

Key Points
The presence of NPM1 mutation is the primary prognostic factor for OS in IDH1- or IDH2R140-mutated AML treated by IC.
In nonfavorable European LeukemiaNet 2010 IDH-mutated AML, patients achieving transplantation in CR1 had longer OS and DFS.

Abstract
In patients with isocitrate dehydrogenase (IDH)–mutated acute myeloid leukemia (AML) treated by intensive chemotherapy (IC), prognostic significance of co-occurring genetic alterations and allogeneic hematopoietic stem cell transplantation (HSCT) are of particular interest with the advent of IDH1/2 mutant inhibitors. We retrospectively analyzed 319 patients with newly diagnosed AML (127 with IDH1, 135 with IDH2R140, and 57 with IDH2R172 mutations) treated with IC in 3 Acute Leukemia French Association prospective trials. In each IDH subgroup, we analyzed the prognostic impact of clinical and genetic covariates, and the role of HSCT. In patients with IDH1 mutations, the presence of NPM1 mutations was the only variable predicting improved overall survival (OS) in multivariate analysis (P < .0001). In IDH2R140-mutated AML, normal karyotype (P = .008) and NPM1 mutations (P = .01) predicted better OS. NPM1 mutations were associated with better disease-free survival (DFS; P = .0009), whereas the presence of DNMT3A mutations was associated with shorter DFS (P = .0006). In IDH2R172-mutated AML, platelet count was the only variable retained in the multivariate model for OS (P = .002). Among nonfavorable European LeukemiaNet 2010–eligible patients, 71 (36%) underwent HSCT in first complete remission (CR1) and had longer OS (P = .03) and DFS (P = .02) than nontransplanted patients. Future clinical trials testing frontline IDH inhibitors combined with IC may consider stratification on NPM1 mutational status, the primary prognostic factor in IDH1- or IDH2R140-mutated AML. HSCT improve OS of nonfavorable IDH1/2-mutated AML and should be fully integrated into the treatment strategy.
Medscape Continuing Medical Education online
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine’s (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider’s responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 2856.
Disclosures
Editor Nancy Berliner and CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC, declare no competing financial interests. Author Lionel Adès has received research support from Celgene and Agios and has received compensation from Celgene for serving on its Advisory Board. The remaining authors declare no competing financial interests.
Learning objectives
Upon completion of this activity, participants will be able to:
Describe the prognostic impact, according to IDH subgroup analysis, of clinical and genetic alterations in a large cohort of patients with newly diagnosed IDH-mutated acute myeloid leukemia (AML) treated with intensive chemotherapy (IC) in 3 prospective ALFA clinical trials
Determine the role of hematopoietic stem cell transplantation (HSCT) in eligible patients with newly diagnosed IDH-mutated AML treated with IC in 3 prospective ALFA clinical trials
Identify the clinical and research implications of the prognostic impact of clinical and genetic alterations, and the role of HSCT in eligible patients with newly diagnosed IDH-mutated AML treated with IC in 3 prospective ALFA clinical trials
Release date: May 20, 2021; Expiration date: May 20, 2022
Introduction
Point mutations in isocitrate dehydrogenase (IDH) genes (IDH1 and IDH2) are found in 7% to 14% and in 8% to 19% of adult patients with acute myeloid leukemia (AML), respectively.1 IDH1/2-mutant enzymes gain neomorphic enzymatic activity, producing an excess of D-2-hydroxyglutarate,2-4 leading to histone and DNA hypermethylation and cell differentiation blockade.5-9 Despite a shared oncogenic mechanism, IDH mutation subtypes (IDH1, IDH2R140, and IDH2R172) have distinct patterns of co-occurring genetic alterations that may correspond to distinct entities.10-13 However, risk stratification within each IDH mutation subtype remains conflicting in patients with AML treated with intensive chemotherapy (IC).11,14-22 One large comprehensive series investigated the mutational landscape of patients with AML that have different IDH mutation subtypes, but it did not investigate the prognostic impact of co-occurring mutations in each group.12 Furthermore, some studies addressed the role of allogeneic hematopoietic stem cell transplantation (HSCT) in limited cohorts of IDH-mutated AML, but none analyzed its impact in specific IDH subgroups.23,24 Oral targeted inhibitors of mutant IDH1 and IDH2 enzymes (ivosidenib25 and enasidenib,26 respectively) were recently approved by the US Food and Drug Administration for the treatment of relapsed/refractory AML as single agents, but also for the treatment of newly diagnosed AML with a susceptible IDH1 mutation for ivosidenib, in patients who are at least 75 years old or who have comorbidities that preclude the use of intensive induction chemotherapy.27-29 In the context of clinical trials evaluating the combination of IDH inhibitors plus IC (#NCT02632708,30 #NCT03839771), it seems important to identify the main prognostic factors for each IDH mutation subtype to guide study design (ie, patients stratification) and data interpretation. Here, we report an IDH subgroup analysis of the prognostic impact of clinical and genetic covariates and the outcome after HSCT in a large cohort of 319 newly diagnosed patients with IDH-mutated AML who were treated with IC in 3 prospective Acute Leukemia French Association (ALFA) clinical trials.
Patients and methods
Patients
We analyzed 319 newly diagnosed patients with IDH1/2-mutated AML treated with IC in 3 ALFA prospective clinical trials (supplemental Figure 1, available on the Blood Web site). Patients provided written consent, and the study was conducted according to the Declaration of Helsinki and approved by institutional review boards. Eight patients with dual IDH1 and IDH2 mutations were excluded. Fifty-eight older patients (50-70 years) were treated between 2008 and 2010 in ALFA-070131 (EudraCT 2007-002933-36), 132 younger patients (18-60 years) were treated between 2009 and 2013 in ALFA-070232 (#NCT00932412), and 129 older patients (>60 years) were treated between 2012 and 2016 in ALFA-120033 (#NCT01966497) trials. All patients received an induction course, including anthracycline and cytarabine. Patients in ALFA-0701 who were randomized to receive gemtuzumab ozogamicin were excluded from survival analyses to obtain a homogenous patient cohort treated with IC. ALFA-0701 and ALFA-0702 trials included a salvage course based on high-dose cytarabine. Patients in ALFA-1200 received a second intermediate-dose course of cytarabine (IDAC), regardless of response. Consolidation included daunorubicin plus cytarabine in ALFA-0701, high-dose cytarabine or clofarabine plus IDAC in ALFA-0702, and IDAC in ALFA-1200. Patients with nonfavorable risk according to the 2010 European LeukemiaNet study (ELN 2010)34 were eligible for HSCT if they had a sibling donor or a fully 10/10 HLA-matched unrelated donor.
Cytogenetic and molecular analyses
Conventional karyotype and fluorescence in situ hybridization were centrally reviewed. Molecular analyses were performed in the ALFA Central Laboratory (Lille University Hospital; C.P.) by high-throughput sequencing on diagnostic peripheral blood or bone marrow samples, as reported previously for the ALFA-0701,35 ALFA-0702,36 and ALFA-120033 studies. Analyses focused on the 37 genes overlapping in the 3 studies: IDH1, IDH2, ASXL1, BCOR, BCORL1, CALR, CBL, CEBPA, CSF3R, DNMT4A, ETV6, EZH2, FLT3, GATA2, JAK2, KIT, KRAS, MPL, NIPBL, NPM1, NRAS, PHF6, PTPN11, RAD21, RIT1, RUNX1, SETBP1, SF3B1, SMC1A, SMC3, SRSF3, STAG2, TET3, TP53, U2AF1, WT1, and ZRSR2 (supplemental Table 1). Screening for NPM1 mutations and FLT3 internal tandem duplications (FLT3-ITD) was also performed by fragment analysis; screening for mutations in CEBPA was performed by Sanger sequencing.
Statistical analyses
Continuous variables are reported as median and interquartile range (IQR), and categorical and ordinal variables are reported as number and proportion. Differences in quantitative variables between groups was assessed using the Kruskal-Wallis test, followed by a pairwise Mann-Whitney U test, if significant. Correlation between genotype and frequent covariates (present in >5% of the whole IDH cohort) was made using point biserial correlation for continuous variables and evaluated with the Φ coefficient and tested with the Fisher test for dichotomic variables. P values were corrected for multiple testing using the Benjamini-Hochberg procedure37 (q values). Standard National Cancer Institute criteria were used to define CR and CRp38 after the first course and salvage therapy in ALFA-0701 and ALFA-0702 and after the first and second courses in ALFA-1200. Patients alive after induction or induction and salvage, but not reaching CR/CRp criteria, were considered to have refractory disease. Relapse was defined as reappearance of circulating leukemic blasts, recurrence of >5% marrow blasts, and/or appearance of extramedullary leukemia. Bivariate analyses for response were done by logistic regression stratified on the trial. Follow-up duration was calculated with the inverse method. Overall survival (OS) analyses were considered from the date of diagnosis to the date of death or last follow up. Analyses of disease-free survival (DFS) were restricted to patients achieving complete remission (CR)/CR with incomplete platelet recovery (CRp) after 2 courses and were considered from the date of response to the date of death, relapse, or last follow-up. OS and DFS were obtained according to the Kaplan-Meier method and censored at HSCT. For survival analysis, frequent variables (present in >10% of each IDH subgroup) were selected using a LASSO penalized regression with the R package glmnet, using the regularization parameter λ-min determined on 100 cross-validations. Selected variables were included in multivariate Cox models, followed by backward regression. The proportional-hazards assumption was tested on the basis of Schoenfeld residuals. To determine the role of HSCT in first CR, HSCT was considered a time-dependent covariate; survival curves for OS and DFS were obtained using the Simon-Makuch method and compared using a time-dependent bivariate Cox model. All tests were 2 sided, and statistical significance was defined as a P value or q value <.05; all analyses for response and survival were stratified on the clinical trial. All analyses were performed with R version 3.5.2.
Results
Characteristics of patients
A total of 319 patients with IDH1/2-mutated AML from ALFA-0701 (n = 58), ALFA-0702 (n = 132), and ALFA-1200 (n = 129) prospective clinical trials was analyzed in the study, including 127 with IDH1-mutated AML (40%), 135 with IDH2R140-mutated AML (42%), and 57 with IDH2R172-mutated AML (18%) (supplemental Figure 1). Characteristics of patients at diagnosis are listed in Table 1. Overall, 164 were men (52%) with a median age of 61 years (IQR, 52-67). Most patients had a normal karyotype (67%); ELN 2010 risk was favorable, intermediate-1, intermediate-2, and high in 29%, 38%, 25%, and 8% of evaluable patients, respectively. Among patients with IDH1, 60 had a p.R132C mutation (47%), 44 had a p.R132H mutation (35%), 11 had a p.R132G mutation (9%), 10 had a p.R132S mutation (8%), and 2 had a p.R132L mutation (1%). Among patients with IDH2R140 mutations, 127 had a p.R140Q mutation (94%), 7 had a p.R140W mutation (5%), and 1 had a p.R140L mutation (1%). The IDH2R172 variants were p.R172K (n = 56) and p.R172S (n = 1). The majority of patients with IDH mutations (>95%) had ≥1 comutation, primarily in DNMT3A (42%), NPM1 (40%), SRSF2 (20%), FLT3-ITD (15%), or NRAS (14%) (Figure 1A-B; supplemental Figure 2). Patiens with IDH2R172 mutations had significantly fewer comutations than did other patients: a median of 2 (IQR, 1-3) for IDH2R172 vs a median of 3 (IQR, 2-4) for IDH1 and a median of 3 (IQR, 2-4) for IDH2R140; q = 0.004 and q = 0.01, respectively (Figure 1B). Patients with IDH2R172 mutations also had a significantly smaller allele burden (median variant allele frequency of 27% [IQR, 18-38] vs 38% [IQR, 15-43] for IDH1 and 42% [IQR, 24-47] for IDH2R140, q = 0.04 and q < 0.001, respectively; Figure 1C).
Characteristics of patients
| All | IDH1R132 | IDH2R140 | IDH2R172 | |
|---|---|---|---|---|
| Patients | 319 (100) | 127 (40) | 135 (42) | 57 (18) |
| Age, median (IQR), y | 61 (52-67) | 61 (52-67) | 61 (50-67) | 62 (56-68) |
| Males | 164 (52) | 68 (54) | 74 (55) | 22 (39) |
| sAML | 16 (5) | 10 (8) | 4 (3) | 2 (4) |
| WBCs, median (IQR), ×109/L | 3.7 (1.6-23.8) | 4.5 (1.6-20.7) | 8.8 (2.3-31.6) | 1.8 (1.2-2.7) |
| Cytogenetics | ||||
| Normal | 198 (67) | 72 (63) | 98 (77) | 28 (52) |
| Complex | 11 (4) | 9 (8) | 2 (2) | 0 |
| Trisomy 8/8q | 31 (10) | 16 (14) | 10 (8) | 5 (9) |
| Monosomy 7 | 12 (3) | 10 (9) | 2 (2) | 0 |
| Trisomy 11/11q | 13 (4) | 3 (3) | 2 (2) | 8 (15) |
| Not available, n | 22 | 12 | 7 | 3 |
| ELN 2010 risk groups | ||||
| Favorable | 86 (29) | 38 (33) | 48 (37) | 0 (0) |
| Intermediate-1 | 112 (38) | 34 (29) | 50 (39) | 28 (52) |
| Intermediate-2 | 75 (25) | 25 (22) | 24 (19) | 26 (48) |
| Adverse | 24 (8) | 18 (16) | 6 (5) | 0 (0) |
| Not available, n | 22 | 12 | 7 | 2 |
| Trials | ||||
| ALFA-0701 | 58 (18) | 26 (20) | 21 (16) | 11 (19) |
| ALFA-0702 | 132 (41) | 54 (43) | 57 (42) | 21 (37) |
| ALFA-1200 | 129 (41) | 47 (37) | 57 (42) | 25 (44) |
| HSCT in CR1 | ||||
| Eligible patients* | 197 (62) | 69 (54) | 77 (57) | 51 (89) |
| HSCT performed | 71 (36) | 23 (33) | 28 (36) | 20 (39) |
| All | IDH1R132 | IDH2R140 | IDH2R172 | |
|---|---|---|---|---|
| Patients | 319 (100) | 127 (40) | 135 (42) | 57 (18) |
| Age, median (IQR), y | 61 (52-67) | 61 (52-67) | 61 (50-67) | 62 (56-68) |
| Males | 164 (52) | 68 (54) | 74 (55) | 22 (39) |
| sAML | 16 (5) | 10 (8) | 4 (3) | 2 (4) |
| WBCs, median (IQR), ×109/L | 3.7 (1.6-23.8) | 4.5 (1.6-20.7) | 8.8 (2.3-31.6) | 1.8 (1.2-2.7) |
| Cytogenetics | ||||
| Normal | 198 (67) | 72 (63) | 98 (77) | 28 (52) |
| Complex | 11 (4) | 9 (8) | 2 (2) | 0 |
| Trisomy 8/8q | 31 (10) | 16 (14) | 10 (8) | 5 (9) |
| Monosomy 7 | 12 (3) | 10 (9) | 2 (2) | 0 |
| Trisomy 11/11q | 13 (4) | 3 (3) | 2 (2) | 8 (15) |
| Not available, n | 22 | 12 | 7 | 3 |
| ELN 2010 risk groups | ||||
| Favorable | 86 (29) | 38 (33) | 48 (37) | 0 (0) |
| Intermediate-1 | 112 (38) | 34 (29) | 50 (39) | 28 (52) |
| Intermediate-2 | 75 (25) | 25 (22) | 24 (19) | 26 (48) |
| Adverse | 24 (8) | 18 (16) | 6 (5) | 0 (0) |
| Not available, n | 22 | 12 | 7 | 2 |
| Trials | ||||
| ALFA-0701 | 58 (18) | 26 (20) | 21 (16) | 11 (19) |
| ALFA-0702 | 132 (41) | 54 (43) | 57 (42) | 21 (37) |
| ALFA-1200 | 129 (41) | 47 (37) | 57 (42) | 25 (44) |
| HSCT in CR1 | ||||
| Eligible patients* | 197 (62) | 69 (54) | 77 (57) | 51 (89) |
| HSCT performed | 71 (36) | 23 (33) | 28 (36) | 20 (39) |
Unless otherwise noted, data are n (%).
sAML, secondary AML; WBCs, white blood cells.
Patients with nonfavorable AML and not receiving gemtuzumab ozogamicin in ALFA-0701.
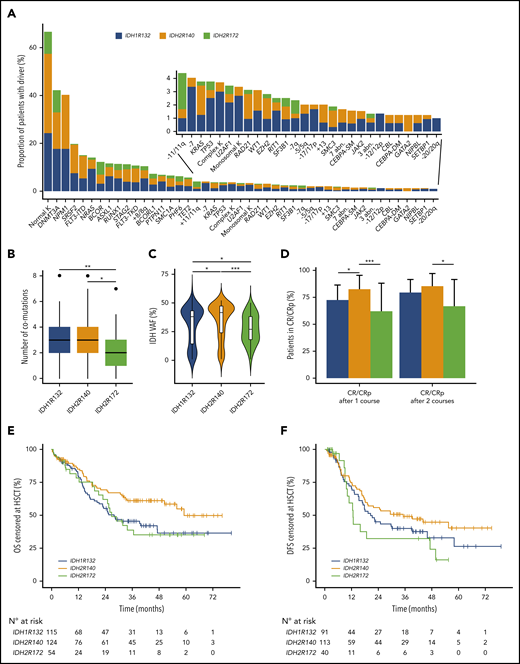
Characteristics of the IDH1/2-mutated AML cohort. (A) Molecular and cytogenetic characteristics of the IDH-mutated AML cohort according to IDH subgroup. (B) Box plots showing the number of co-occurring mutations in each IDH subgroup. (C) Violin plots showing the variant allele frequency of the IDH variant in each IDH subgroup. (D) Bar graph showing the CR/CRp rates after 1 and 2 courses of IC in each IDH subgroup. The error bars represent the upper limit of the 95% confidence interval. OS (E) and DFS (F) censored at HSCT according to IDH subgroup. *q < 0.05; **q < 0.01; ***q < 0.001.
Characteristics of the IDH1/2-mutated AML cohort. (A) Molecular and cytogenetic characteristics of the IDH-mutated AML cohort according to IDH subgroup. (B) Box plots showing the number of co-occurring mutations in each IDH subgroup. (C) Violin plots showing the variant allele frequency of the IDH variant in each IDH subgroup. (D) Bar graph showing the CR/CRp rates after 1 and 2 courses of IC in each IDH subgroup. The error bars represent the upper limit of the 95% confidence interval. OS (E) and DFS (F) censored at HSCT according to IDH subgroup. *q < 0.05; **q < 0.01; ***q < 0.001.
After exclusion of the 26 patients who received gemtuzumab ozogamicin, 224 patients (76%) and 244 patients (83%) achieved a CR/CRp after 1 and 2 courses of IC, respectively. Upon bivariate analyses stratified on the trial, IDH2R140 mutations predicted higher response rates after 1 course of IC (86% vs 74% for IDH1 mutations and 59% for IDH2R172 mutations, q = 0.03 and q < 0.001, respectively) and after 2 courses of IC (91% vs 79% for IDH1 mutations and 74% for IDH2R172 mutations, q = 0.02 and q = 0.02, respectively; Figure 1D). Of note, concomitant NPM1 mutations were associated with significantly higher CR/CRp rates for IDH1 mutations (94% [50/53] vs 66% [41/62] for NPM1 wild-type; P = .0002) and for IDH2R140 mutations (100% [62/62] vs 82% [51/62] for NPM1 wild-type; P = .0003).
Median follow-up was 46.0 months (IQR, 38.0-58.4). Median OS censored at HSCT was 39.7 months (IQR, 14.7-not reached); in bivariate analyses stratified on the clinical trial, patients with IDH2R140 mutations had better OS censored at HSCT than did patients with IDH1 mutations (3-year OS, 61% vs 46%; hazard ratio [HR], 0.60; 95% confidence interval [CI], 0.40-0.93; P = .02), but it was not better than for patients with IDH2R172 mutations (3-year OS, 61% vs 39%; HR, 0.65; 95% CI, 0.38-1.09; P = .10; Figure 1E). Median DFS censored at HSCT was 22.3 months (IQR, 10.8-not reached), and there was no significant difference according to IDH subtype (Figure 1F). Of note, the ELN 2010 and the most recent ELN 2017 risk stratification poorly discriminated intermediate-risk and adverse-risk patients with IDH-mutated AML (supplemental Figure 3).
IDH1R132-mutated cohort
Only frequent covariates (>5% of the entire IDH cohort; supplemental Table 2) were included in the correlation analyses. The only covariate associated with IDH1 mutations was NRAS mutation (24% vs 8% in IDH1 wild-type; q = 0.003). We investigated the associations of the 2 most frequent IDH1 variants (ie, p.R132H and p.R132C) within the IDH1-mutated cohort. p.R132H variants were present in younger patients (median age, 56 years vs 63 years for other IDH1 variants; q = 0.02) and were associated with higher rates of NPM1 mutations (64% vs 34%; q = 0.01) and FLT3-ITD (27% vs 6%; q = 0.01), as well as higher white blood cell (WBC) counts (17.9 × 109/L vs 2.4 × 109/L; q = 0.0006). p.R132C variants were significantly associated with higher rates of trisomy 8 (23% vs 5%; q = 0.01), as well as mutations in PHF6 (13% vs 0%; q = 0.01), BCOR (15% vs 1%; q = 0.03), and BCORL1 (13% vs 1%, q = 0.048; Figure 2A). Despite these 2 distinct mutational patterns, there was no difference in survival between patients with p.R132C or p.R132H variants (Figure 2B).
![Characteristics of patients with IDH1-mutated AML. (A) Volcano plot representing the association between IDH1, IDH1R132C, and IDH1R132H variants and covariates (estimate of the point-biserial correlation [continuous variables] or Φ [dichotomous variables] on the x-axis) and the significance of the difference. The P value was calculated using the Mann-Whitney U test (for continuous variables) or Fisher’s exact (dichotomous) test, expressed on an inverted logarithmic scale on the y-axis). The size of the circle corresponds to the frequency of the variable in the cohort. Only covariates with q < 0.05 are highlighted. (B) OS censored at HSCT according to the type of IDH1 variant. (C) OS censored at HSCT according to the mutational status of NPM1.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020010165/1/m_bloodbld2020010165f2.png?Expires=1767695769&Signature=3IYixuhVZs5-g8Xc2zDG2TXHsvLEBj3gB1d3l~4QjUVPGqte-cJ3hH1Db8pttkW7IkfeNQb43FplljDbzWWuToPCuTh3Fv-rk8j7Cq2f9Hiu-6558bqObYNoUv5mFY6mnPI~BluEB~cRSH07dxQTe2El~ZDprHPj2piAF4CStA5eFNNXv8u9QtBtJ9iDIAtpfHHYizf4rFRccqvnXUQFc1cdo8J9gEM-LtxMAP7u9ka4jjt3gQuns5Mre7PF7Oxz7cvzZvRdxguvT~YolESrmFO88akjDlV4~R212xDigMBy8MqSDIJNa-M8oixT7WbXWw79cUPnB~8Eg4PLCyI8ug__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Characteristics of patients with IDH1-mutated AML. (A) Volcano plot representing the association between IDH1, IDH1R132C, and IDH1R132H variants and covariates (estimate of the point-biserial correlation [continuous variables] or Φ [dichotomous variables] on the x-axis) and the significance of the difference. The P value was calculated using the Mann-Whitney U test (for continuous variables) or Fisher’s exact (dichotomous) test, expressed on an inverted logarithmic scale on the y-axis). The size of the circle corresponds to the frequency of the variable in the cohort. Only covariates with q < 0.05 are highlighted. (B) OS censored at HSCT according to the type of IDH1 variant. (C) OS censored at HSCT according to the mutational status of NPM1.
Characteristics of patients with IDH1-mutated AML. (A) Volcano plot representing the association between IDH1, IDH1R132C, and IDH1R132H variants and covariates (estimate of the point-biserial correlation [continuous variables] or Φ [dichotomous variables] on the x-axis) and the significance of the difference. The P value was calculated using the Mann-Whitney U test (for continuous variables) or Fisher’s exact (dichotomous) test, expressed on an inverted logarithmic scale on the y-axis). The size of the circle corresponds to the frequency of the variable in the cohort. Only covariates with q < 0.05 are highlighted. (B) OS censored at HSCT according to the type of IDH1 variant. (C) OS censored at HSCT according to the mutational status of NPM1.
To identify the prognostic impact of covariates in IDH1-mutated AML, we included all covariates present in ≥10% of IDH1 patients (list in supplemental Table 3) in a LASSO penalized regression for OS and DFS censored at HSCT. The final multivariate Cox models stratified on the trial are summarized in Table 2. NPM1 mutational status was the only variable predicting prolonged OS (3-year OS of 65% in mutated NPM1 vs 28% in wild-type NPM1; HR, 0.29; 95% CI, 0.16-0.54; P < .0001; Figure 2C). No variable was retained in the multivariate analysis for DFS.
Multivariate analyses for survival
| OS censored at HSCT, stratified on the trial | DFS censored at HSCT, stratified on the trial | |||||
|---|---|---|---|---|---|---|
| IDH1 | IDH2R140 | IDH2R172 | IDH1 | IDH2R140 | IDH2R172 | |
| NPM1, mutated | 0.29 (0.16-0.54) P < .0001 | 0.41 (0.21-0.81) P = .01 | — | — | 0.33 (0.17-0.63) P = .0009 | — |
| DNMT3A, mutated | — | — | — | — | 3.05 (1.61-5.78) P = .0006 | — |
| Karyotype, normal | — | 0.38 (0.19-0.78) P = .008 | — | — | — | — |
| Log10(platelets) | — | — | 0.19 (0.07-0.53) P = .002 | — | — | — |
| OS censored at HSCT, stratified on the trial | DFS censored at HSCT, stratified on the trial | |||||
|---|---|---|---|---|---|---|
| IDH1 | IDH2R140 | IDH2R172 | IDH1 | IDH2R140 | IDH2R172 | |
| NPM1, mutated | 0.29 (0.16-0.54) P < .0001 | 0.41 (0.21-0.81) P = .01 | — | — | 0.33 (0.17-0.63) P = .0009 | — |
| DNMT3A, mutated | — | — | — | — | 3.05 (1.61-5.78) P = .0006 | — |
| Karyotype, normal | — | 0.38 (0.19-0.78) P = .008 | — | — | — | — |
| Log10(platelets) | — | — | 0.19 (0.07-0.53) P = .002 | — | — | — |
Data are shown as HR (95% CI).
—, variable not retained in the multivariate analysis.
IDH2R140-mutated cohort
Compared with other IDH mutations, IDH2R140 variants were associated with higher rates of NPM1 mutations (53% vs 30%; q = 0.0005), FLT3-ITD (22% vs 10%; q = 0.01), SRSF2 mutations (28% vs 14%; q = 0.01), normal karyotypes (77% vs 59%; q = 0.01), and higher WBC counts (median, 8.80 × 109/L vs 2.60 × 109/L; q = 0.0005; Figure 3A).
![Characteristics of patients with IDH2R140-mutated AML. (A) Volcano plot representing the association between IDH2R140 variants and covariates (estimate of the point-biserial correlation [continuous variables] or Φ [dichotomous variables] on the x-axis) and the significance of the difference. The P value was calculated using a Mann-Whitney (continuous variables) U test or Fisher’s exact (dichotomous) test, expressed on an inverted logarithmic scale on the y-axis. The size of the circle corresponds to the frequency of the variable in the cohort. Only covariates with q < 0.05 are highlighted. (B) OS censored at HSCT in IDH2R140-mutated patients according to the karyotype and NPM1 mutational status. (C) DFS censored at HSCT according to the mutational statuses of NPM1 and DNMT3A.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020010165/1/m_bloodbld2020010165f3.png?Expires=1767695769&Signature=TO8sgJYBzn5gzhcgQkSBcyt8dZvPYEyUJSh~fJxsYidU9stSHcfXnwYDqC~aaWPOld0fhY3CfJYOPBa15HT5amgnbUMBJwNNtDvYGdejrm5VmK7VQQC64q-oNS953eEXRt4jeDIPJL8jzZwyQLCiqbAEyyWtd7YeblZXczOxRwL2OOQyd~q1ZuiIA0IzAb7-m2RIcA~k-iMLS7KItKQSmpT4KykuWLfuRlSu3SYd5fubA~Eq9eUjNhNqL96GgTstU~ZX2dAD~zGw0q3joDu~JPjWV6HfkiHKnohZud1h1yJxqNrFKN7QeCri7M9ahG~xZJ0gIpOZI2M3C1ATX9ZfAQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Characteristics of patients with IDH2R140-mutated AML. (A) Volcano plot representing the association between IDH2R140 variants and covariates (estimate of the point-biserial correlation [continuous variables] or Φ [dichotomous variables] on the x-axis) and the significance of the difference. The P value was calculated using a Mann-Whitney (continuous variables) U test or Fisher’s exact (dichotomous) test, expressed on an inverted logarithmic scale on the y-axis. The size of the circle corresponds to the frequency of the variable in the cohort. Only covariates with q < 0.05 are highlighted. (B) OS censored at HSCT in IDH2R140-mutated patients according to the karyotype and NPM1 mutational status. (C) DFS censored at HSCT according to the mutational statuses of NPM1 and DNMT3A.
Characteristics of patients with IDH2R140-mutated AML. (A) Volcano plot representing the association between IDH2R140 variants and covariates (estimate of the point-biserial correlation [continuous variables] or Φ [dichotomous variables] on the x-axis) and the significance of the difference. The P value was calculated using a Mann-Whitney (continuous variables) U test or Fisher’s exact (dichotomous) test, expressed on an inverted logarithmic scale on the y-axis. The size of the circle corresponds to the frequency of the variable in the cohort. Only covariates with q < 0.05 are highlighted. (B) OS censored at HSCT in IDH2R140-mutated patients according to the karyotype and NPM1 mutational status. (C) DFS censored at HSCT according to the mutational statuses of NPM1 and DNMT3A.
Results of the multivariate analyses for OS and DFS censored at HSCT are summarized in Table 2. Normal karyotype (3-year OS of 67% vs 28%; HR, 0.38; 95% CI, 0.19-0.78; P = .008) and NPM1 mutations (3-year OS of 77% vs 40%; HR, 0.41; 95% CI, 0.21-0.81; P = .01) predicted prolonged OS (Figure 3B). NPM1 mutations predicted prolonged DFS (3-year DFS of 63% vs 23%; HR, 0.33; 95% CI, 0.17-0.63; P = .0009), whereas DNMT3A mutations were detrimental (3-year DFS of 22% vs 67%; HR, 3.05; 95% CI, 1.61-5.78; P = .0006; Figure 3C).
IDH2R172-mutated cohort
IDH2R172 variants were associated with higher rates of BCOR mutations (30% vs 8%; q < 0.001), no NPM1 mutations (0% vs 49%; q < 0.001), fewer SRSF2 mutations (2% vs 24%; q < 0.001) and FLT3-ITD (2% vs 18%; q < 0.001), and lower WBC count (median, 1.80 × 109/L vs 6.25 × 109/L; q < 0.001; Figure 4A).
![Characteristics of the patients with IDH2R172-mutated AML. (A) Volcano plot representing the association between IDH2R172 variants and covariates (estimate of the point-biserial correlation [continuous variables] or Φ [dichotomous variables] on the x-axis) and the significance of the difference, expressed on an inverted logarithmic scale on the y-axis. The P value was calculated using the Mann-Whitney U test (continuous variables) or Fisher’s exact (dichotomous) test. The size of the circle corresponds to the frequency of the variable in the cohort. Only covariates with q < 0.05 are highlighted. (B) OS censored at HSCT in patients with IDH2R172 mutations, according to the platelet counts at diagnosis (arbitrary cutoff at 100 × 109/L).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020010165/1/m_bloodbld2020010165f4.png?Expires=1767695769&Signature=ARYyJq7j0jc2nPqSnAFalWQABg-n27vA849nRsrK9drTWCGxQ8h163atBWBtSoXIpaNqD9XXx8CTi4WgUP~pg8-Ytm5vH167IXucIkDzgRc78tDBCmzdxLEZayUy-gDlC8nNioRG9HXM-JZPIy8M2CUITFtoyQEFSaqYgPo9alCQl1lGm38b~V-mjq6y9VduNf4uhNRBMEXgBLmxwJcG1QyqR0b7oLQHvGA7bqIEH~J4eorjDcTalQXq0PxiwhoG7ZbnsWw1EhWcDpJ4uAPhO0MfRWUE-YnKe2ga5q2sdBpmXeOwiiS2A3KLctwvC7UYJYLblKXLa5IMaRaSWSmRaQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Characteristics of the patients with IDH2R172-mutated AML. (A) Volcano plot representing the association between IDH2R172 variants and covariates (estimate of the point-biserial correlation [continuous variables] or Φ [dichotomous variables] on the x-axis) and the significance of the difference, expressed on an inverted logarithmic scale on the y-axis. The P value was calculated using the Mann-Whitney U test (continuous variables) or Fisher’s exact (dichotomous) test. The size of the circle corresponds to the frequency of the variable in the cohort. Only covariates with q < 0.05 are highlighted. (B) OS censored at HSCT in patients with IDH2R172 mutations, according to the platelet counts at diagnosis (arbitrary cutoff at 100 × 109/L).
Characteristics of the patients with IDH2R172-mutated AML. (A) Volcano plot representing the association between IDH2R172 variants and covariates (estimate of the point-biserial correlation [continuous variables] or Φ [dichotomous variables] on the x-axis) and the significance of the difference, expressed on an inverted logarithmic scale on the y-axis. The P value was calculated using the Mann-Whitney U test (continuous variables) or Fisher’s exact (dichotomous) test. The size of the circle corresponds to the frequency of the variable in the cohort. Only covariates with q < 0.05 are highlighted. (B) OS censored at HSCT in patients with IDH2R172 mutations, according to the platelet counts at diagnosis (arbitrary cutoff at 100 × 109/L).
Results of the multivariate analyses for OS and DFS censored at HSCT are summarized in Table 2. Log-transformed platelet count was the only variable retained in the multivariate model for OS (HR, 0.19; 95% CI, 0.07-0.53; P = .002). Using an arbitrary cutoff for platelets of 100 × 109/L, 3-year OS was 17% for patients with thrombocytopenia vs 52% for those without (Figure 4B). Patients with IDH2R172 mutations that have low platelet counts (<100 × 109/L) suffered more early deaths (3/20 vs 0/34 with high platelet counts) and had significantly higher rates of induction failure after 1 course of IC (CR/CRp rates of 25% vs 79%; P = .001) or 2 courses of IC (CR/CRp rates of 45% vs 91%; P = .001). No variable was retained as an independent prognostic factor for DFS. Of note, the 16 patients with IDH2R172 mutations without other classifying abnormalities, as defined by Papaemmanuil et al,11 did not have distinct comutations or prolonged outcomes (supplemental Figure 4).
Impact of HSCT in patients with IDH mutations
Finally, we investigated the role of HSCT in first complete remission (CR1) for the 197 eligible patients (ie, patients with nonfavorable AML according to ELN 2010 risk and who did not receive gemtuzumab ozogamicin). Overall, 71 (36%) eventually received an HSCT after a median time from diagnosis of 5.3 months (IQR, 4.5-6.1), including 23 (33%), 28 (36%), and 20 (39%) patients in the IDH1R132-mutated, IDH2R140-mutated, and IDH2R172-mutated groups, respectively (Table 1). The main characteristics were similar between the transplant and notransplant cohorts; however, most transplanted patients came from the ALFA-0702 trial, resulting in a younger age in this group (supplemental Table 4). When we considered HSCT as a time-dependent covariate in bivariate analyses stratified on the clinical trial, transplantation in CR1 was associated with a prolonged OS (HR, 0.60; 95% CI, 0.37-0.96; P = .03; Figure 5A) and a prolonged DFS (HR, 0.55; 95% CI, 0.34-0.89; P = .02; Figure 5B). Analyses in IDH subtypes revealed a benefit in OS only for patients with IDH1R132 mutations (HR, 0.48; 95% CI, 0.23-0.99; P = .048) and a trend toward better DFS for patients with IDH2R172 mutations (HR, 0.41; 95% CI, 0.14-1.20; P = .10; supplemental Figure 5).
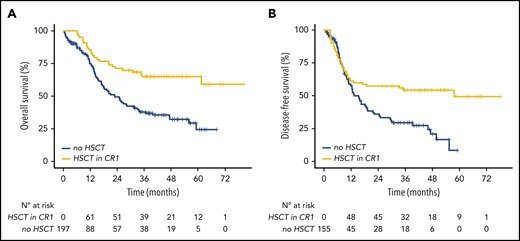
Impact of HSCT in first CR/CRp in patients with IDH-mutated AML. Simon-Makuch plot of OS (A) and DFS (B) according to achievement of HSCT. HSCT was considered a time-dependent variable.
Impact of HSCT in first CR/CRp in patients with IDH-mutated AML. Simon-Makuch plot of OS (A) and DFS (B) according to achievement of HSCT. HSCT was considered a time-dependent variable.
Discussion
In recent years, there has been renewed interest in IDH1/2 mutations in AML with the advent of specific inhibitors. However, the prognostic impact of IDH1/2 mutations and their comutations remained unclear because, in most studies, IDH1, IDH2R140, and IDH2R172 mutations were analyzed together12,20,21 and in cohorts treated with different-intensity regimens.21,39 The present study represents one of the largest series of patients with IDH1/2-mutated AML prospectively enrolled in 3 clinical trials with sequencing data on 37 genes; we assessed the prognostic impact of clinical and genetic covariates, as well as the impact of HSCT in CR1, for each IDH mutation subtype.
Our cohort included 127 patients with IDH1 mutations (40%), 135 patients with IDH2R140 mutations (42%), and 57 (18%) patients with IDH2R172 mutations; these proportions are in line with previous reports.12,17,40 Clinical and genetic covariates differed between IDH mutation subtypes. Among patients with IDH1 mutations, we could emphasize that p.R132H variants were more frequently associated with NPM1 and FLT3-ITD mutations than were other IDH1 subtypes, as reported previously.13 In line with previous reports,11,12 patients with IDH2R140 mutations had significantly higher WBC counts, as well as more frequent normal karyotypes and NPM1, FLT3-ITD, and SRSF2 mutations, whereas patients with IDH2R172 mutations had lower WBC counts, more frequent BCOR mutations, and no NPM1 mutations. These differences in clinical and genetic covariates suggest that IDH mutation subtypes are distinct entities that should be considered separately; which is supported by biological differences.41,42
After intensive treatment, CR/CRp rates were significantly higher in patients with IDH2R140 mutations (86%) than in patients with IDH1 (74%) and IDH2R172 (59%) mutations, as reported previously.40 NPM1 mutations were associated with remarkably high CR/CRp rates in IDH1R132-mutated (94%) and IDH2R140-mutated (100%) patients. Considering long-term outcome, patients with IDH2R140 mutations had a significantly prolonged OS compared with patients who had IDH1 mutations (P = .02), as well as a trend toward prolonged OS compared with patients who had IDH2R172 mutations (P = .1). Higher rates of NPM1 mutations and normal karyotypes may account for the better outcome in patients with IDH2R140 mutations. In keeping with studies on patients accrued to the ALFA,16 MRC,40 and CALGB10 clinical trials, patients with IDH2R172 mutations did not have a prolonged survival compared with other patients who had IDH mutations in our cohort, even in the 16 patients with IDH2R172 mutations without other classifying abnormalities, as defined by Papaemmanuil et al11 (supplemental Figure 4). These results conflict with 2 other studies,11,12 perhaps owing to the greater treatment heterogeneity in them.
Most importantly, our study identified the main prognostic factors for long-term outcome in each IDH mutation subtype. NPM1 was the only mutation predicting OS in multivariate analysis for patients with IDH1 and IDH2R140 mutations. Surprisingly, other stratifying mutations, such as FLT3 mutations11,43,44 (supplemental Figure 6), did not have any significant prognostic impact in our cohort; however, we acknowledge that the number of patients with a high allelic ratio of FLT3-ITD was low (n = 11). We report for the first time the negative impact of cytogenetic abnormalities in IDH2R140 on OS, such as in NPM1-mutated AML.45 We also found that platelet count was the only variable retained in the multivariate model for OS in IDH2R172. Patients with IDH2R172 mutations who had low platelet counts (<100 × 109/L) had a more advanced and resistant disease with higher rates of induction failure. Interestingly, no variable was retained as an independent prognostic factor for DFS in patients with IDH1 or IDH2R172 mutations. In patients with IDH2R140 mutations, NPM1 mutations were associated with prolonged DFS only in patients with wild-type DNMT3A. These results are in line with previous results in NPM1-mutated AML.46 In patients with both IDH and DNMT3A mutations (n = 135), cancer cell fractions of IDH and DNMT3A mutations were similar across all 3 IDH subgroups (supplemental Table 5). However, in patients with IDH1 and IDH2R140 mutations, NPM1 mutations had a lower allelic burden (P < .001), suggesting that they are later events. The poorer prognosis related to the association between IDH2R140 and DNMT3A mutations has already been reported.11 Both mutations are early events and are associated with clonal dominance in single-cell genotyping studies,47 a feature that is associated with a poorer prognosis in AML.48 Mouse models suggest an epigenetic cooperation between the 2 alterations, leading to activation of a stem cell–like gene signature.49 Following previous reports in refractory/relapsing IDH-mutated AML,50,51 it will be interesting to further study the genetic landscape and molecular predictors in patients with relapsed IDH-mutated AML, to compare trials conducted in this population.
In the 3 prospective trials, HSCT in CR1 was only recommended for patients with intermediate- or adverse-risk AML, according to the ELN 2010 classification, when a fully 10/10 HLA-matched donor could be identified. ELN 2010 is a valid stratification for allogeneic HSCT, and the more recent ELN 2017 reclassifies <5% of patients considered as favorable.52 Seventy-one (36%) of the eligible patients aged 70 years or younger underwent transplantation in CR1, and we were able to compare transplant and no-transplant cohorts with similar primary characteristics. As expected, HSCT in CR1 was associated with improved OS and DFS. However, analyses in IDH-mutated subtypes were impaired by limited power and only revealed a benefit in OS for patients with IDH1R132 mutations.
Combining IDH inhibitors with intensive induction and consolidation chemotherapy could be synergistic because primary resistance to IDH inhibitors is related to the number of co-occurring mutations,28,50 as well as to the expansion of a clone harboring mutations in receptor tyrosine kinase pathway genes (ie, NRAS, KRAS, PTPN11, KIT, and FLT3), which are associated with significantly lower CR with partial hematologic recovery rates.28,50 In the present cohorts, mutations in signaling pathway genes, including FLT3 mutations, did not have any impact on the outcome after IC (supplemental Figure 6). Thus, the combination of IC plus IDH inhibitors might prevent primary resistance and relapses conveyed by signaling mutations. This is supported by a recent report of a phase 1 study evaluating the association between IDH inhibitors and IC in newly diagnosed AML,30 in which FLT3 and RAS mutations were cleared after induction chemotherapy. It has yet to be proven whether combination therapies might also prevent secondary resistance to IDH inhibitors related to second site mutations hampering the binding of the drugs or a mutational switch.50,53,54
Thus, future clinical trials testing frontline IDH inhibitors with IC should consider stratification on NPM1 mutational status. HSCT should be fully integrated into the treatment strategy, with important implications for trial design (guidelines for HSCT indications, censoring at HSCT in survival outcomes analyses, post-HSCT maintenance). Future studies should also evaluate alternative strategies (ie, HSCT indications based on minimum residual disease levels, maintenance therapy with IDH inhibitors).
Data sharing requests should be sent to Stéphane de Botton (stephane.debotton@gustaveroussy.fr).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This study was supported by the French Cancer National Institute (InCa - PHRC 2007/1911 and PRTK TRANSLA10-060).
Authorship
Contribution: M.D. and S.d.B. designed the study, analyzed the data, and wrote the manuscript; R.I., H.D., and S.d.B. supervised the study; M.D. performed statistical analyses; N.D. and C. Preudhomme performed genotyping; C.T. performed cytogenetic analyses; J.-B.M., E.R., X.T., J.-P.M., T.B., L.A., S.C., E.L., C.B., J.-V.M., C. Pautas, J.L., N.B., K.C.-L., D.C., P.T., N.V., A.P., C.R., C.G., R.I., H.D., and S.d.B. provided clinical data; and all authors edited the manuscript and approved its final version.
Conflict-of-interest disclosure: L.A. has received research support from Celgene and Agios and has received compensation from Celgene for serving on its Advisory Board. The remaining authors declare no competing financial interests.
Correspondence: Stéphane de Botton, Département d'Hématologie, Gustave Roussy, Université Paris-Saclay,114 Rue Edouard Vaillant, 94805 Villejuif, France, e-mail: stephane.debotton@gustaveroussy.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal