.7 macrophages but only IRAK1 in human MDSL cells (Figure 2B). Lack of TRAF6 inhibition by C-miR146a is likely related to a mutation in one of the miR-146a–binding sites in TRAF6 3′UTR in human MDSL cells (supplemental Figure 5). Activated NF-κB translocates to the nucleus; we therefore examined the effect of C-miR146a on the localization of NF-κB in lipopolysaccharide (LPS)-treated RAW264.7 cells expressing NF-κB/p65-eGFP fusion protein.27 Confocal microscopy confirmed that C-miR146a, but not C-scrRNA, efficiently blocked the nuclear translocation of NF-κB (Figure 2C). Correspondingly, C-miR146a reduced the NF-κB DNA-binding activity in nuclear extracts from RAW264.7 and MDSL cells as measured by using gel shift assays (Figure 2D). These C-miR146a effects reduced LPS-induced transcriptional NF-κB activity in RAW-Blue macrophages (Figure 2E) and secretion of IL-6 by parental RAW264.7 cells (Figure 2F).
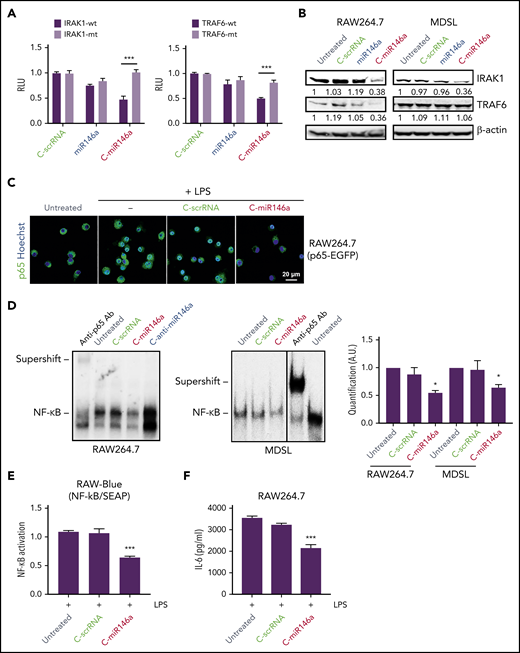
C-miR146a mimic targets upstream regulators of NF-κB signaling. (A) C-miR146a inhibits IRAK1 and TRAF6 expression through a sequence-dependent, on-target effect. HEK293T cells were incubated for 24 hours with 500 nM of C-miR146a, control C-scrRNA, or miR146a alone, and then transfected with WT or mutated IRAK1 or TRAF6 3′UTR luciferase reporter and control Renilla luciferase plasmids. The reporter luciferase activities were evaluated after 48 hours. (B) Reduced protein levels of IRAK1 and TRAF6 in C-miR146a–treated mouse macrophages and human leukemia cells. IRAK1 and TRAF6 protein levels were assessed by using western blot in RAW264.7 and MDSL cells after 48 hours of incubation with 500 nM of the indicated oligonucleotides. Shown are representative results; band intensities were quantified with normalization to β-actin as a loading control. (C) miR-146a mimic delivery prevents nuclear translocation of NF-κB. RAW264.7 cells stably expressing p65-eGFP fusion protein were incubated overnight with 500 nM of C-miR146a or control C-scrRNA and then stimulated with 100 ng/mL of LPS for 4 hours. Translocation of NF-κB/p65 (green) into nuclei (blue) was visualized by using confocal microscopy. (D) C-miR146a inhibits NF-κB DNA binding in target myeloid cells, RAW264.7, or MDSL. Cells were incubated with 500 nM of C-miR146a, C-anti-miR146a, or control C-scrRNA for 48 hours. The NF-κB DNA binding was assessed in nuclear extracts and verified by using p65-specific antibody supershift. Representative blots (left/middle) and the quantification of band intensities combined from 3 experiments (right). (E-F) C-miR146a reduces transcriptional activity of NF-κB in macrophages. RAW-Blue (E) or RAW264.7 (F) cells were incubated overnight with 500 nM of C-miR146a or control C-scrRNA and then stimulated with 100 ng/mL of LPS for 4 hours. The expression of the NF-κB–dependent reporter gene (panel E) or IL-6 secretion (panel F) was assessed by using Quanti-Blue assay or ELISA, respectively. Shown are representative results obtained in 3 independent experiments; means ± SEM. ***P < .001 and *P < .05 compared to untreated groups or as indicated. RLU, relative luciferase unit; SEAP, secreted alkaline phosphatase.
C-miR146a mimic targets upstream regulators of NF-κB signaling. (A) C-miR146a inhibits IRAK1 and TRAF6 expression through a sequence-dependent, on-target effect. HEK293T cells were incubated for 24 hours with 500 nM of C-miR146a, control C-scrRNA, or miR146a alone, and then transfected with WT or mutated IRAK1 or TRAF6 3′UTR luciferase reporter and control Renilla luciferase plasmids. The reporter luciferase activities were evaluated after 48 hours. (B) Reduced protein levels of IRAK1 and TRAF6 in C-miR146a–treated mouse macrophages and human leukemia cells. IRAK1 and TRAF6 protein levels were assessed by using western blot in RAW264.7 and MDSL cells after 48 hours of incubation with 500 nM of the indicated oligonucleotides. Shown are representative results; band intensities were quantified with normalization to β-actin as a loading control. (C) miR-146a mimic delivery prevents nuclear translocation of NF-κB. RAW264.7 cells stably expressing p65-eGFP fusion protein were incubated overnight with 500 nM of C-miR146a or control C-scrRNA and then stimulated with 100 ng/mL of LPS for 4 hours. Translocation of NF-κB/p65 (green) into nuclei (blue) was visualized by using confocal microscopy. (D) C-miR146a inhibits NF-κB DNA binding in target myeloid cells, RAW264.7, or MDSL. Cells were incubated with 500 nM of C-miR146a, C-anti-miR146a, or control C-scrRNA for 48 hours. The NF-κB DNA binding was assessed in nuclear extracts and verified by using p65-specific antibody supershift. Representative blots (left/middle) and the quantification of band intensities combined from 3 experiments (right). (E-F) C-miR146a reduces transcriptional activity of NF-κB in macrophages. RAW-Blue (E) or RAW264.7 (F) cells were incubated overnight with 500 nM of C-miR146a or control C-scrRNA and then stimulated with 100 ng/mL of LPS for 4 hours. The expression of the NF-κB–dependent reporter gene (panel E) or IL-6 secretion (panel F) was assessed by using Quanti-Blue assay or ELISA, respectively. Shown are representative results obtained in 3 independent experiments; means ± SEM. ***P < .001 and *P < .05 compared to untreated groups or as indicated. RLU, relative luciferase unit; SEAP, secreted alkaline phosphatase.
For an additional verification that the observed effects were sequence-specific, we used an analogical approach to deliver the miR-146a–targeting antisense oligonucleotide, C-anti-miR146a (supplemental Figure 6A).28 C-anti-miR146a exhibited similar cell selectivity as the C-miR146a mimic (supplemental Figure 6B) but opposite biological activity. C-anti-miR146a knocked-down miR-146a in mouse splenocytes and myeloid cells (supplemental Figure 6C), thereby upregulating IRAK1 and TRAF6 3′UTR activities (supplemental Figure 6D-E), NF-κB DNA binding (Figure 2D), and nuclear translocation (supplemental Figure 6F). Taken together, the flexibility of our strategy permits positive or negative regulation of NF-κB activity by using SR/TLR9-mediated miR-146a mimic or antisense delivery, respectively.
Systemic C-miR146a mimic delivery restored control over IRAK1 and TRAF6 in miR-146a–deficient myeloid cells in vivo
Treatment of NF-κB–related disorders requires efficient delivery of miR-146a mimic to target myeloid cells in various organs. To directly quantify the dose- and time-dependent biodistribution of C-miR146a mimic in the absence of endogenous miR-146a, we used the real-time qPCR to detect miR-146a guide-strand in various organs of miR-146a–deficient mice.9 Our initial pharmacokinetic analysis found that C-miR146a reaches the maximum plasma level ∼30 minutes after IV injection, with 144 minutes of circulatory half-life (supplemental Figure 7). Consistently, we found dose-dependent accumulation of miR146a guide-strand in bone marrow, spleens, and lymph nodes of miR-146a−/− mice after IV injection of C-miR146a (Figure 3A). In the 2.5 to 20 mg/kg dose range, C-miR146a delivered miR146a-5p guide-strand preferentially to CD11b+ myeloid cells from bone marrow (Figure 3B). For more precise identification of target cells, we injected IV fluorescently labeled C-miR146aCy3 or miR146aCy3 (5 mg/kg) into AML-bearing mice and analyzed their cellular uptake after 3 hours by using flow cytometry. C-miR146aCy3 was internalized by ∼30% to 50% of various myeloid cells and leukemic cells, ∼20% of long-term hematopoietic stem cells, and minimally by B cells and natural killer cells but not by T cells (Figure 3C). As expected, cellular uptake of miR146aCy3 alone was negligible.
![C-miR146a targets myeloid cell populations and restores miR-146a levels and activity in miR-146a−/−mice. Dose-dependent biodistribution of miR-146a mimic in organs (A) and in bone marrow (BM) CD11b+ myeloid cells and CD11b– cells (B) of miR-146a–deficient mice. Mice were injected intravenously by using various doses of C-miR146a and euthanized 3 hours later to assess miR-146a levels in BM, spleen, lymph nodes (LN), blood, and enriched BM CD11b+ myeloid cells and CD11b– cells by using qPCR. (C) Biodistribution of systemically injected C-miR146a and miR-146a. C57BL/6 mice engrafted with Cbfb/Myh11/Mpl leukemia were injected intravenously with 5 mg/kg of C-miR146aCy3 or miR-146aCy3 and euthanized 3 hours later. The percentages of Cy3+ cells in BM were assessed by using flow cytometry (T cells, CD3+; natural killer [NK] cells, NK1.1+; B cells, CD19+; short-term hematopoietic stem cells [ST-HSC], Lin–cKit+Sca1+Flt3–CD150–CD48–; long-term hematopoietic stem cells [LT-HSC], Lin–cKit+Sca1+Flt3–CD150+CD48–; monocytes, CD11b+CD11c–; macrophages, CD11b+F4/80+; dendritic cells [DCs], CD11b+CD11c+; myeloid-derived suppressor cells [MDSCs], CD11b+Gr1+; and AML cells, GFP+). Shown are results representative for 2 independent experiments; means ± SEM (n = 3 mice/group). (D-E) Myeloid cell–selective delivery of miR-146a in vivo. miR-146a−/− or WT mice were injected intravenously with 5 mg/kg of C-miR146a and euthanized 3 hours later. miR-146a levels in BM and splenocytes and enriched CD11b+ myeloid cells, CD19+ B cells, and CD3+ T cells from spleen were assessed by using qPCR and compared with the same populations in untreated WT mice. Shown are representative results obtained in 3 independent experiments; means ± SEM (n = 3). (F) Single injection of C-miR146a results in transient downregulation of IRAK1 and TRAF6 protein levels. miR-146a−/− mice were injected intravenously with 5 mg/kg of C-miR146a and euthanized at indicated times. Protein levels of IRAK1 and TRAF6 in BM cells were assessed by using western blot, and the quantified band intensities normalized to β-actin are shown. Shown are results representative for two independent experiments.***P < .001; **P < .01; and *P < .05 compared to untreated or as indicated.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/3/10.1182_blood.2019002045/2/m_bloodbld2019002045f3.png?Expires=1769087287&Signature=i6LBFlm0n7vfeu5VH0jRaQwP2xukl6vh2FQDEThFSlp-LOrPzXg9~NzO-5t2QeCvojb958EGXlxj1t~xdCjrIMURgD-bEvc9uG5BldOdap6zwyGES6LsKWw7KqRgP79C5nAGvSvuDwvTkzvuuHG360C9hvVSpH4wMiMqiL9I1ogHItvLyUMLEbdqaMSQZYzWgkBxnV8bbUmB5hZWUmmJGzIrD2O8RE~UIaCaXTe09P9KtNlD0qhcOxNKVVKX6nnIgoUveN2Ff0H9Z4SzrT~rspVysbK79YBerkGgra-kb~nmvMBlxXogLdKgR2hVhw0fpOq3d8Q8zSx-sb6RlWoBfw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
C-miR146a targets myeloid cell populations and restores miR-146a levels and activity in miR-146a−/−mice. Dose-dependent biodistribution of miR-146a mimic in organs (A) and in bone marrow (BM) CD11b+ myeloid cells and CD11b– cells (B) of miR-146a–deficient mice. Mice were injected intravenously by using various doses of C-miR146a and euthanized 3 hours later to assess miR-146a levels in BM, spleen, lymph nodes (LN), blood, and enriched BM CD11b+ myeloid cells and CD11b– cells by using qPCR. (C) Biodistribution of systemically injected C-miR146a and miR-146a. C57BL/6 mice engrafted with Cbfb/Myh11/Mpl leukemia were injected intravenously with 5 mg/kg of C-miR146aCy3 or miR-146aCy3 and euthanized 3 hours later. The percentages of Cy3+ cells in BM were assessed by using flow cytometry (T cells, CD3+; natural killer [NK] cells, NK1.1+; B cells, CD19+; short-term hematopoietic stem cells [ST-HSC], Lin–cKit+Sca1+Flt3–CD150–CD48–; long-term hematopoietic stem cells [LT-HSC], Lin–cKit+Sca1+Flt3–CD150+CD48–; monocytes, CD11b+CD11c–; macrophages, CD11b+F4/80+; dendritic cells [DCs], CD11b+CD11c+; myeloid-derived suppressor cells [MDSCs], CD11b+Gr1+; and AML cells, GFP+). Shown are results representative for 2 independent experiments; means ± SEM (n = 3 mice/group). (D-E) Myeloid cell–selective delivery of miR-146a in vivo. miR-146a−/− or WT mice were injected intravenously with 5 mg/kg of C-miR146a and euthanized 3 hours later. miR-146a levels in BM and splenocytes and enriched CD11b+ myeloid cells, CD19+ B cells, and CD3+ T cells from spleen were assessed by using qPCR and compared with the same populations in untreated WT mice. Shown are representative results obtained in 3 independent experiments; means ± SEM (n = 3). (F) Single injection of C-miR146a results in transient downregulation of IRAK1 and TRAF6 protein levels. miR-146a−/− mice were injected intravenously with 5 mg/kg of C-miR146a and euthanized at indicated times. Protein levels of IRAK1 and TRAF6 in BM cells were assessed by using western blot, and the quantified band intensities normalized to β-actin are shown. Shown are results representative for two independent experiments.***P < .001; **P < .01; and *P < .05 compared to untreated or as indicated.
C-miR146a targets myeloid cell populations and restores miR-146a levels and activity in miR-146a−/−mice. Dose-dependent biodistribution of miR-146a mimic in organs (A) and in bone marrow (BM) CD11b+ myeloid cells and CD11b– cells (B) of miR-146a–deficient mice. Mice were injected intravenously by using various doses of C-miR146a and euthanized 3 hours later to assess miR-146a levels in BM, spleen, lymph nodes (LN), blood, and enriched BM CD11b+ myeloid cells and CD11b– cells by using qPCR. (C) Biodistribution of systemically injected C-miR146a and miR-146a. C57BL/6 mice engrafted with Cbfb/Myh11/Mpl leukemia were injected intravenously with 5 mg/kg of C-miR146aCy3 or miR-146aCy3 and euthanized 3 hours later. The percentages of Cy3+ cells in BM were assessed by using flow cytometry (T cells, CD3+; natural killer [NK] cells, NK1.1+; B cells, CD19+; short-term hematopoietic stem cells [ST-HSC], Lin–cKit+Sca1+Flt3–CD150–CD48–; long-term hematopoietic stem cells [LT-HSC], Lin–cKit+Sca1+Flt3–CD150+CD48–; monocytes, CD11b+CD11c–; macrophages, CD11b+F4/80+; dendritic cells [DCs], CD11b+CD11c+; myeloid-derived suppressor cells [MDSCs], CD11b+Gr1+; and AML cells, GFP+). Shown are results representative for 2 independent experiments; means ± SEM (n = 3 mice/group). (D-E) Myeloid cell–selective delivery of miR-146a in vivo. miR-146a−/− or WT mice were injected intravenously with 5 mg/kg of C-miR146a and euthanized 3 hours later. miR-146a levels in BM and splenocytes and enriched CD11b+ myeloid cells, CD19+ B cells, and CD3+ T cells from spleen were assessed by using qPCR and compared with the same populations in untreated WT mice. Shown are representative results obtained in 3 independent experiments; means ± SEM (n = 3). (F) Single injection of C-miR146a results in transient downregulation of IRAK1 and TRAF6 protein levels. miR-146a−/− mice were injected intravenously with 5 mg/kg of C-miR146a and euthanized at indicated times. Protein levels of IRAK1 and TRAF6 in BM cells were assessed by using western blot, and the quantified band intensities normalized to β-actin are shown. Shown are results representative for two independent experiments.***P < .001; **P < .01; and *P < .05 compared to untreated or as indicated.
We next compared the miR-146a levels in oligonucleotide-treated miR-146a−/− mice vs those in the untreated WT mice to quantify the relative miR-146a restoration in specific immune cell populations. As shown in Figure 3D, the single injection of 5 mg/kg C-miR146a to miR-146a−/− mice replaced ∼61% and ∼53% of WT miR-146a levels in bone marrow and spleens, respectively. miR146a was restored to ∼47%, ∼28%, and ∼5% of the WT levels in splenic CD11b+ myeloid cells, CD19+ B cells, and CD3+ T cells, respectively (Figure 3E), confirming preferential myeloid cell targeting. Finally, we tested whether partial restoration of miR-146a will have sufficient biological activity to inhibit target IRAK1 and TRAF6 proteins. In fact, within 12 to 24 hours after a single IV injection of C-miR146a (5 mg/kg), both IRAK1 and TRAF6 were reduced close to the WT levels, returning to baseline after 2 to 3 days (Figure 3F). These results indicate that systemic delivery of C-miR146a using relatively low dosing can transiently restore levels and full biological activity of miR-146a in target myeloid cells in vivo.
C-miR146a abrogates myeloproliferation and exaggerated inflammatory responses in miR-146a–deficient mice
Expansion of myeloid cells is one of the critical phenotypic features of the miR-146a–deficient mice.29 Compared with bone marrow–derived macrophages (BMDMs) from WT mice, BMDMs from miR-146a–deficient mice exhibit enhanced proliferation accompanied by elevated expression of colony-stimulating factor–1 receptor (CSF1R).30 We therefore examined whether delivery of C-miR146a to miR-146a−/− BMDMs prevents aberrant myeloproliferation. In fact, the C-miR146a but not C-scrRNA treatment reduced the proliferative rate of miR-146a−/− BMDMs close to the WT BMDMs (Figure 4A). Together with the reduced proliferation, the elevated cell surface levels of CSF1R on miR-146a−/− BMDMs were reduced by C-miR146a to baseline WT levels (Figure 4B). Consistently, repeated 2-week C-miR146a treatment of aged miR-146a−/− mice reduced the expansion of immature CD11b+Gr1+ myeloid cells, together with their proliferation and CSF1R expression on macrophages (Figure 4C-E).
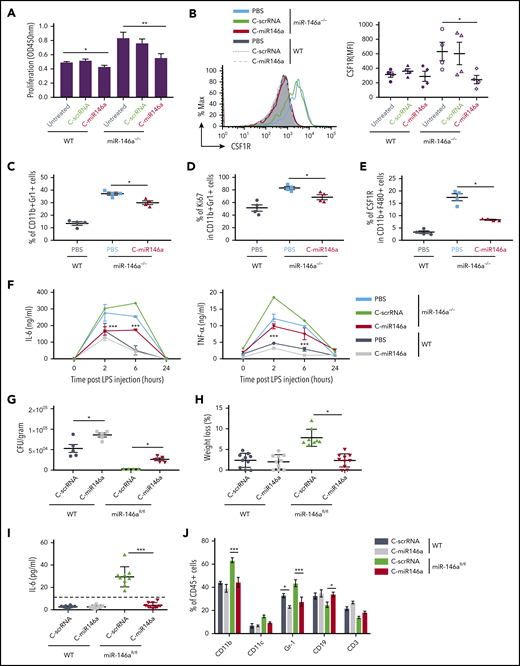
C-miR146a corrects the aberrant myeloproliferation and inflammatory responses in miR-146a–deficient mice. (A-B) Reduced proliferation of miR-146a−/− BMDMs treated with C-miR146a. Bone marrow cells from WT and miR-146a−/− mice were cultured in the presence of 50 ng/mL of M-CSF for 7 days and treated by using 1 µM of C-miR146a or C-scrRNA. The cell proliferation was measured by using colorimetric XTT assay (A), and the CSF1R expression on CD11b+F4/80+ cells was quantified by using flow cytometry (B). (C-E) Systemic C-miR146a injections reduced aberrant myeloproliferation of miR-146a−/− mice in vivo. Ten-month-old WT or miR-146a−/− female mice (n = 4/group) were injected intravenously by using 10 mg/kg of C-miR146a or PBS daily for 2 weeks and euthanized 1 day after the last injection. Percentages of splenic CD11b+Gr1+ (C), Ki67+CD11b+Gr1+ (D), and the CSF1R expression on macrophages (E) were analyzed by using flow cytometry. (F) Systemic injections of C-miR146a alleviate exaggerated response to endotoxin in miR-146a−/− mice. WT or miR-146a−/− mice were injected intravenously with 5 mg/kg of C-miR146a or C-scrRNA daily for 3 days before LPS challenge (1 mg/kg, intraperitoneally). Blood was collected at the indicated times to analyze IL-6 and TNF-α levels using ELISA. (G-J) C-miR146a treatment restored tolerance to L monocytogenes infection in mice with myeloid cell–specific miR-146a deletion. WT or miR-146afl/fl mice injected daily intravenously by using 5 mg/kg of C-miR146a or C-scrRNA were infected with L monocytogenes on day 3 and euthanized on day 6. The liver bacterial load (G), the percentage of weight loss (H), plasma levels of IL-6 (I), and percentages of various hematopoietic cell populations in circulation (J) were assessed. Representative results from at least 2 independent experiments are shown; means ± SEM (n = 5/group). ***P < .001; **P < .01; and *P < .05 compared to untreated or as indicated. CFU, colony-forming unit.
C-miR146a corrects the aberrant myeloproliferation and inflammatory responses in miR-146a–deficient mice. (A-B) Reduced proliferation of miR-146a−/− BMDMs treated with C-miR146a. Bone marrow cells from WT and miR-146a−/− mice were cultured in the presence of 50 ng/mL of M-CSF for 7 days and treated by using 1 µM of C-miR146a or C-scrRNA. The cell proliferation was measured by using colorimetric XTT assay (A), and the CSF1R expression on CD11b+F4/80+ cells was quantified by using flow cytometry (B). (C-E) Systemic C-miR146a injections reduced aberrant myeloproliferation of miR-146a−/− mice in vivo. Ten-month-old WT or miR-146a−/− female mice (n = 4/group) were injected intravenously by using 10 mg/kg of C-miR146a or PBS daily for 2 weeks and euthanized 1 day after the last injection. Percentages of splenic CD11b+Gr1+ (C), Ki67+CD11b+Gr1+ (D), and the CSF1R expression on macrophages (E) were analyzed by using flow cytometry. (F) Systemic injections of C-miR146a alleviate exaggerated response to endotoxin in miR-146a−/− mice. WT or miR-146a−/− mice were injected intravenously with 5 mg/kg of C-miR146a or C-scrRNA daily for 3 days before LPS challenge (1 mg/kg, intraperitoneally). Blood was collected at the indicated times to analyze IL-6 and TNF-α levels using ELISA. (G-J) C-miR146a treatment restored tolerance to L monocytogenes infection in mice with myeloid cell–specific miR-146a deletion. WT or miR-146afl/fl mice injected daily intravenously by using 5 mg/kg of C-miR146a or C-scrRNA were infected with L monocytogenes on day 3 and euthanized on day 6. The liver bacterial load (G), the percentage of weight loss (H), plasma levels of IL-6 (I), and percentages of various hematopoietic cell populations in circulation (J) were assessed. Representative results from at least 2 independent experiments are shown; means ± SEM (n = 5/group). ***P < .001; **P < .01; and *P < .05 compared to untreated or as indicated. CFU, colony-forming unit.
We next examined whether miR-146a mimic delivery can attenuate the exaggerated inflammatory response to a systemic bacterial endotoxin challenge resulting from the miR-146a ablation.30 Age- and sex-matched WT and miR-146a−/− mice were injected with a sublethal dose of LPS, and the plasma levels of proinflammatory cytokines IL-6 and tumor necrosis factor-α (TNF-α) were measured by using ELISA. As expected, LPS induced a higher and more prolonged cytokine response in miR-146a−/− mice than in WT mice, peaking at 2 to 6 hours before returning to baseline at 24 hours. In contrast, C-miR146a treatment reduced IL-6 and TNF-α upregulation in miR-146a−/− mice at both 2 and 6 hours compared with C-scrRNA control treatment (Figure 4F). The effect of C-miR146a on the inflammatory cytokine secretion in WT mice was minimal, indicating adequate negative control of cytokine production by the endogenous miR-146a.
Following on these results, we used mice with the myeloid cell–specific miR-146a ablation (LyzM-Cre/miR-146afl/fl) to assess the effect of miR-146a restoration on the IL-6–controlled response to a bacterial infection with L monocytogenes.20,31,32 Both WT and miR-146afl/fl mice were injected daily with C-miR146a or control C-scrRNA (5 mg/kg) starting 3 days before the sublethal infection with L monocytogenes. The C-miR146a treatment interfered with the bacterial clearance in miR-146a–deficient and WT mice, increasing bacterial burden in the liver and spleen (Figure 4G; supplemental Figure 8A). The reduced bacterial load in control-treated miR-146afl/fl mice correlated with weight loss (Figure 4H) and elevated plasma levels of IL-6 (Figure 4I). All these effects of miR-146a deletion were completely alleviated by C-miR146a treatment. Finally, miR-146a restoration decreased the expansion of CD11b+ myeloid cells, including Gr1+ granulocytes, in blood and spleen of control miR-146afl/fl mice (Figure 4J; supplemental Figure 8B). Taken together, our results indicate that myeloid cell–targeted miR-146a restoration using C-miRNA mimic can reverse the key phenotypic features of miR-146a deletion in mice. It is worth noting that we observed no significant effect of C-miRNA mimic on other immune cell populations in WT mice, such as dendritic cells, B cells, or natural killer cells, even after repeated 2-week daily IV injections (supplemental Figures 9-10). Finally, our C-miR146a two-week dose escalation study (5-20 mg/kg per day) found no evidence of toxicities to hematopoietic cells or organ gross abnormalities (supplemental Figure 11).
Monocyte-targeted miR-146a delivery alleviates CRS triggered by CD19 CAR T cells without compromising antitumor effects
CAR T-cell immunotherapy has emerged as a revolutionary treatment of various hematologic cancers; however, many patients experience severe side effects of CRS.23,33 Recent studies linked CRS to CAR T-cell–dependent activation of CD40 signaling in monocytes/macrophages, thereby inducing IL-1 and IL-6, the key cytokine storm mediators. Although CD40 triggers canonical and noncanonical NF-κB signaling, it does not upregulate miR-146a levels.29 Thus, we tested whether delivery of miR-146a mimic will dampen the proinflammatory effects of human monocytes induced by the CD19-specific CAR T cells. Predictably, CD19 CAR T cells exhibited low uptake of fluorescently labeled C-miR146aCy3 compared with monocytes (supplemental Figure 12A). C-miR146a did not affect CAR T-cell viability or cytotoxicity against target CD19+ Nalm6 leukemia cells (Figure 5A). We next tested C-miR146a in a 3-component CRS model coculturing in vitro CD19+ Nalm6 cells with CD19-specific CAR T cells (generated from peripheral blood mononuclear cells from 4 different donors) and donor-matched CD14+ monocytes (Figure 5B). The upregulation of IL-1 and IL-6 depended on the presence of monocytes and was induced only by CD19+ leukemia-specific and not mock-transfected CAR T cells. Importantly, C-miR146a reduced IL-1 secretion by one-half, while bringing IL-6 levels close to baseline compared with control treatments. Similar results were obtained by using THP-1 monocyte-like cells instead of primary monocytes (supplemental Figure 12B-C).
![Targeting monocytes using C-miR146a mimic alleviates CRS induced by CD19 CAR T cells without compromising antitumor effects. (A-B) C-miR146a does not affect cytotoxic activity of CD19 CAR T cells against target leukemia cells. Mock- or CD19 CAR-transduced T cells were cocultured at a 1:1:1 ratio with donor-matched CD14+ monocytes and target CD19+ Nalm6 B-cell leukemia for 48 hours in the presence of 500 nM of C-miR146a or control C-scrRNA. (A) Shown are the percentages of live CD19+ target cells (left) or CAR T cells (right) vs untreated control. (B) The IL-1 and IL-6 levels in cultured supernatants as measured by using ELISA. Shown are results combined from 4 different peripheral blood mononuclear cell donors. (C) The experimental design for in vivo studies on the CAR T-cell–induced CRS using a xenotransplanted lymphoma model. SCID-Beige mice were engrafted with luciferase-expressing Raji lymphoma cells (intraperitoneally [IP]), and after 2 weeks were injected daily by using 5 mg/kg (IP) of C-miR146a or PBS. A total of 12.5 × 106 mock or CD19 CAR T cells were injected IP on day 18 before euthanizing mice on day 22. (D-E) Tumor progression was monitored by using bioluminescent imaging; means ± SEM. (F) The intracellular miR-146a levels were measured by using qPCR in CD11b+ myeloid and CD11b– nonmyeloid cells derived from peritoneal lavage. (G) Serum human cytokine levels and mouse IL-6 and G-CSF were measured by using ELISA after 24 hours from CAR T-cell transfer. Representative results from at least 2 independent experiments are shown as means ± SEM (n = 4). ***P < .001; **P < .01; and *P < .05 compared to untreated or as indicated. mG-CSF, mouse G-CSF; p/s, photons per second; ROI, regions of interest.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/3/10.1182_blood.2019002045/2/m_bloodbld2019002045f5.png?Expires=1769087287&Signature=gwd3XduGpJUk9Pu98U5j28~jUr2jzCzXQnKAaWcdi~afLUClhvEoUVcsHG6KVt-osw9-rTWFnJLVboIlTuRq5NHqt5XPgilzPpCqGSj-L8iIROXRn7-zY5TFOhBmwaOZCgH-5yboacHca568wWNu~zT5RNI3TsXPiLt0lpTuvZW6MAK0SXNpaaisu9HfJdWPub~z~4-~og0zQVf9BYo4HMdWYwii65GwxjKPsDEi5nK00hL-I7iql7~MYeqcPUJbSKKlHiUTVbKOeGBShPa2K7fakAVOc~WFwy5Ru3UaR~YVcbUlWqH1gClW9aKckdV7tW~lEPLt~oT~f46~q2mydw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Targeting monocytes using C-miR146a mimic alleviates CRS induced by CD19 CAR T cells without compromising antitumor effects. (A-B) C-miR146a does not affect cytotoxic activity of CD19 CAR T cells against target leukemia cells. Mock- or CD19 CAR-transduced T cells were cocultured at a 1:1:1 ratio with donor-matched CD14+ monocytes and target CD19+ Nalm6 B-cell leukemia for 48 hours in the presence of 500 nM of C-miR146a or control C-scrRNA. (A) Shown are the percentages of live CD19+ target cells (left) or CAR T cells (right) vs untreated control. (B) The IL-1 and IL-6 levels in cultured supernatants as measured by using ELISA. Shown are results combined from 4 different peripheral blood mononuclear cell donors. (C) The experimental design for in vivo studies on the CAR T-cell–induced CRS using a xenotransplanted lymphoma model. SCID-Beige mice were engrafted with luciferase-expressing Raji lymphoma cells (intraperitoneally [IP]), and after 2 weeks were injected daily by using 5 mg/kg (IP) of C-miR146a or PBS. A total of 12.5 × 106 mock or CD19 CAR T cells were injected IP on day 18 before euthanizing mice on day 22. (D-E) Tumor progression was monitored by using bioluminescent imaging; means ± SEM. (F) The intracellular miR-146a levels were measured by using qPCR in CD11b+ myeloid and CD11b– nonmyeloid cells derived from peritoneal lavage. (G) Serum human cytokine levels and mouse IL-6 and G-CSF were measured by using ELISA after 24 hours from CAR T-cell transfer. Representative results from at least 2 independent experiments are shown as means ± SEM (n = 4). ***P < .001; **P < .01; and *P < .05 compared to untreated or as indicated. mG-CSF, mouse G-CSF; p/s, photons per second; ROI, regions of interest.
Targeting monocytes using C-miR146a mimic alleviates CRS induced by CD19 CAR T cells without compromising antitumor effects. (A-B) C-miR146a does not affect cytotoxic activity of CD19 CAR T cells against target leukemia cells. Mock- or CD19 CAR-transduced T cells were cocultured at a 1:1:1 ratio with donor-matched CD14+ monocytes and target CD19+ Nalm6 B-cell leukemia for 48 hours in the presence of 500 nM of C-miR146a or control C-scrRNA. (A) Shown are the percentages of live CD19+ target cells (left) or CAR T cells (right) vs untreated control. (B) The IL-1 and IL-6 levels in cultured supernatants as measured by using ELISA. Shown are results combined from 4 different peripheral blood mononuclear cell donors. (C) The experimental design for in vivo studies on the CAR T-cell–induced CRS using a xenotransplanted lymphoma model. SCID-Beige mice were engrafted with luciferase-expressing Raji lymphoma cells (intraperitoneally [IP]), and after 2 weeks were injected daily by using 5 mg/kg (IP) of C-miR146a or PBS. A total of 12.5 × 106 mock or CD19 CAR T cells were injected IP on day 18 before euthanizing mice on day 22. (D-E) Tumor progression was monitored by using bioluminescent imaging; means ± SEM. (F) The intracellular miR-146a levels were measured by using qPCR in CD11b+ myeloid and CD11b– nonmyeloid cells derived from peritoneal lavage. (G) Serum human cytokine levels and mouse IL-6 and G-CSF were measured by using ELISA after 24 hours from CAR T-cell transfer. Representative results from at least 2 independent experiments are shown as means ± SEM (n = 4). ***P < .001; **P < .01; and *P < .05 compared to untreated or as indicated. mG-CSF, mouse G-CSF; p/s, photons per second; ROI, regions of interest.
To further confirm that the combination of C-miR146a with CD19 CAR T cells has potential for alleviating CRS-related toxicities, we adopted a model of CAR T-cell–induced CRS in human CD19+ Raji B-cell lymphoma in partly immunodeficient SCID-Beige mice, similar to that recently published.15 Mice with heavy intraperitoneal lymphoma burden developed CRS and acute inflammation within 3 days after CD19 CAR T-cell injection (Figure 5C). Mice were treated by using C-miR146a or control vehicle 3 days before CAR T-cell transfer. As shown in Figure 5D-E, the C-miR146a did not interfere with the CAR T-cell–mediated inhibition of lymphoma progression. However, the C-miR146a mimic doubled the amount of endogenous miR146a in target peritoneal myeloid cells (Figure 5F). As a result, C-miR146a dramatically reduced major CRS-related cytokines, IL-6 and granulocyte colony-stimulating factor (G-CSF), likely derived from mouse monocytes in this model as suggested by others (Figure 5G). Our findings underscore the potential of using C-miR146a mimic to alleviate adverse effects of CD19 CAR T-cell therapy without impeding on-target therapeutic effects.
C-miR146a targets NF-κB signaling and inhibits del(5q) leukemia progression
Low expression of miR-146a located on chromosome 5q has been reported in both MDS and AML as del(5q) aberrations are common in high-risk leukemia.7 However, constitutive activation of NF-κB survival signaling is not limited to del(5q) MDS/AML and occurs in up to 40% of AML cases.34 We tested C-miR146a mimic effect on viability of HL-60, MDSL del(5q) leukemia cells, and also FLT3ITD+/N(5q) MV4-11 AML cells, which have all been found to be sensitive to small-molecule NF-κB inhibitors (supplemental Figure 13). As shown in Figure 6A, C-miR146a induced cell death in all 3 models, although it was most effective against miR-146a–deficient MDSL and HL-60 cells. In initial testing, we confirmed that local delivery of C-miR146a into subcutaneously engrafted leukemia models effectively inhibited growth of both HL-60 (supplemental Figure 14A) and MDSL (supplemental Figure 14B). We next examined the efficacy of systemic C-miR146a administration (10 mg/kg) against disseminated HL-60 leukemia. As shown in Figure 6B-D, compared with control mice, C-miR146a reduced AML progression and thereby significantly prolonged mice survival. Focused gene expression analysis of a panel of 84 NF-κB–related genes confirmed that C-miR146a downregulated key elements of NF-κB signaling in HL-60 cells, such as RELA, MYD88, NFKB1/2, TRAF3, and TRAF6, or mediators IL1B and TNF (Figure 6E; supplemental Figure 15). Altogether, these results illustrate the direct antitumor effect of C-miR146a, both in vitro and in vivo, against miR-146a–deficient MDS/AML, likely through the inhibition of NF-κB–mediated survival signaling. These conclusions are also supported by analysis of The Cancer Genome Atlas dataset from 164 patients with AML. As expected, low miR-146a expression was associated with worse overall patient survival (Figure 6F). The gene set enrichment analysis indicated significant inverse correlation of miR-146a levels in AML with 2 different sets of NF-κB–related genes, including IRAK1, NFKB1, and NFKB2 but not TRAF6 (Figure 6G-H; supplemental Figure 16). Overall, these results underscore the therapeutic potential of C-miR146a for the treatment of del(5q) MDS/AML and potentially other NF-κB–dependent types of leukemia.
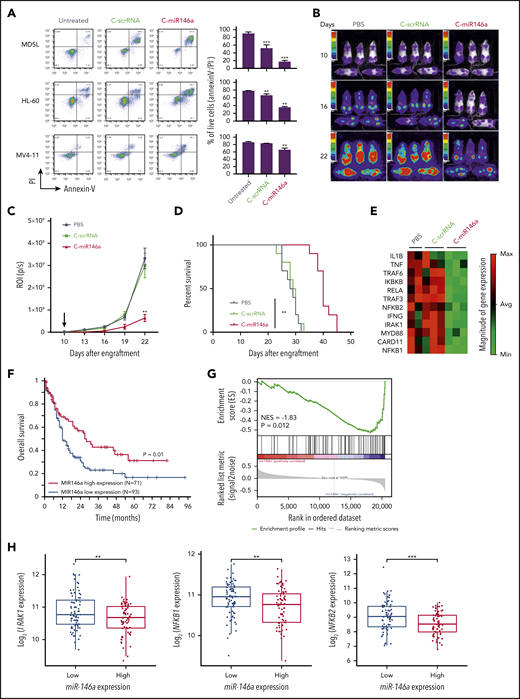
C-miR146a targets NF-κB signaling and inhibits progression of del(5q) MDS and AML. (A) Del(5q) HL-60 or MDSL cells or Flt3 mutation MV4-11 leukemia cells were treated in vitro by using 500 nM of C-miR146a or C-scrRNA for 6 days, and the percentages of live cells were assessed by using flow cytometry. Representative results obtained from 3 independent experiments; means ± SEM. Systemic administration of C-miR146a extended survival of human HL-60 AML-bearing mice. NSG-SGM3 mice engrafted with disseminated HL-60-luc cells were injected daily intravenously by using 10 mg/kg of C-miR146a or C-scrRNA, and leukemia progression was monitored by using bioluminescent imaging (B), leukemia progression (C), and the Kaplan-Meier survival curves (D). Shown are representative results from at least 2 independent experiments; means ± SEM (n = 10). (E) NF-κB pathway gene analysis in HL-60 AML-bearing mice after 10 injections of C-miR146a, C-scrRNA, or PBS (n = 3/group). Total RNA isolated from bone marrow was analyzed by using RT2 Profiler PCR arrays. The clustergram of significantly (>1.5 fold) downregulated genes. (F) Low miR-146a expression is associated with worse overall survival of patients with AML based on The Cancer Genome Atlas data set. The optimal cut-point was identified by using log-rank statistics in a survfit model. The log-rank test P value is shown. (G) Gene Set Enrichment Analysis in AML patients with high versus low miR-146a expression based on The Cancer Genome Atlas data set with a NF-κB gene set from Gene Ontology (70 genes). Samples having both miRNA and mRNA expression data (n = 151) were applied to this analysis. Normalized enrichment score (NES) and P value are shown. (H) Standard boxplots were applied to visualize the distribution of log2-transformed expression of IRAK1, NFKB1, and NFKB2 with the low and high miR-146a expression groups. ***P < .001 and **P < .01 compared to untreated groups or as indicated.
C-miR146a targets NF-κB signaling and inhibits progression of del(5q) MDS and AML. (A) Del(5q) HL-60 or MDSL cells or Flt3 mutation MV4-11 leukemia cells were treated in vitro by using 500 nM of C-miR146a or C-scrRNA for 6 days, and the percentages of live cells were assessed by using flow cytometry. Representative results obtained from 3 independent experiments; means ± SEM. Systemic administration of C-miR146a extended survival of human HL-60 AML-bearing mice. NSG-SGM3 mice engrafted with disseminated HL-60-luc cells were injected daily intravenously by using 10 mg/kg of C-miR146a or C-scrRNA, and leukemia progression was monitored by using bioluminescent imaging (B), leukemia progression (C), and the Kaplan-Meier survival curves (D). Shown are representative results from at least 2 independent experiments; means ± SEM (n = 10). (E) NF-κB pathway gene analysis in HL-60 AML-bearing mice after 10 injections of C-miR146a, C-scrRNA, or PBS (n = 3/group). Total RNA isolated from bone marrow was analyzed by using RT2 Profiler PCR arrays. The clustergram of significantly (>1.5 fold) downregulated genes. (F) Low miR-146a expression is associated with worse overall survival of patients with AML based on The Cancer Genome Atlas data set. The optimal cut-point was identified by using log-rank statistics in a survfit model. The log-rank test P value is shown. (G) Gene Set Enrichment Analysis in AML patients with high versus low miR-146a expression based on The Cancer Genome Atlas data set with a NF-κB gene set from Gene Ontology (70 genes). Samples having both miRNA and mRNA expression data (n = 151) were applied to this analysis. Normalized enrichment score (NES) and P value are shown. (H) Standard boxplots were applied to visualize the distribution of log2-transformed expression of IRAK1, NFKB1, and NFKB2 with the low and high miR-146a expression groups. ***P < .001 and **P < .01 compared to untreated groups or as indicated.
Discussion
Results of the current study show the feasibility of systemic delivery of miRNA mimic specifically to human and mouse myeloid cells for therapeutic modulation of their immune activity or neoplastic growth. The C-miR146a injected intravenously restored miR-146a-5p in target myeloid cells to levels sufficient for complete elimination of exacerbated NF-κB activity in miR-146a−/− mice, thereby preventing the exaggerated inflammatory responses and aberrant myeloproliferation. Our miR-146a mimic delivery strategy proved effective also in human models of NF-κB–driven inflammation and in myeloid leukemia (MDS/AML). Importantly, unlike standard anti-inflammatory strategies (eg, steroid hormones), the myeloid cell–specific miR-146a mimic delivery proved effective without interfering with antitumor activity of CAR T cells or any signs of toxicity even with repeated, long-term administration.
Despite broad clinical potential, only a few miRNA therapeutic agents entered clinical trials, and the majority represented antisense molecules (anti-miRs or antagomirs), which as single-stranded oligonucleotides proved easier to optimize forin vivo use.19,35 One of the best examples is targeting lymphoma cell addiction to oncogenic miR-155.36 In contrast, the therapeutic replacement/restoration of tumor suppressor miRNAs proved challenging. The chemical modification of miRNA to ensure nuclease resistance can interfere with intracellular processing, RISC loading, and/or mRNA targeting. Pharmacologic formulation of miRNA mimics for systemic administration can result in severe immune toxicities.37 Cell-selective delivery of chemically stabilized, naked miRNA mimics is gaining attention with the success of small interfering RNA delivery using hepatocyte-specific GalNAc-conjugates,38 and a recent study revisited folate as targeting moiety to miRNA delivery to breast and lung cancer cells.39
Our previous studies focused on myeloid cell–selective delivery of oligonucleotides to disrupt immunosuppression in the tumor microenvironment. The conjugation of CpG ODNs with inhibitors of STAT3, a master immune checkpoint regulator, resulted in potent antitumor immune responses. Although it seems counterintuitive, the SR/TLR9-mediated delivery strategy can be adapted for dampening excessive immune activity of myeloid cells in autoinflammatory diseases or myeloid malignancies. Unlike type B CpG sequences, the type A CpG ODNs are poor activators of NF-κB signaling and cytokines such as IL-6 and IL-10.40 Although a recent study showed that injections of CpG(A) alleviated severity of sepsis in mice by reducing blood clotting,41 the control CpG(A)-scrRNA did not exhibit anti-inflammatory activity in our experiments. The presence of a functional miR-146a-5p guide strand in the CpG conjugate was key to the biological activity of this oligonucleotide. The more extensive chemical modifications of miR-146a guide strand affect its activity, likely interfering with unwinding of the duplex while in RISC.42 Importantly, even partial restoration of miR-146a-5p levels in target immune cells in vivo was sufficient for near complete and durable inhibition of target IRAK1 and TRAF6. The systemic administration of C-miR146a effectively reversed key features of the miR-146a deletion related to excessive NF-κB signaling in both mouse and human myeloid cells.
Cytokine storm or CRS occur in response to various conditions, from bacterial infections to antibody-based therapies to immunomodulatory drugs. In context of CAR T-cell cancer therapies, especially CD19-specific CAR T cells, CRS is one of the most common and potentially fatal adverse effects. The lower incidence of CRS correlates with prolonged survival of patients receiving CAR T-cell therapy.43 Despite the routine use of an IL-6 receptor antagonist (tocilizumab), high-dose steroid hormones are frequently required to control severe CRS but at the same time they potentially curb the antitumor efficacy of CAR T cells.43,44 Thus, the need exists for more precise and safer immunomodulatory strategies addressing the complexity of immune responses. Myeloid cell–selective miR-146a mimic acts at the nodal point of the immune cell network, preventing the release of cytokines driving CRS from monocytes and macrophages without interfering with CAR T-cell activity.12,15
The simplicity and flexibility of the C-miRNA mimic design, adaptable to delivery of both miRNA mimics and anti-miRs, provide an opportunity for the development of miRNA therapeutic agents for immunomodulation and/or therapy of myeloproliferative diseases and leukemia. The pleiotropic effect of a single miRNA on multiple protein targets benefits their potency and reduces development of drug resistance, whereas the myeloid cell–targeted delivery can improve the safety of miRNA therapeutic agents. The growing list of myeloid cell–specific anti-inflammatory miRNAs extends beyond miR-146a, including recently described miR-125b and miR-203b or tolerance-inducing miR-221 and miR-222.45,46 In addition to the immunoregulatory role, the miR-146a is a well-established tumor suppressor, and the genetic loss of miR-146a or downregulation is common in patients with MDS and AML.6 miR-146a replacement using SR/TLR9–targeted delivery provides therapeutic opportunity in patients with MDS/AML. However, given the genetic instability of AML, C-miR146a therapy could likely benefit from combination with targeting additional oncogenic or tumor suppressor miRNAs. The upregulation of oncogenic miR-155 is common in FMS-like tyrosine kinase 3 internal tandem duplication–associated AML.47 Noteworthy, miR-155 can potentially antagonize the inhibitory effects of miR-146a on NF-κB in mouse myeloid cells.20 It is not known whether miR-155 plays a similar role, lessening the tumor suppressive effect of miR-146a in human leukemia, but our strategy can easily be adapted to delivery of anti-miR155 to AML cells and leukemia stem cells.48 Further studies should explore the possibility of personalizing miRNA mimic/antisense combinations to an AML patient–specific miRNA profile using the same SR/TLR9 delivery platform. The emerging CAR T-cell strategies for AML, such as CD123-specific CAR design, underscore the potential to combine AML-specific CAR T cells with C-miR146a, thereby alleviating potential immunotoxicities as well as reducing AML cell survival.49 We believe that SR/TLR9–targeted delivery of miR-146a mimic provides an outline for the development of miRNA therapeutic agents for a variety of inflammatory disorders and myeloid malignancies.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE141402).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank and acknowledge the dedication of staff members at the DNA/RNA Synthesis, Analytical Cytometry, Light Microscopy Cores and Animal Resources Center (City of Hope).
This work was supported in part by the National Cancer Institute/National Institutes of Health awards R01CA213131 (M.K.), Lymphoma Specialized Program of Research Excellence P50CA107399 (S.J.F.), and P30CA033572 (to the City of Hope). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: Y.-L.S. and M.K. were responsible for study design; Y.-L.S., X.W., M.M., T.P.A., D.F.M., Z.Z., D.W., P.M.S., and M.K. performed data acquisition; Y.-L.S., M.M., T.P.A., D.F.M., D.W., and C.O. analyzed and interpreted the data; Y.-L.S., X.W., X.H., B.Z., P.M.S., S.J.F., D.B., L.L., G.M., M.P.B., and M.K. were responsible for administrative, technical, or material support; and Y.-L.S. and M.K. wrote the manuscript.
Conflict-of-interest disclosure: M.K., P.M.S., and G.M. have a patent on C-miRNA conjugates and uses thereof compositions for the treatment of cancers and other diseases. The remaining authors declare no competing financial interests.
Correspondence: Marcin Kortylewski, Beckman Research Institute at City of Hope Comprehensive Cancer Center, 1500 East Duarte Rd, Beckman Center, Room 3111, Duarte, CA 91010; e-mail: mkortylewski@coh.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal