

Key Points
D-RVd improved sCR rates and MRD negativity vs RVd, both of which deepened over time.
No new safety concerns were observed with D-RVd, and no clinically significant impact on stem cell mobilization or engraftment was noted.

Abstract
Lenalidomide, bortezomib, and dexamethasone (RVd) followed by autologous stem cell transplantation (ASCT) is standard frontline therapy for transplant-eligible patients with newly diagnosed multiple myeloma (NDMM). The addition of daratumumab (D) to RVd (D-RVd) in transplant-eligible NDMM patients was evaluated. Patients (N = 207) were randomized 1:1 to D-RVd or RVd induction (4 cycles), ASCT, D-RVd or RVd consolidation (2 cycles), and lenalidomide or lenalidomide plus D maintenance (26 cycles). The primary end point, stringent complete response (sCR) rate by the end of post-ASCT consolidation, favored D-RVd vs RVd (42.4% vs 32.0%; odds ratio, 1.57; 95% confidence interval, 0.87-2.82; 1-sided P = .068) and met the prespecified 1-sided α of 0.10. With longer follow-up (median, 22.1 months), responses deepened; sCR rates improved for D-RVd vs RVd (62.6% vs 45.4%; P = .0177), as did minimal residual disease (MRD) negativity (10−5 threshold) rates in the intent-to-treat population (51.0% vs 20.4%; P < .0001). Four patients (3.8%) in the D-RVd group and 7 patients (6.8%) in the RVd group progressed; respective 24-month progression-free survival rates were 95.8% and 89.8%. Grade 3/4 hematologic adverse events were more common with D-RVd. More infections occurred with D-RVd, but grade 3/4 infection rates were similar. Median CD34+ cell yield was 8.2 × 106/kg for D-RVd and 9.4 × 106/kg for RVd, although plerixafor use was more common with D-RVd. Median times to neutrophil and platelet engraftment were comparable. Daratumumab with RVd induction and consolidation improved depth of response in patients with transplant-eligible NDMM, with no new safety concerns. This trial was registered at www.clinicaltrials.gov as #NCT02874742.
Introduction
Recommended frontline treatment of transplant-eligible newly diagnosed multiple myeloma (MM; NDMM) includes induction therapy, high-dose melphalan and autologous stem cell transplantation (ASCT), and maintenance.1-3 However, this approach is noncurative in the vast majority of patients. Therefore, novel strategies with manageable toxicity are needed to improve depth of response, progression-free survival (PFS), and overall survival (OS). The combination of lenalidomide, bortezomib, and dexamethasone (RVd) is a standard induction regimen for NDMM.4,5 This regimen improved PFS and OS in NDMM patients not undergoing ASCT6 and demonstrated a median PFS of 50 months and a 4-year OS rate of 81% when used as induction and consolidation therapy with frontline ASCT.7
Although advances in frontline therapy have yielded unprecedented improvement in long-term patient outcomes, surrogate markers of PFS and OS are needed. In this regard, achievement of stringent complete response (CR; sCR) after ASCT is associated with improvement in median PFS and OS.8,9 Similarly, minimal residual disease (MRD) testing has emerged as a sensitive measure of depth of response that strongly correlates with PFS and OS.7,10-13
Daratumumab is a human immunoglobulin G kappa (IgGκ) monoclonal antibody targeting CD38.14-20 In randomized phase 3 studies in NDMM and relapsed/refractory MM, daratumumab-based regimens improved depth of response, including MRD negativity, which translated into longer median PFS.21-27 Recently, the CASSIOPEIA study demonstrated the benefit of adding daratumumab to triplet induction and consolidation therapy consisting of a proteasome inhibitor (bortezomib), immunomodulatory agent (thalidomide), and dexamethasone in transplant-eligible NDMM.21
The GRIFFIN study was designed to examine whether the addition of daratumumab to RVd (D-RVd) and ASCT improves depth of response, including rates of sCR and MRD negativity, in NDMM patients. The safety run-in phase demonstrated that D-RVd was safe and well-tolerated, and depth of response continued to improve over the course of therapy.28 Herein, we report the primary efficacy and updated secondary efficacy and safety results of the randomized phase of the trial.
Methods
Trial design and oversight
This multicenter, randomized, open-label, active-controlled, phase 2 study was designed by Janssen Oncology in partnership with Alliance Foundation Trials and enrolled patients between 20 December 2016 and 10 April 2018 at 35 sites across the United States. The protocol and all amendments were approved by the institutional review board or independent ethics committee at each participating site, and all patients gave written informed consent. The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines, the principles originating from the Declaration of Helsinki, and study site-specific regulations.
Patients
Eligible patients were 18 to 70 years of age at study entry, had NDMM as defined by International Myeloma Working Group (IMWG) criteria,29 had an Eastern Cooperative Oncology Group (ECOG) performance status score of 0 to 2, and were candidates for high-dose therapy and ASCT. Other inclusion criteria included: absolute neutrophil count, ≥1.0 × 109/L; hemoglobin level, >7.5 g/dL; platelet count, ≥75 × 109/L (≥50 × 109/L if ≥50% of the bone marrow was infiltrated with MM cells); alanine aminotransferase and aspartate aminotransferase levels <2.5 times the upper limit of normal; total bilirubin level, <1.5 times the upper limit of normal; creatinine clearance, ≥30 mL/min; and corrected serum calcium, ≤14.0 mg/dL (≤3.5 mmol/L). Additional eligibility criteria are listed in the supplemental Appendix (available on the Blood Web site).
Trial treatments
We randomly assigned patients in a 1:1 ratio to D-RVd or RVd (supplemental Figure 1). Randomization was stratified by International Staging System (ISS) disease stage (I, II, or III) and creatinine clearance (30-50 or >50 mL/min).
All patients received four 21-day induction cycles and two 21-day consolidation cycles of oral lenalidomide (25 mg daily on days 1-14), subcutaneous bortezomib (1.3 mg/m2 on days 1, 4, 8, and 11), and oral dexamethasone (20 mg on days 1, 2, 8, 9, 15, and 16). Patients in the D-RVd group received IV daratumumab (16 mg/kg) on days 1, 8, and 15 of cycles 1 through 4 and day 1 of post-ASCT consolidation cycles (cycles 5 and 6). Starting at cycle 7, all patients received maintenance therapy (28-day cycles) consisting of oral lenalidomide (10 mg daily on days 1-21; increasing to 15 mg after 3 cycles, if tolerated). Patients in the D-RVd group also received IV daratumumab (16 mg/kg) on day 1 every 8 weeks or every 4 weeks per protocol amendment 2 (approved 5 February 2018), based on emerging pharmacokinetic results from other studies. Maintenance therapy on study will continue until disease progression or up to 2 years. Following completion of study maintenance therapy, patients can continue lenalidomide per local standard of care. The supplemental Appendix includes details regarding pre- and postinfusion medications and dose modifications.
After induction cycle 4, patients underwent stem cell mobilization with granulocyte colony-stimulating factor with or without plerixafor, according to institutional standards; if unsuccessful, cyclophosphamide-based mobilization was permitted. Patients then received high-dose therapy (melphalan 200 mg/m2) followed by ASCT. Consolidation began 60 to 100 days after ASCT, when the patient had recovered sufficiently per investigator’s discretion. Maintenance therapy began within 14 days following postconsolidation disease evaluation.
End points and assessments
The primary end point was the sCR rate by the end of post-ASCT consolidation, systematically assessed by a validated computer algorithm in accordance with IMWG criteria9,29 (supplemental Table 1) within 7 days after completion of consolidation. As daratumumab may interfere with serum immunofixation, reflex assays were performed to confirm responses of CR or better.30 Patients with confirmed daratumumab interference on serum M-protein electrophoresis and immunofixation who demonstrated all other criteria for CR or sCR were regarded as having CR or sCR, respectively.
The secondary end point of the MRD-negativity rate was defined as the proportion of patients who achieved MRD negativity, using a minimum sensitivity threshold of 1 in 105 nucleated cells or higher, in accordance with IMWG criteria.9 MRD was evaluated by next-generation sequencing (NGS; clonoSEQ Assay 2.0)31 of bone marrow aspirates at baseline and first evidence of suspected CR or sCR as recommended per IMWG criteria (including patients with very good partial response [VGPR] or better and suspected daratumumab interference). Following protocol amendment 2, MRD assessments were scheduled for the end of induction and consolidation, and after 12 and 24 months of maintenance, regardless of response. Patients were considered MRD positive if the MRD assessment was positive, indeterminate, or unavailable. MRD was analyzed in the intent-to-treat population, in the subgroup of patients who achieved a response of CR or better, and in the MRD-evaluable population (patients who had both baseline [clone identified/calibrated] and post-baseline MRD [with negative, positive, or indeterminate result] samples taken). Additional secondary end points, including complete definitions, are provided in the supplemental Appendix.
Adverse events (AEs) were monitored continuously from informed consent through 30 days after last study treatment and graded according to National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03.
Statistical analysis
The prespecified primary analysis occurred after all randomized patients completed the post-ASCT consolidation disease evaluation or discontinued from study treatment before this time point. A second protocol-specified analysis will be performed after all randomized patients complete the maintenance phase or discontinue from study treatment before completing maintenance.
The primary hypothesis was that patients in the D-RVd group would have an improved rate of sCR by the end of post-ASCT consolidation compared with the RVd group (primary end point), tested at a 1-sided α of 0.10. All secondary analyses were evaluated using a 2-sided P value (α 0.05) and were not adjusted for multiplicity. Historical data suggest that the sCR rate by the end of consolidation is ∼35% for RVd therapy. To detect an absolute 15% increase in the sCR rate with 80% power using a 1-sided likelihood ratio test with a 0.1 significance level, 200 patients were required for 1:1 randomization, assuming a 5% nonevaluable rate.
Results
Patients and treatment
Among 292 patients screened, 16 were enrolled in the safety run-in phase28 (results previously reported) and 207 were randomized in part 2 and are reported in this analysis (D-RVd, n = 104; RVd, n = 103; supplemental Figure 2). Patients who failed screening (n = 69) did not meet inclusion and/or exclusion criteria predefined by the protocol; the most common reasons (occurring in >5 patients) were failure to provide signed informed consent, inability to demonstrate documented MM/measurable disease at baseline, clinically significant cardiac disease, and abnormal laboratory test results. In part 2 of GRIFFIN, 201 patients received ≥1 dose of study treatment and were analyzed for safety (D-RVd, n = 99; RVd, n = 102). The primary efficacy analysis group consisted of 196 patients (D-RVd, n = 99; RVd, n = 97) who had measurable disease at baseline, received ≥1 study treatment dose, and underwent ≥1 disease assessment. Demographics and clinical characteristics were well balanced between randomized groups (Table 1). Median age was 60 years (range, 29-70 years); median time since diagnosis was 0.8 months (range, 0-61 months). Nearly one-half of all patients (47.8%) had ISS stage I disease, and 30 patients (15.4%) had high-risk cytogenetic abnormalities (del17p, t[4;14], or t[14;16] by fluorescence in situ hybridization [FISH]).
Patient demographic and disease characteristics in the intent-to-treat population at baseline
| D-RVd | RVd | |
|---|---|---|
| Age, y | n = 104 | n = 103 |
| Median (range) | 59 (29-70) | 61 (40-70) |
| Category, n (%) | ||
| <65 | 76 (73.1) | 75 (72.8) |
| ≥65 | 28 (26.9) | 28 (27.2) |
| Sex, n (%) | n = 104 | n = 103 |
| Male | 58 (55.8) | 60 (58.3) |
| Female | 46 (44.2) | 43 (41.7) |
| ECOG performance status, n (%)* | n = 101 | n = 102 |
| 0 | 39 (38.6) | 40 (39.2) |
| 1 | 51 (50.5) | 52 (51.0) |
| 2 | 11 (10.9) | 10 (9.8) |
| ISS disease stage, n (%)† | n = 104 | n = 103 |
| I | 49 (47.1) | 50 (48.5) |
| II | 40 (38.5) | 37 (35.9) |
| III | 14 (13.5) | 14 (13.6) |
| Missing | 1 (1.0) | 2 (1.9) |
| Baseline creatinine clearance, mL/min, n (%) | n = 104 | n = 103 |
| 30-50 | 9 (8.7) | 9 (8.7) |
| >50 | 95 (91.3) | 94 (91.3) |
| Cytogenetic risk profile, n (%)‡ | n = 98 | n = 97 |
| Standard | 82 (83.7) | 83 (85.6) |
| High risk | 16 (16.3) | 14 (14.4) |
| Time since diagnosis of MM, mo | n = 103 | n = 102 |
| Median (range) | 0.7 (0-12) | 0.9 (0-61) |
| D-RVd | RVd | |
|---|---|---|
| Age, y | n = 104 | n = 103 |
| Median (range) | 59 (29-70) | 61 (40-70) |
| Category, n (%) | ||
| <65 | 76 (73.1) | 75 (72.8) |
| ≥65 | 28 (26.9) | 28 (27.2) |
| Sex, n (%) | n = 104 | n = 103 |
| Male | 58 (55.8) | 60 (58.3) |
| Female | 46 (44.2) | 43 (41.7) |
| ECOG performance status, n (%)* | n = 101 | n = 102 |
| 0 | 39 (38.6) | 40 (39.2) |
| 1 | 51 (50.5) | 52 (51.0) |
| 2 | 11 (10.9) | 10 (9.8) |
| ISS disease stage, n (%)† | n = 104 | n = 103 |
| I | 49 (47.1) | 50 (48.5) |
| II | 40 (38.5) | 37 (35.9) |
| III | 14 (13.5) | 14 (13.6) |
| Missing | 1 (1.0) | 2 (1.9) |
| Baseline creatinine clearance, mL/min, n (%) | n = 104 | n = 103 |
| 30-50 | 9 (8.7) | 9 (8.7) |
| >50 | 95 (91.3) | 94 (91.3) |
| Cytogenetic risk profile, n (%)‡ | n = 98 | n = 97 |
| Standard | 82 (83.7) | 83 (85.6) |
| High risk | 16 (16.3) | 14 (14.4) |
| Time since diagnosis of MM, mo | n = 103 | n = 102 |
| Median (range) | 0.7 (0-12) | 0.9 (0-61) |
The intent-to-treat population was defined as all randomized patients. Informal testing showed no differences between the 2 treatment groups in the characteristics evaluated at baseline.
ECOG performance status is scored on a scale from 0 to 5, with 0 indicating no symptoms and higher scores indicating increasing disability.
The ISS disease stage is based on the combination of serum β2-microglobulin and albumin levels. Higher stages indicate more advanced disease.
Cytogenetic risk was assessed by FISH (local testing); high risk was defined as the presence of del17p, t(4;14), or t(14;16) among patients with available cytogenetic risk data.
Ninety-eight patients (94.2%) in the D-RVd group and 94 (91.3%) in the RVd group completed induction, 95 (91.3%) and 80 (77.7%) underwent stem cell mobilization, 94 (90.4%) and 78 (75.7%) underwent ASCT, and 91 (87.5%) and 72 (69.9%) completed consolidation, respectively (supplemental Figure 2). Ninety patients (86.5%) and 70 patients (68.0%) in the D-RVd and RVd groups, respectively, entered maintenance. Seventeen patients (16.3%) in the D-RVd group discontinued study treatment, compared with 44 (42.7%) in the RVd group. The most common reasons for discontinuation were investigator-assessed progressive disease (PD; D-RVd, 6.7%; RVd, 10.7%), patient withdrawal (3.8%; 9.7%), AEs (2.9%; 10.7%), and investigator discretion (2.9%; 8.7%). In the D-RVd group, fewer patients discontinued during induction (1.9% vs 6.8%) and after induction but before mobilization (2.9% vs 13.6%) than in the RVd group (supplemental Figure 2). All 4 response-evaluable patients in the D-RVd group who completed induction but did not undergo ASCT achieved VGPR or better. Of the 15 response-evaluable patients in the RVd group who completed induction but did not undergo ASCT, 3 achieved VGPR or better, 4 had a partial response (PR), and 8 had stable disease (SD). Most patients who discontinued study treatment with RVd received subsequent therapy (supplemental Table 2).
The prespecified primary analysis occurred at a median follow-up of 13.5 months. At the last follow-up, with a median of 22.1 months (range, 0-30.0 months), the median duration of treatment was 21.9 months (range, 1.1-29.7 months) in the D-RVd group and 19.5 months (range, 0.5-29.7 months) in the RVd group. Median relative dose intensities for RVd were 91.6% or greater and similar in both groups (supplemental Table 3).
Efficacy
The primary end point of sCR by the end of post-ASCT consolidation was achieved in 42 patients (42.4%) in the D-RVd group and 31 patients (32.0%) in the RVd group (odds ratio, 1.57; 95% confidence interval [CI], 0.87-2.82; 1-sided P = .068; Table 2), which was statistically significant at the preset 1-sided α of 0.10. Secondary end points of overall response rate (ORR; 99.0% vs 91.8%; P = .0160) and rate of VGPR or better (90.9% vs 73.2%; P = .0014) by the end of consolidation were higher in the D-RVd group (Table 2). Responses continued to deepen over time, and at the last follow-up (median follow-up, 22.1 months; Figure 1A), the percentage of patients with sCR was 62.6% in the D-RVd group and 45.4% in the RVd group (odds ratio, 1.98; 95% CI, 1.12-3.49; P = .0177). Moreover, the rate of CR or better was 79.8% in the D-RVd group vs 60.8% in the RVd group (odds ratio, 2.53; 95% CI, 1.33-4.81; P = .0045).
Primary analysis: summary of responses by the end of consolidation
| D-RVd, n (%) | RVd, n (%) | Odds ratio (95% CI)* | P | |
|---|---|---|---|---|
| Best response† | n = 99 | n = 97 | ||
| ORR | 98 (99.0) | 89 (91.8) | 8.75 (1.08, 71.01) | .0160‡ |
| sCR | 42 (42.4) | 31 (32.0) | 1.57 (0.87, 2.82) | .0680‡,§ |
| ≥CR | 51 (51.5) | 41 (42.3) | ||
| CR | 9 (9.1) | 10 (10.3) | ||
| ≥VGPR | 90 (90.9) | 71 (73.2) | 3.53 (1.55, 8.00) | .0014‡ |
| VGPR | 39 (39.4) | 30 (30.9) | ||
| PR | 8 (8.1) | 18 (18.6) | ||
| SD | 1 (1.0) | 7 (7.2) | ||
| PD | 0 | 1 (1.0) |
| D-RVd, n (%) | RVd, n (%) | Odds ratio (95% CI)* | P | |
|---|---|---|---|---|
| Best response† | n = 99 | n = 97 | ||
| ORR | 98 (99.0) | 89 (91.8) | 8.75 (1.08, 71.01) | .0160‡ |
| sCR | 42 (42.4) | 31 (32.0) | 1.57 (0.87, 2.82) | .0680‡,§ |
| ≥CR | 51 (51.5) | 41 (42.3) | ||
| CR | 9 (9.1) | 10 (10.3) | ||
| ≥VGPR | 90 (90.9) | 71 (73.2) | 3.53 (1.55, 8.00) | .0014‡ |
| VGPR | 39 (39.4) | 30 (30.9) | ||
| PR | 8 (8.1) | 18 (18.6) | ||
| SD | 1 (1.0) | 7 (7.2) | ||
| PD | 0 | 1 (1.0) |
The primary analysis occurred on 25 January 2019 (median follow-up, 13.5 months) after all randomized patients completed the post-ASCT disease evaluation or discontinued from study treatment by this time point.
Mantel-Haenszel estimate of the common odds ratio for stratified tables is used. The stratification factors are ISS stage (I, II, III) and creatinine clearance (CrCl [30-50 mL/min or >50 mL/min]) at randomization. An odds ratio >1 indicates an advantage for the daratumumab group.
Responses were assessed according to the IMWG recommendations by computer algorithm (details on the response criteria are provided in supplemental Table 1). This analysis included all patients in the response-evaluable population of all randomized patients who had a confirmed diagnosis of MM, measurable disease at baseline, received ≥1 dose of study treatment, and had ≥1 postbaseline disease assessment.
P values were calculated with the use of the Cochran-Mantel-Haenszel χ2 test.
A 1-sided P value is reported for the primary end point (postconsolidation sCR); all other responses were calculated using a 2-sided P value not adjusted for multiplicity.
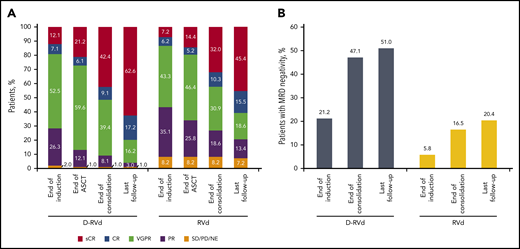
Summary of response rates and MRD-negativity (10−5) rates over time. (A) Response rates over time are shown. Data for the end of induction, end of ASCT, and end of consolidation are from the primary analysis. Response data with longer median follow-up of 22.1 months are also shown (last follow-up). (B) MRD-negativity (10−5) rates in the intent-to-treat population by the end of induction therapy, end of consolidation, and last follow-up. All MRD data are from the analysis with a median follow-up of 22.1 months. MRD was evaluated at baseline, first evidence of suspected CR or sCR, at the end of induction and consolidation, and after 12 and 24 months of maintenance, regardless of response (per protocol amendment 2).
Summary of response rates and MRD-negativity (10−5) rates over time. (A) Response rates over time are shown. Data for the end of induction, end of ASCT, and end of consolidation are from the primary analysis. Response data with longer median follow-up of 22.1 months are also shown (last follow-up). (B) MRD-negativity (10−5) rates in the intent-to-treat population by the end of induction therapy, end of consolidation, and last follow-up. All MRD data are from the analysis with a median follow-up of 22.1 months. MRD was evaluated at baseline, first evidence of suspected CR or sCR, at the end of induction and consolidation, and after 12 and 24 months of maintenance, regardless of response (per protocol amendment 2).
At a median follow-up of 22.1 months, the MRD-negativity (10−5) rate was higher in the D-RVd group than in the RVd group in the intent-to-treat population (51.0% [53 of 104 patients] vs 20.4% [21 of 103 patients]; P < .0001), as well as among patients who achieved CR or better (62.0% [49 of 79 patients] vs 32.2% [19 of 59 patients]; P = .0006), and among the MRD-evaluable population (68.8% [53 of 77 patients] vs 32.3% [21 of 65 patients]; P < .0001; Table 3). The proportion of patients in the intent-to-treat population who were MRD negative (10−5) and achieved CR or better also favored the D-RVd group (47.1% [49 of 104 patients] vs 18.4% [19 of 103 patients]; P < .0001; Table 3).
MRD status at last follow-up
| MRD-negative (10−5) status* | D-RVd, n (%) | RVd, n (%) | Odds ratio (95% CI)† | P‡ |
|---|---|---|---|---|
| Intent-to-treat population | ||||
| MRD-negative | 53/104 (51.0) | 21/103 (20.4) | 4.07 (2.18-7.59) | <.0001 |
| MRD-negative with ≥CR | 49/104 (47.1) | 19/103 (18.4) | 3.89 (2.07-7.33) | <.0001 |
| In patients achieving ≥CR | 49/79 (62.0) | 19/59 (32.2) | 3.57 (1.72-7.44) | .0006 |
| MRD-evaluable population | 53/77 (68.8) | 21/65 (32.3) | 4.47 (2.19-9.11) | <.0001 |
| MRD-negative (10−5) status* | D-RVd, n (%) | RVd, n (%) | Odds ratio (95% CI)† | P‡ |
|---|---|---|---|---|
| Intent-to-treat population | ||||
| MRD-negative | 53/104 (51.0) | 21/103 (20.4) | 4.07 (2.18-7.59) | <.0001 |
| MRD-negative with ≥CR | 49/104 (47.1) | 19/103 (18.4) | 3.89 (2.07-7.33) | <.0001 |
| In patients achieving ≥CR | 49/79 (62.0) | 19/59 (32.2) | 3.57 (1.72-7.44) | .0006 |
| MRD-evaluable population | 53/77 (68.8) | 21/65 (32.3) | 4.47 (2.19-9.11) | <.0001 |
MRD data are from last follow-up at a median follow-up of 22.1 months.
The threshold of MRD negativity was defined as 1 tumor cell per 105 white cells. MRD status is based on assessment of bone marrow aspirates by NGS in accordance with IMWG criteria.9 The MRD-evaluable population included patients who had both baseline (with clone identified/calibrated) and postbaseline MRD (with negative, positive, or indeterminate result) samples taken (D-RVd group, n = 77; RVd group, n = 65). Patients with a missing or inconclusive assessment were considered positive for MRD.
Mantel-Haenszel estimate of the common odds ratio for stratified tables is used. The stratification factors are ISS stage (I, II, III) and creatinine clearance (CrCl [30-50 mL/min or > 50 mL/min]) at randomization. An odds ratio >1 indicates an advantage for the daratumumab group.
P values were calculated from the Fisher exact test.
To assess the impact of the higher rate of early treatment discontinuation in the RVd group on MRD negativity, an analysis was performed for patients who underwent ASCT. MRD-negativity (10−5) rates were higher in D-RVd patients undergoing ASCT compared with RVd patients undergoing ASCT (55.3% [52 of 94 patients] vs 25.6% [20 of 78 patients]; P < .0001).
The higher MRD-negativity (10−5) rates of the D-RVd regimen were observed as early as the end of induction in the intent-to-treat population (21.2% [22 of 104 patients] vs 5.8% [6 of 103 patients]; P = .0019; Figure 1B) and among patients who achieved CR or better by the end of induction (47.4% [9 of 19 patients] vs 7.7% [1 of 13 patients]; P = .0237). Moreover, MRD-negativity rates using a higher sensitivity threshold of 10−6 also favored the D-RVd group at a median of 22.1 months follow-up: 18.3% (19 of 104 patients) vs 6.8% (7 of 103 patients; P = .0197), including among patients who reached CR or better (24.1% [19 of 79 patients] vs 10.2% [6 of 59 patients]; P = .0447).
Subgroup analyses for prespecified factors were conducted for the primary efficacy end point at a median follow-up of 13.5 months (Figure 2A) and for MRD negativity at a median follow-up of 22.1 months (Figure 2B). For the prespecified primary end point, D-RVd vs RVd improved the sCR rate by the end of post-ASCT consolidation in all subgroups except for patients with ISS stage III disease or high-risk cytogenetic abnormalities (Figure 2A). At both the time of the primary analysis (data not shown) and after 22.1 months of median follow-up, the subgroup analysis of MRD negativity (10−5) favored D-RVd in all analyzed subgroups (Figure 2B) but was not statistically significant for ISS stage III disease or high-risk cytogenetic abnormalities.
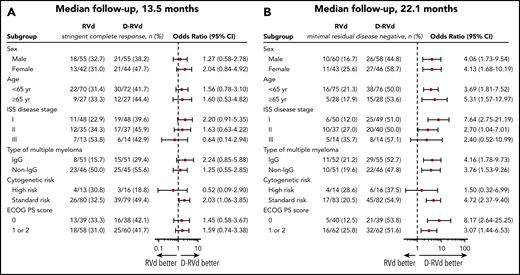
Subgroup analysis of sCR by the end of post-ASCT consolidation (primary end point; median follow-up, 13.5 months) and subgroup analysis of MRD negativity (10−5) by last follow-up (median follow-up, 22.1 months). (A) sCR by the end of post-ASCT consolidation (primary end point; median follow-up, 13.5 months). Analysis of sCR for the primary end point in prespecified subgroups of the response-evaluable population that were defined according to baseline characteristics. (B) MRD negativity by last follow-up (median follow-up, 22.1 months). Analysis of MRD by last follow-up in subgroups of the intent-to-treat population. The ISS disease stage is derived based on the combination of serum β2-microglobulin and albumin levels, with higher stages indicating more advanced disease. The subgroup analysis for the type of multiple myeloma was performed on data from patients who had measurable disease in serum. A high-risk cytogenetic profile was defined by the detection of a del17p, t(4;14), and/or t(14;16) cytogenetic abnormality on FISH testing. ECOG performance status (PS) is scored on a scale from 0 to 5, with 0 indicating no symptoms and higher scores indicating increasing disability.
Subgroup analysis of sCR by the end of post-ASCT consolidation (primary end point; median follow-up, 13.5 months) and subgroup analysis of MRD negativity (10−5) by last follow-up (median follow-up, 22.1 months). (A) sCR by the end of post-ASCT consolidation (primary end point; median follow-up, 13.5 months). Analysis of sCR for the primary end point in prespecified subgroups of the response-evaluable population that were defined according to baseline characteristics. (B) MRD negativity by last follow-up (median follow-up, 22.1 months). Analysis of MRD by last follow-up in subgroups of the intent-to-treat population. The ISS disease stage is derived based on the combination of serum β2-microglobulin and albumin levels, with higher stages indicating more advanced disease. The subgroup analysis for the type of multiple myeloma was performed on data from patients who had measurable disease in serum. A high-risk cytogenetic profile was defined by the detection of a del17p, t(4;14), and/or t(14;16) cytogenetic abnormality on FISH testing. ECOG performance status (PS) is scored on a scale from 0 to 5, with 0 indicating no symptoms and higher scores indicating increasing disability.
At a median follow-up of 22.1 months, 4 events (3.8%) of disease progression occurred in the D-RVd group compared with 7 events (6.8%) for the RVd group. Median PFS was not reached in either group. The Kaplan-Meier estimate of the 24-month PFS rate was 95.8% (95% CI, 89.2-98.4) in the D-RVd group and 89.8% (95% CI, 77.1-95.7) in the RVd group (Figure 3). Median OS was not reached in either group, and follow-up for long-term survival is ongoing.
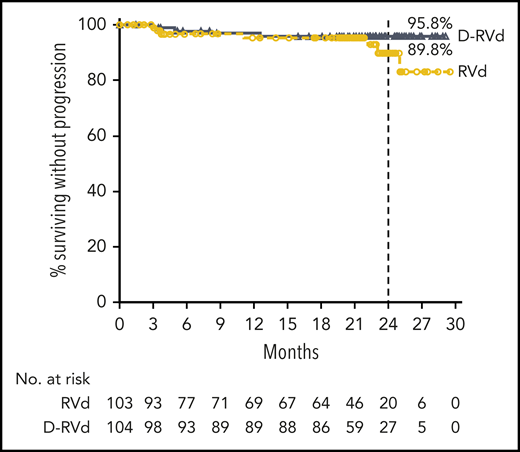
PFS. Results of the Kaplan-Meier estimates of PFS are shown among patients in the intent-to-treat population. Median PFS was not reached for either group, and Kaplan-Meier estimates of 24-month PFS rates are shown.
PFS. Results of the Kaplan-Meier estimates of PFS are shown among patients in the intent-to-treat population. Median PFS was not reached for either group, and Kaplan-Meier estimates of 24-month PFS rates are shown.
Safety
The most common AEs of any grade (occurring in ≥30% of patients in either group) are summarized in Table 4. The most common (≥10%) grade 3/4 AEs were neutropenia (D-RVd, 41.4%; RVd, 21.6%), lymphopenia (23.2%; 21.6%), thrombocytopenia (16.2%; 8.8%), leukopenia (16.2%; 6.9%), and pneumonia (8.1%; 10.8%; Table 4).
Most common adverse events reported during treatment in the safety population
| Adverse event, n (%) | D-RVd, n = 99 | RVd, n = 102 | |||
|---|---|---|---|---|---|
| Any grade | Grade 3/4 | Any grade | Grade 3/4 | ||
| Hematologic | |||||
| Neutropenia | 57 (57.6) | 41 (41.4) | 36 (35.3) | 22 (21.6) | |
| Thrombocytopenia | 43 (43.4) | 16 (16.2) | 36 (35.3) | 9 (8.8) | |
| Leukopenia | 36 (36.4) | 16 (16.2) | 29 (28.4) | 7 (6.9) | |
| Anemia | 35 (35.4) | 9 (9.1) | 33 (32.4) | 6 (5.9) | |
| Lymphopenia | 30 (30.3) | 23 (23.2) | 28 (27.5) | 22 (21.6) | |
| Nonhematologic | |||||
| Fatigue | 68 (68.7) | 6 (6.1) | 62 (60.8) | 6 (5.9) | |
| Upper respiratory tract infection | 62 (62.6) | 1 (1.0) | 45 (44.1) | 2 (2.0) | |
| Peripheral neuropathy* | 59 (59.6) | 7 (7.1) | 74 (72.5) | 8 (7.8) | |
| Diarrhea | 59 (59.6) | 7 (7.1) | 51 (50.0) | 4 (3.9) | |
| Constipation | 51 (51.5) | 2 (2.0) | 40 (39.2) | 1 (1.0) | |
| Cough | 50 (50.5) | 0 | 27 (26.5) | 0 | |
| Nausea | 49 (49.5) | 2 (2.0) | 50 (49.0) | 1 (1.0) | |
| Pyrexia | 45 (45.5) | 2 (2.0) | 28 (27.5) | 3 (2.9) | |
| Insomnia | 42 (42.4) | 2 (2.0) | 31 (30.4) | 1 (1.0) | |
| Back pain | 36 (36.4) | 1 (1.0) | 34 (33.3) | 4 (3.9) | |
| Peripheral edema | 34 (34.3) | 2 (2.0) | 35 (34.3) | 3 (2.9) | |
| Arthralgia | 33 (33.3) | 0 | 33 (32.4) | 2 (2.0) | |
| Infusion-related reaction | 42 (42.4) | 6 (6.1)† | NA | NA | |
| Adverse event, n (%) | D-RVd, n = 99 | RVd, n = 102 | |||
|---|---|---|---|---|---|
| Any grade | Grade 3/4 | Any grade | Grade 3/4 | ||
| Hematologic | |||||
| Neutropenia | 57 (57.6) | 41 (41.4) | 36 (35.3) | 22 (21.6) | |
| Thrombocytopenia | 43 (43.4) | 16 (16.2) | 36 (35.3) | 9 (8.8) | |
| Leukopenia | 36 (36.4) | 16 (16.2) | 29 (28.4) | 7 (6.9) | |
| Anemia | 35 (35.4) | 9 (9.1) | 33 (32.4) | 6 (5.9) | |
| Lymphopenia | 30 (30.3) | 23 (23.2) | 28 (27.5) | 22 (21.6) | |
| Nonhematologic | |||||
| Fatigue | 68 (68.7) | 6 (6.1) | 62 (60.8) | 6 (5.9) | |
| Upper respiratory tract infection | 62 (62.6) | 1 (1.0) | 45 (44.1) | 2 (2.0) | |
| Peripheral neuropathy* | 59 (59.6) | 7 (7.1) | 74 (72.5) | 8 (7.8) | |
| Diarrhea | 59 (59.6) | 7 (7.1) | 51 (50.0) | 4 (3.9) | |
| Constipation | 51 (51.5) | 2 (2.0) | 40 (39.2) | 1 (1.0) | |
| Cough | 50 (50.5) | 0 | 27 (26.5) | 0 | |
| Nausea | 49 (49.5) | 2 (2.0) | 50 (49.0) | 1 (1.0) | |
| Pyrexia | 45 (45.5) | 2 (2.0) | 28 (27.5) | 3 (2.9) | |
| Insomnia | 42 (42.4) | 2 (2.0) | 31 (30.4) | 1 (1.0) | |
| Back pain | 36 (36.4) | 1 (1.0) | 34 (33.3) | 4 (3.9) | |
| Peripheral edema | 34 (34.3) | 2 (2.0) | 35 (34.3) | 3 (2.9) | |
| Arthralgia | 33 (33.3) | 0 | 33 (32.4) | 2 (2.0) | |
| Infusion-related reaction | 42 (42.4) | 6 (6.1)† | NA | NA | |
The safety analysis population included all randomized patients who received ≥1 dose of study treatment; analysis was according to treatment received. Adverse events of any grade that are listed are those that occurred in 30% or more of the patients in either group. The safety analysis occurred at a median follow-up of 22.1 months.
NA, not applicable.
Includes patients with neuropathy peripheral and peripheral sensory neuropathy.
There were no grade 4 infusion-related reactions.
Serious AEs were reported in 39 patients (39.4%) in the D-RVd group and 52 (51.0%) in the RVd group; the most common (occurring in ≥5% in either group) were pyrexia (D-RVd, 10.1%; RVd, 7.8%) and pneumonia (9.1%; 10.8%). The percentage of patients with AEs leading to treatment discontinuation was 15.2% in the D-RVd group and 20.6% in the RVd group, most commonly (≥5%) peripheral sensory neuropathy (D-RVd, 5.1%; RVd, 3.9%). One AE in the RVd group had an outcome of death, which was not related to study drug. In the D-RVd group, no AEs led to death.
Infections of any grade were more common in the D-RVd group compared with the RVd group (90.9% vs 61.8%), largely due to more grade 1/2 upper respiratory tract infections in the D-RVd group (supplemental Table 4); however, the incidence of grade 3/4 infections was similar between groups (23.2% vs 21.6%), as was the percentage of patients with infections leading to discontinuation (D-RVd, 2.0%; RVd, 2.9%). Four second primary malignancies were observed in the D-RVd group (3 squamous cell carcinoma and 1 prostate cancer) and 1 secondary primary malignancy was observed in the RVd group (melanoma).
Infusion-related reactions (IRRs) to daratumumab occurred in 42 patients (42.4%) and were generally mild, with grade 3 events occurring in 6 patients (6.1%); no grade 4/5 events occurred (Table 4). Most patients with an IRR experienced the reaction during the first infusion (35 of 99; 35.4%), with 2 patients (2.0%) and 11 patients (11.1%) experiencing reactions during the second and subsequent infusions, respectively. Three patients (3.0%) experienced an IRR in the first D-RVd consolidation cycle following ASCT. No IRRs led to treatment discontinuation.
Median CD34+ cell yield was 8.2 × 106/kg (range, 2.6 × 106 to 33.0 × 106) for the D-RVd group and 9.4 × 106/kg (range, 4.1 × 106 to 28.7 × 106) for the RVd group. Median CD34+ cells transplanted were 4.2 × 106 in the D-RVd group and 4.8 × 106 in the RVd group. Hematopoietic reconstitution was comparable for both groups; the median number of days for neutrophil and platelet engraftment were 12 and 13 for the D-RVd group, and 12 and 12 for the RVd group, respectively. More patients undergoing stem cell mobilization and collection in the D-RVd group received plerixafor compared with the RVd group (69.5% [66 of 95 patients] vs 56.3% [45 of 80 patients]); cyclophosphamide use was low in both groups (≤5.3%).
Discussion
In this randomized phase 2 study, addition of daratumumab to RVd for transplant-eligible NDMM improved response rates and depth of response, including sCR and MRD-negativity (10−5) rates. Responses deepened with continued therapy in both groups, in accordance with previous observations.21,22,25,27,32
These findings are consistent with the benefit demonstrated in CASSIOPEIA, in which addition of daratumumab to bortezomib, thalidomide, and dexamethasone improved the rates of sCR and MRD negativity (10−5) in transplant-eligible NDMM.21 In GRIFFIN and CASSIOPEIA, the odds ratios for sCR were nearly identical (1.57 and 1.60, respectively), indicating that the addition of daratumumab to a standard-of-care regimen including an immunomodulatory drug, proteasome inhibitor, and dexamethasone achieves deeper responses. Improved long-term outcomes of OS and PFS have been previously reported with the achievement of sCR after ASCT in MM patients.8 It should be noted that the improvement in sCR seen in the daratumumab arm of CASSIOPEIA was associated with an improvement in median PFS; however, differences in study design, including a second randomization to maintenance or observation in CASSIOPEIA, confound any comparisons between the 2 studies. Longer follow-up is needed to confirm whether a PFS benefit will be achieved in GRIFFIN.
Reported CR rates from prior clinical studies including RVd for induction and/or consolidation therapy with ASCT were generally higher compared with the rate in the RVd arm of GRIFFIN; CR (or ≥CR) rates ranged from 50% to 78% after consolidation.7,33,34 However, it is difficult to make cross-trial comparisons due to differences in study designs, patient populations, and assessment methodologies. For example, these studies varied in the duration of induction therapy (ranging from three 21-day cycles to six 28-day cycles), use of chemomobilization with cyclophosphamide (permitted in GRIFFIN only if mobilization with granulocyte colony-stimulating factor with or without plerixafor was not successful), and the duration and timing of initiation of consolidation. Differences in duration and timing of induction and consolidation impact the duration of follow-up at the postconsolidation assessment, which is an important point, as responses are expected to deepen over time. Nevertheless, the randomized GRIFFIN study demonstrates that the addition of daratumumab to RVd improves and deepens responses compared with RVd therapy.
Achievement of MRD negativity has also been associated with superior long-term patient outcomes, resulting in increasing interest in this end point as a surrogate marker for PFS and OS.7,10-13 Importantly, GRIFFIN demonstrated that addition of daratumumab to RVd and ASCT improved MRD negativity at the standard sensitivity threshold of 10−5 and the more stringent threshold of 10−6. Improvements in MRD-negativity rates for the D-RVd group vs the RVd group were seen as early as at the end of induction (21.2% vs 5.8%), and continued to deepen at the end of consolidation (47.1% vs 16.5%) and by the last follow-up (51.0% vs 20.4%). The results from GRIFFIN confirm previous findings that the addition of daratumumab to standard-of-care regimens across lines of therapy improves MRD negativity in myeloma,21-23,25,27 which has consistently translated into improved PFS, whether the backbone regimen was used for a defined duration21,22,26,27 or until disease progression.23-25
One limitation of our study is that the proportion of patients with ISS stage I disease in GRIFFIN was higher than in other studies of NDMM patients eligible for transplant.7,21,33 Additionally, the number of patients with high-risk cytogenetics was low. Nonetheless, the proportions of high-risk patients in GRIFFIN were well balanced between treatment groups and similar to other clinical studies that evaluated patients with transplant-eligible NDMM.7,21,34 In this context, subgroup analyses in GRIFFIN demonstrated that D-RVd improved sCR rates across most of the subgroups for both the primary end point and at a median follow-up time of 22.1 months, except for patients with ISS stage III disease or high-risk cytogenetics. The subgroup analysis for MRD negativity showed that D-RVd favored all subgroups. Although these findings are informative, the small number of patients in these categories limits interpretation of the effect of daratumumab for these groups. Additional studies of D-RVd as therapy for patients with high-risk NDMM are needed to better understand its potential impact in these patients.
The MRD-negativity rate for the RVd arm of the GRIFFIN trial was lower than that reported in the IFM 2009 trial, although direct comparisons are confounded by differences in the methods and timing of MRD assessments, as well as eligibility criteria and study design. The primary IFM 2009 analysis reported an MRD-negativity rate of 79% but used a flow cytometric-based MRD assay with a sensitivity of 10−4.7 A subsequent IFM 2009 MRD analysis using NGS (10−6 threshold) by the end of lenalidomide therapy reported an MRD-negativity rate of 30%, but this included only a subset of patients who entered the maintenance phase of the trial.13 Moreover, all IFM 2009 patients in VGPR or better were tested for MRD, whereas testing in GRIFFIN was initially only performed at the time of suspected CR or sCR in accordance with IMWG guidelines (additional time points added in amendment 2). Although GRIFFIN critically demonstrates that the addition of daratumumab to RVd improves the rate of MRD negativity in transplant-eligible NDMM patients, longer follow-up is needed to determine whether this results in a survival advantage, as in other studies.
A lower percentage of patients underwent transplant in the RVd arm (75.7%) vs the D-RVd arm (90.4%). It is worth noting that this imbalance was due to a higher rate of treatment discontinuation in the RVd group compared with the D-RVd group. Thirteen patients (12.6%) assigned to RVd discontinued therapy prior to ASCT due to investigator-assessed PD or investigator discretion, both of which can be viewed as treatment failures. The high proportion of RVd patients with SD or PR who did not undergo ASCT on study further highlights the increased rate of suboptimal responses in the RVd arm of the trial. Furthermore, D-RVd already achieved higher rates of sCR (12.1% vs 7.2%), VGPR or better (71.7% vs 56.7%), and MRD negativity (21.2% vs 5.8%) at the end of induction. Lastly, in an analysis of patients who completed ASCT, MRD negativity remained superior for D-RVd (55.3% vs 25.6%). These results suggest that daratumumab played a critical role in deepening responses.
Adding daratumumab to RVd did not lead to unexpected safety concerns. AEs were consistent with the known safety profiles for RVd4-7 as well as daratumumab,21-24,26,35,36 and consistent with observations from the safety run-in.28 The D-RVd group had higher rates of grade 3/4 hematologic events, consistent with previous phase 3 studies. Interestingly, the RVd group experienced a larger number of serious AEs. There is no clear explanation for this finding, as no single serious AE or group of serious AEs occurred more frequently in the RVd group vs the D-RVd group. However, it is possible that the open-label study design and relatively small number of patients in this randomized phase 2 study may have been contributing factors. The rate of discontinuation was lower in the D-RVd group, and no AEs led to death in the D-RVd group. The incidence and severity of IRRs was consistent with other daratumumab studies.35,36 Although more neutropenia and grade 1/2 infections were observed in the D-RVd arm, there was no difference in grade 3/4 infections or treatment discontinuations due to infection, confirming previous study findings that any increased infection risk with daratumumab is clinically manageable.
The results from GRIFFIN demonstrate that stem cell mobilization and ASCT are feasible when daratumumab is added to induction therapy for myeloma. These results are consistent with those in CASSIOPEIA; however, differences in mobilization strategies in these 2 studies limit direct comparison of stem cell yield data. Stem cell mobilization in GRIFFIN occurred as per institutional guidelines but without chemotherapy unless the initial mobilization was unsuccessful. The study allowed centers to follow institutional guidelines for the use of plerixafor, which was more common in patients undergoing stem cell mobilization in the D-RVd group compared with the RVd group (69.5% vs 56.3%). A similarly low number of patients in both treatment groups received cyclophosphamide for mobilization (5.3% vs 5.0%), indicating that mobilization without chemotherapy was successful. The median stem cell yield, although numerically lower for the D-RVd group (8.2 vs 9.4 × 106 cells/kg), was sufficient to allow cryopreservation of enough stem cells for a potential future second transplant. In addition, time to engraftment and hematopoietic recovery were not impacted by daratumumab.
Study results from GRIFFIN are promising and practice informing; this randomized phase 2 study was designed to expediently provide efficacy and safety information on a new regimen of great interest to myeloma clinicians. The aim of this study was to inform clinical decision-making, as well as design of a definitive registrational trial. As a phase 2 study, GRIFFIN was not intended to evaluate and does not provide sufficient power to analyze small patient subgroups, such as those with high-risk cytogenetics. The positive, practice-informing results from GRIFFIN support and provide a strong rationale for the ongoing phase 3 PERSEUS registration study (ClinicalTrials.gov Identifier, NCT03710603), which will determine whether subcutaneous daratumumab in combination with RVd improves PFS in transplant-eligible NDMM.
In summary, the addition of daratumumab to RVd in transplant-eligible NDMM patients resulted in improved rates of sCR and MRD negativity, an acceptable safety profile, and no clinically significant impact on stem cell mobilization or engraftment. These results indicate that the combination of D-RVd may be a potential new standard of care for transplant-eligible NDMM.
The data-sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through the Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The GRIFFIN team acknowledges the contributions of all principal investigators who participated in the study: Carlos Alemany, Larry D. Anderson Jr, Ashraf Badros, Ajai Chari, Robert Cornell, Luciano J. Costa, Caitlin Costello, Andrew J. Cowan, Binod Dhakal, Yvonne A. Efebera, Laura Finn, Cristina Gasparetto, Sarah A. Holstein, Andrzej Jakubowiak, Jonathan L. Kaufman, Hakan Kaya, Sarah Larson, Jacob Laubach, Tomer Mark, Nitya Nathwani, Ruben Niesvizky, Joanne Filicko-O’Hara, Robert Z. Orlowski, Brandi Reeves, Paul G. Richardson, Cesar Rodriguez, Douglas W. Sborov, Nina Shah, Kenneth H. Shain, Leyla Shune, Rebecca Silbermann, Pallawi Torka, Peter M. Voorhees, Andrew Whiteley, Tanya M. Wildes, and Jeffrey Zonder. The team also acknowledges the Alliance Multiple Myeloma Committee, particularly Phil McCarthy and patient advocate Jim Omel, for their input and support of the study. Lastly, the team thanks the Alliance Foundation Trials team for their key role in the conduct of this trial (https://acknowledgments.alliancefound.org). The authors thank the patients who volunteered to participate in this trial, their families, and the staff members at the trial sites who cared for them; the members of the data review committee (Tracy McGowan [Chair], Leslie Killion, Daniela Hoehn, and Wayne Langholff) and safety monitoring committee (Carol Ann Huff [Chair], Adam Cohen, Weichung Shih, and Parexel International, LLC [Statistical Support Group]); representatives of the sponsor who were involved in data collection and analyses; and Charlotte Majerczyk and Melissa Brunckhorst of MedErgy for editorial assistance funded by the sponsor.
This work was supported by research funding from Janssen Oncology.
Authorship
Contribution: P.M.V., N.S., A.J., D.H., J.U., and P.G.R. contributed to study design and data acquisition and contributed to data analysis or interpretation; J.L.K., D.W.S., C.R., A.C., R.S., L.J.C., L.D.A., Y.A.E., S.A.H., C.C., T.M.W., R.Z.O., K.H.S., A.J.C., and J.V. contributed to data acquisition and contributed to data analysis or interpretation; S.M. and T.S.L. contributed to study design and contributed to data analysis or interpretation; Y.L., H.P., and C.d.B. contributed to data analysis or interpretation; J.L. contributed to study design; B.R. and N.N. contributed to data acquisition; and all authors reviewed the manuscript, approved the final version, decided to publish this report, and vouch for the data accuracy and completeness.
Conflict-of-interest disclosure: P.M.V. served in a consultancy or advisory role and received honoraria from Adaptive Biotechnologies, Bristol-Myers Squibb, Celgene, Janssen, Takeda, and TeneoBio; served in a consultancy or advisory role for Novartis, and Oncopeptides; served on a speakers’ bureau for Janssen and Amgen; and received research funding from Amgen, Celgene, Janssen, GlaxoSmithKline, Takeda, and TeneoBio. J.L.K. has held membership on an entity’s board of directors or advisory committee for Pharmacyclics and Karyopharm; has received honoraria from Janssen; served as a consultant for AbbVie, Takeda, Celgene, Amgen, Bristol-Myers Squibb, Incyte, and TG Therapeutics; and is an employee of Winship Cancer Institute of Emory University. D.W.S. served in a consultancy or advisory role for Amgen, Celgene, and Janssen; and received honoraria from Celgene. B.R. has served as a consultant for Incyte, Takeda, and Seattle Genetics; received honoraria from Celgene, Incyte, Takeda, and Seattle Genetics; and served on a speakers’ bureau for Celgene. C.R. has served in a consultancy role or on a speakers’ bureau for Celgene, Takeda, Janssen, Kite, Sanofi, and Bristol-Myers Squibb. A.C. has received research funding from, served as a consultant for, and served on an advisory board for Janssen, Celgene, Novartis, and Amgen; has served in a consultancy role for Bristol-Myers Squibb; has served on an advisory board for Karyopharm, Sanofi, and Oncopeptides; has received research funding and served on an advisory board for Seattle Genetics and Millennium Pharmaceuticals/Takeda; and has received research funding from Pharmacyclics. R.S. has served on an advisory committee for Janssen and on an advisory board for Sanofi. L.J.C. has received research funding from Janssen, Celgene, GlaxoSmithKline, and Amgen; received honoraria from Amgen, Celgene, Sanofi, GlaxoSmithKline, and Janssen; has served in a consultancy or advisory role for AbbVie, Amgen, Celgene, Sanofi, GlaxoSmithKline, and Karyopharm; and has served on a speakers’ bureau for Amgen, Sanofi, and Janssen. L.D.A. has served in a consultancy or advisory role for, served on a speakers’ bureau for, and received honoraria from Celgene, Takeda, Janssen, and Amgen. N.S. has received research funding from Celgene, Janssen, Bluebird Bio, Sutro Biopharma, and Poseida; has served in an advisory role or held membership on an entity’s board of directors for Bristol-Myers Squibb, Amgen, Kite, Nkarta, TeneoBio, Genentech, Seattle Genetics, Oncopeptides, Karyopharm, Surface Oncology, Precision BioSciences, GlaxoSmithKline, Nektar, Amgen, Indapta Therapeutics, and Sanofi; owns stock in Indapta Therapeutics; and has received research funding from Celgene, Janssen, Bluebird Bio, and Sutro Biopharma. Y.A.E. has served on a speakers’ bureau for Janssen; received honoraria from and served on an independent adjudication committee for Takeda; and served on an advisory board and speakers’ bureau for Akcea. S.A.H. has served in a consultancy or advisory role for and received honoraria from Adaptive Biotechnologies, Bristol-Myers Squibb, Celgene, Genentech, Oncopeptides, Sorrento, and Takeda; and has received research funding from Oncopeptides. C.C. has received honoraria from Takeda and Celgene; has served as a consultant for Celgene; and has received research funding from Takeda, Janssen, and Celgene. A.J. served as a consultant for, received honoraria from, or held membership on an entity’s board of directors or advisory committee for AbbVie, Amgen, Bristol-Myers Squibb, Celgene, GlaxoSmithKline, Janssen, Karyopharm Therapeutics, Millennium Pharmaceuticals, Sanofi, SkylineDx, and Takeda; and has served as a consultant for and received honoraria from Adaptive Biotechnologies and Juno. T.M.W. has received research funding from Janssen and served as a consultant for Carevive. R.Z.O. has received honoraria from or held membership on an entity’s board of directors or advisory committees for Amgen, Janssen, Bristol-Myers Squibb, Kite Pharma, Celgene, Ionis Pharmaceuticals, Legend Biotech, Molecular Partners, Sanofi-Aventis, Servier, Takeda, and Pharmaceuticals North America; and received research funding from Amgen, BioTheryX, and Spectrum Pharmaceuticals. K.H.S. has received research funding from AbbVie; has served as a consultant for Adaptive Biotechnologies; and has held membership on an entity’s board of directors or served in an advisory role for Celgene, Bristol-Myers Squibb, Amgen, Takeda, Janssen, and Sanofi Genzyme. A.J.C. has received research funding from and served as a consultant for Janssen and Celgene; has received research funding from AbbVie and Juno Therapeutics, a subsidiary of Celgene; and served in a consultancy role for Cellectar and Sanofi. Y.L. is an employee of Janssen. S.M., H.P., J.U., J.V., C.d.B., D.H., and T.S.L. are employees of Janssen and have equity ownership. P.G.R. has received research funding from Oncopeptides, Celgene, Takeda, and Bristol-Myers Squibb; and has served as a member of an advisory committee or received honoraria from Karyopharm, Oncopeptides, Celgene, Takeda, Amgen, Janssen, and Sanofi. The remaining authors declare no competing financial interests.
A complete list of investigators in the GRIFFIN trial is provided in the supplemental appendix.
Correspondence: Peter M. Voorhees, Levine Cancer Institute, Atrium Health, 1021 Morehead Medical Dr, Charlotte, NC 28204; e-mail: peter.voorhees@atriumhealth.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal