

Key Points
ECD patients have a very high frequency of clonal hematopoiesis and concomitant overt myeloid malignancies.
ECD patients with clonal hematopoiesis are older and have more frequent retroperitoneal involvement and BRAFV600E mutations.

Abstract
Erdheim-Chester disease (ECD) is a clonal hematopoietic disorder characterized by the accumulation of foamy histiocytes within organs (in particular, frequent retroperitoneal involvement) and a high frequency of BRAFV600E mutations. Although ECD is not commonly recognized to have overt peripheral blood (PB) or bone marrow (BM) disease, we recently identified that ECD patients have a high frequency of a concomitant myeloid malignancy. We thus conducted a systematic clinical and molecular analysis of the BM from 120 ECD patients. Surprisingly, 42.5% of ECD patients (51 of 120) had clonal hematopoiesis whereas 15.8% of patients (19 of 120) developed an overt hematologic malignancy (nearly all of which were a myeloid neoplasm). The most frequently mutated genes in BM were TET2, ASXL1, DNMT3A, and NRAS. ECD patients with clonal hematopoiesis were more likely to be older (P < .0001), have retroperitoneal involvement (P = .02), and harbor a BRAFV600E mutation (P = .049) than those without clonal hematopoiesis. The presence of the TET2 mutation was associated with a BRAFV600E mutation in tissue ECD lesions (P = .0006) and TET2-mutant ECD patients were more likely to have vascular involvement than TET2 wild-type ECD patients. Clonal hematopoiesis mutations in ECD were detected in cells derived from CD34+CD38− BM progenitors and PB monocytes but less frequently present in PB B and T lymphocytes. These data identify a heretofore unrecognized high frequency of clonal hematopoiesis in ECD patients, reaffirm the development of additional high risk of myeloid neoplasms in ECD, and provide evidence of a BM-based precursor cell of origin for many patients with ECD.
Introduction
Erdheim-Chester disease (ECD) is a histiocytic disorder characterized by an accumulation of foamy CD68+, CD1a− histiocytes in various organs.1,2 Although the etiology of ECD was unclear for decades, following the discovery that >60% of ECD patients harbor somatic BRAF V600E mutations3 and that almost all ECD patients harbor a mutation activating the RAS-RAF-MEK-ERK pathway,4 ECD is now considered to be a clonal hematopoietic malignancy.5 Moreover, despite the fact ECD is not currently recognized to typically harbor overt clinical abnormalities in the blood or bone marrow, we recently identified that ECD is associated with a surprisingly high frequency of concomitant myeloid malignancies (including myeloproliferative neoplasms [MPNs], myelodysplastic syndromes [MDSs], and mixed MDS/MPN disorders6 ).
Recent large-scale sequencing studies have revealed that the clonal mutations observed in myeloid malignancy–associated genes are not limited to individuals with myeloid neoplasms: they may also be detected in those without hematologic malignancies.7 This situation, termed clonal hematopoiesis of indeterminate potential (CHIP), and defined by the presence of an expanded somatic blood cell clone in people without hematologic malignancy, is increasingly common as people age.8-10 CHIP is associated with increased risk of hematologic malignancies, cardiovascular disease, and death.11
Due to the myeloid origin of ECD,12 we aimed to determine the prevalence and patterns of clonal hematopoiesis in ECD patients, and its relationship with phenotypes and outcomes.
Methods
Patient selection
We enrolled 120 patients with ECD seen at least once in our specialized center (French National Reference Center for Histiocytoses). ECD was diagnosed according to international consensus criteria.1,2 All samples were centrally reviewed and confirmed the diagnosis. The study was approved by the ethics committee (Comité de Protection des Personnes Ile de France III [#2011-A00447-34]) and was conducted in accordance with the Declaration of Helsinki. All patients underwent BRAFV600E mutation detection by pyrosequencing as previously described.3 All cases without mutation by pyrosequencing were then analyzed by multiplex picodroplet digital polymerase chain reaction (PCR) as previously described.13 The total amount and quality of the tumor DNA available was limited; however, 85% of the samples that were considered negative for BRAF mutations had at least 1000 native copies of the BRAF codon V600 amplified. Cardiac and central nervous system involvement was previously defined.14 Patients were followed according to usual care. Because ECD is considered a myeloid neoplasm, bone marrow (BM) aspiration was performed in routine care.
NGS of clonal hematopoiesis and myeloid malignancy–associated genes
We performed molecular analysis of BM mononuclear cells from ECD patients using high-throughput sequencing of 36 genes recurrently mutated in myeloid malignancies. Libraries were prepared using the Ampliseq system according to the manufacturer’s instructions and run on Ion Proton (ThermoFisher). Raw data were analyzed with both Torrent Browser (ThermoFisher) and SeqNext (JSI Medical System). A high depth of coverage (2500×) was obtained for all genes, allowing detection of mutations with a variant allele frequency (VAF) up to 1%. The genes sequenced were: ASXL1, BCOR, BCORL1, CALR, CBL, CSF3R, DNMT3A, ETV6, EZH2, FLT3, GATA2, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NIPBL1, NPM1, NRAS, PHF6, PTPN11, RAD21, RIT1, RUNX1, SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1, ZRSR2. All detected variants were retained with a sensitivity threshold of 1%. Variants between 1% and 2% were confirmed with another next-generation sequencing (NGS) technology with a library preparation using the Haloplex target enrichment system (Agilent Technologies) and run on MiSeq (Illumina). Variant interpretation was performed considering their absence in public databases of polymorphisms (especially GnomAD) and their status in our in-house database of >8000 samples clinically validated (including acute myeloid leukemia [AML] samples from the Acute Leukemia French Association and MDS samples from the Groupe Français des Myélodysplasies). COSMIC was not considered sufficiently robust for clinical validation due to too many misinterpretations in this database. Additional in silico predictions were performed when it was possible. Frameshift and nonsense variants were always considered as relevant mutations. Single-nucleotide-variant effects on protein function of single-nucleotide variants were predicted using SIFT and PolyPhen-2.
Supplemental Figure 1 (available on the Blood Web site) shows the mean depth of coverage for all genes. Overall, a depth higher than 2000× was obtained for all genes except NPM1 and SRSF2. However, the mean depth of coverage at the proline 95 hotspot in the SRSF2 gene was 2218× ± 368. Additionally, Sanger sequencing of the full SRSF2 gene was performed in all samples. Of note, the depth of coverage for the NPM1 gene was considered sufficient because NPM1 mutations are not reported outside of AML.
Sequencing analyses in PB subpopulations and BM progenitors
For 36 patients with available material (BM and/or peripheral blood [PB] samples), targeted deep sequencing of the exonic regions of 40 genes frequently mutated in myeloid neoplasms (CSF3R, MPL, NRAS, RIT1, DNMT3A, SF3B1, IDH1, MYD88, RHOA, GATA2, STAG1, KIT, TET2, BRAF, EZH2, RAD21, JAK2, WT1, CBL, ETV6, KRAS, PTPN11, FLT3, IDH2, TP53, STAT3, SRSF2, SETBP1, CEBPA, ASXL1, RUNX1, ZRSR2, BCOR, KDM6A, OGT, ATRX, STAG2, BCORL1, PHF6, U2AF1) was performed on flow-sorted CD14+ (monocyte) and CD3+ (T-lymphocyte) cells. Targeted regions were amplified using an AmpliSeq technology (Life Technologies) and PCR products were sequenced with a MiSeq instrument (Illumina). We used a VAF minimal threshold of 5% to detect mutation. We analyzed, by mutation-specific PCRs followed by deep sequencing, the mutational status of CD19+ (B-lymphocyte), CD34+38− (multipotent progenitor/long-term hematopoietic stem cell) and CD34+38+ (myeloid progenitor) fractions of patients who harbored mutations in CD14+ but not CD3+ fractions.
For 19 patients with available material (CD34+CD38− sorted BM cells), single CD34+38− cells were expanded in liquid culture for 10 days. DNA extracted from single-cell–derived colonies was analyzed by mutation-specific PCRs followed by Sanger sequencing.
For the ASXL1 c.1934dupG, the somatic variants were identified and discriminated from background “noise” using a simple methodology, including VAF threshold, strand bias, and insertion vs deletion reads.
Comparison of BM and tissue samples
For 28 patients with available material, tissue NGS was performed in a tissue biopsy, which allowed the diagnosis of ECD.
Statistical analyses
Qualitative variables were expressed as numbers and percentages. Quantitative variables are expressed are means (or medians) and standard deviations (SDs) (or ranges).
Differences between the groups were tested using the Student t test or Mann-Whitney test as appropriate for the continuous data and the Fisher exact test for the qualitative data. Survival curves were built using the Kaplan-Meier method considering the time of complete blood count measurement to death or last follow-up. Follow-up ended in August 2018. For hierarchical ascendant clustering, gene mutation associations were obtained after inclusion of all the variables and represented in a dendrogram plot (“pvclust packages” in R 3.3) generated by a Ward method. Hierarchical cluster analysis was performed using the “Euclidian” method to create the distance matrix and the Ward minimum variance method. All of the tests were 2-sided, and a P value <.05 was considered statistically significant. Statistical analyses were performed using the GraphPad Prism V 6.0 (GraphPad Software), and R software, version 3.3.
Results
Clonal hematopoiesis in ECD
A total of 120 ECD patients were analyzed, 17% of whom had mixed histiocytosis with a concomitant diagnoses of ECD and Langerhans cell histiocytosis (LCH). The mean age at ECD diagnosis was 57 years (SD, 14). The characteristics of the patients are detailed in the Table 1. Sixty-three percent of the population harbored the BRAFV600E mutation. Interestingly, 51 patients (42.5%) had at least 1 mutation in BM defining clonal hematopoiesis (Table 2; Figure 1). Among these 51 patients, 17 (33%) had hematological malignancy, compared with 2 patients (3%) who had a myeloid neoplasm without any mutation in the panel among the 69 ECD patients (P < .0001) (supplemental Table 1). Clonal hematopoiesis was found in 34 of 101 ECD patients (34%) without hematologic malignancy (supplemental Table 1).
Demographic, clinical characteristics, and outcome of ECD patients
| All, n = 120 | CH, n = 51 | No CH, n = 69 | P* | |
|---|---|---|---|---|
| Sex, M/F | 82/38 | 38/13 | 44/25 | .24 |
| Age at first symptoms (mean, SD), y | 53.12 (15.51) | 59.39 (13.12) | 48.48 (15.60) | <.0001† |
| Age at diagnosis (mean, SD), y | 57.12 (13.88) | 63.25 (10.80) | 52.51 (14.21) | <.0001† |
| Age at BM aspirate (mean, SD), y | 61.58 (13.00) | 67.00 (9.83) | 58.00 (13.71) | <.0001† |
| BRAF V600E, (%) | 70/111 (63) | 36/49 (73) | 34/62 (55) | .049† |
| Mixed histiocytosis, (%) | 20 (17) | 8 (16) | 12 (17) | 1.00 |
| Long bone involvement, (%) | 104 (87) | 42 (82) | 62 (90) | .28 |
| Cardiac involvement, (%) | 59 (49) | 27 (53) | 32 (46) | .46 |
| Vascular involvement, (%) | 67 (56) | 34 (67) | 33 (48) | .04† |
| Xanthelasma, (%) | 24 (20) | 10 (20) | 14 (20) | 1.00 |
| Diabetes insipidus, (%) | 34 (28) | 11 (22) | 23 (33) | .22 |
| CNS involvement, (%) | 42 (35) | 15 (29) | 27 (39) | .33 |
| Retro-orbital involvement, (%) | 24 (20) | 13 (25) | 11 (16) | .25 |
| Retroperitoneal involvement | 75 (63) | 38 (75) | 37 (54) | .02† |
| Myeloid malignancies, (%) | 18 (15) | 16 (31) | 2 (3) | <.0001 |
| Deaths, (%) | 8 (7) | 4 (8) | 4 (6) | .72 |
| Follow-up since diagnosis, mean (range), mo | 59 (1-236) | 52 (1-196) | 59 (1-273) | .39 |
| Follow-up since BM aspirate, mean (SD), mo | 13.44 (4.46) | 13.96 (4.83) | 13.06 (4.16) | .21 |
| IFN-α or PEG-IFN-α, (%) | 74 (62) | 31 (61) | 43 (62) | 1.00 |
| Targeted therapy, (%) | ||||
| BRAF inhibitor | 50 (42) | 28 (55) | 22 (33) | .02† |
| MEK inhibitor | 21 (18) | 9 (18) | 12 (17) | 1.00 |
| All, n = 120 | CH, n = 51 | No CH, n = 69 | P* | |
|---|---|---|---|---|
| Sex, M/F | 82/38 | 38/13 | 44/25 | .24 |
| Age at first symptoms (mean, SD), y | 53.12 (15.51) | 59.39 (13.12) | 48.48 (15.60) | <.0001† |
| Age at diagnosis (mean, SD), y | 57.12 (13.88) | 63.25 (10.80) | 52.51 (14.21) | <.0001† |
| Age at BM aspirate (mean, SD), y | 61.58 (13.00) | 67.00 (9.83) | 58.00 (13.71) | <.0001† |
| BRAF V600E, (%) | 70/111 (63) | 36/49 (73) | 34/62 (55) | .049† |
| Mixed histiocytosis, (%) | 20 (17) | 8 (16) | 12 (17) | 1.00 |
| Long bone involvement, (%) | 104 (87) | 42 (82) | 62 (90) | .28 |
| Cardiac involvement, (%) | 59 (49) | 27 (53) | 32 (46) | .46 |
| Vascular involvement, (%) | 67 (56) | 34 (67) | 33 (48) | .04† |
| Xanthelasma, (%) | 24 (20) | 10 (20) | 14 (20) | 1.00 |
| Diabetes insipidus, (%) | 34 (28) | 11 (22) | 23 (33) | .22 |
| CNS involvement, (%) | 42 (35) | 15 (29) | 27 (39) | .33 |
| Retro-orbital involvement, (%) | 24 (20) | 13 (25) | 11 (16) | .25 |
| Retroperitoneal involvement | 75 (63) | 38 (75) | 37 (54) | .02† |
| Myeloid malignancies, (%) | 18 (15) | 16 (31) | 2 (3) | <.0001 |
| Deaths, (%) | 8 (7) | 4 (8) | 4 (6) | .72 |
| Follow-up since diagnosis, mean (range), mo | 59 (1-236) | 52 (1-196) | 59 (1-273) | .39 |
| Follow-up since BM aspirate, mean (SD), mo | 13.44 (4.46) | 13.96 (4.83) | 13.06 (4.16) | .21 |
| IFN-α or PEG-IFN-α, (%) | 74 (62) | 31 (61) | 43 (62) | 1.00 |
| Targeted therapy, (%) | ||||
| BRAF inhibitor | 50 (42) | 28 (55) | 22 (33) | .02† |
| MEK inhibitor | 21 (18) | 9 (18) | 12 (17) | 1.00 |
CH, clonal hematopoiesis; CNS, central nervous system; F, female; IFN, interferon; M, male.
P is a comparison between patients with and without CH.
Statistically significant.
Clonal hematopoiesis mutational frequency and genes mutated in ECD patients
| No. of patients with mutations, n (% among 51 patients) | No. of mutations, n (% among 123 mutations) | |
|---|---|---|
| TET2 | 27 (53) | 47 (38) |
| ASXL1 | 11 (22) | 13 (11) |
| DNMT3A | 9 (18) | 10 (8) |
| NRAS | 7 (14) | 9 (7) |
| CBL | 6 (12) | 6 (5) |
| JAK2 | 5 (10) | 5 (4) |
| KRAS | 4 (8) | 5 (4) |
| SRSF2 | 4 (8) | 4 (3) |
| ZRSR2 | 4 (8) | 4 (3) |
| CALR | 3 (6) | 3 (2) |
| TP53 | 3 (6) | 3 (2) |
| EZH2 | 2 (4) | 2 (2) |
| SF3B1 | 2 (4) | 2 (2) |
| SMC1A | 2 (4) | 2 (2) |
| U2AF1 (U2AF35) | 2 (4) | 2 (2) |
| IDH2 | 1 (2) | 1 (1) |
| ETV6 | 1 (2) | 1 (1) |
| MPL | 1 (2) | 1 (1) |
| NMP1 | 1 (2) | 1 (1) |
| PHF6 | 1 (2) | 1 (1) |
| RIT1 | 1 (2) | 1 (1) |
| BCOR | 0 | 0 |
| BCORL1 | 0 | 0 |
| CSF3R | 0 | 0 |
| FLT3 (-TKD) | 0 | 0 |
| GATA | 0 | 0 |
| IDH1 | 0 | 0 |
| KIT | 0 | 0 |
| NIPBL | 0 | 0 |
| PTPN11 | 0 | 0 |
| RAD21 | 0 | 0 |
| RUNX1 (AML1) | 0 | 0 |
| SETBP1 | 0 | 0 |
| SMC3 | 0 | 0 |
| STAG2 | 0 | 0 |
| WT1 | 0 | 0 |
| Total | 97 | 123 |
| No. of patients with mutations, n (% among 51 patients) | No. of mutations, n (% among 123 mutations) | |
|---|---|---|
| TET2 | 27 (53) | 47 (38) |
| ASXL1 | 11 (22) | 13 (11) |
| DNMT3A | 9 (18) | 10 (8) |
| NRAS | 7 (14) | 9 (7) |
| CBL | 6 (12) | 6 (5) |
| JAK2 | 5 (10) | 5 (4) |
| KRAS | 4 (8) | 5 (4) |
| SRSF2 | 4 (8) | 4 (3) |
| ZRSR2 | 4 (8) | 4 (3) |
| CALR | 3 (6) | 3 (2) |
| TP53 | 3 (6) | 3 (2) |
| EZH2 | 2 (4) | 2 (2) |
| SF3B1 | 2 (4) | 2 (2) |
| SMC1A | 2 (4) | 2 (2) |
| U2AF1 (U2AF35) | 2 (4) | 2 (2) |
| IDH2 | 1 (2) | 1 (1) |
| ETV6 | 1 (2) | 1 (1) |
| MPL | 1 (2) | 1 (1) |
| NMP1 | 1 (2) | 1 (1) |
| PHF6 | 1 (2) | 1 (1) |
| RIT1 | 1 (2) | 1 (1) |
| BCOR | 0 | 0 |
| BCORL1 | 0 | 0 |
| CSF3R | 0 | 0 |
| FLT3 (-TKD) | 0 | 0 |
| GATA | 0 | 0 |
| IDH1 | 0 | 0 |
| KIT | 0 | 0 |
| NIPBL | 0 | 0 |
| PTPN11 | 0 | 0 |
| RAD21 | 0 | 0 |
| RUNX1 (AML1) | 0 | 0 |
| SETBP1 | 0 | 0 |
| SMC3 | 0 | 0 |
| STAG2 | 0 | 0 |
| WT1 | 0 | 0 |
| Total | 97 | 123 |
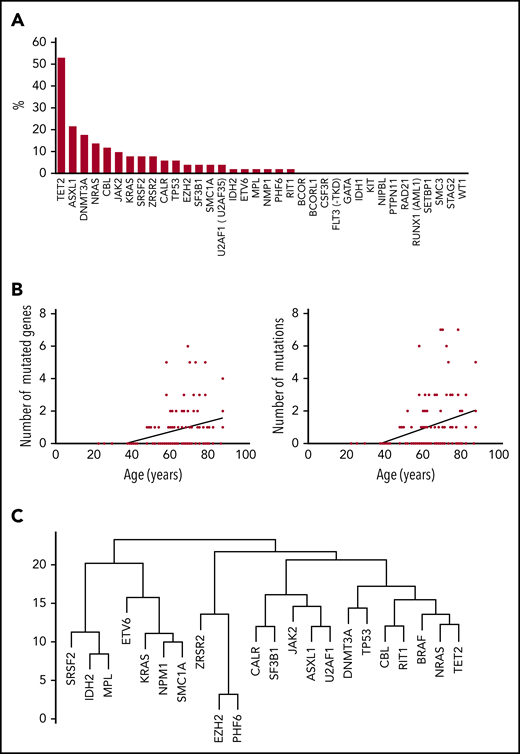
Clonal hematopoiesis in ECD. (A) Mutations (%) on each of the genes of the myeloid panel (BM samples), among patients with clonal hematopoiesis (n = 51). (B) Correlation between the number of mutated genes (y-axis, left) and total number of mutations (y-axis, right) and age at BM aspirate (x-axis). (C) Dendrogram plot of mutations in 51 patients.
Clonal hematopoiesis in ECD. (A) Mutations (%) on each of the genes of the myeloid panel (BM samples), among patients with clonal hematopoiesis (n = 51). (B) Correlation between the number of mutated genes (y-axis, left) and total number of mutations (y-axis, right) and age at BM aspirate (x-axis). (C) Dendrogram plot of mutations in 51 patients.
ECD patients with clonal hematopoiesis were older than those without. Interestingly, the age at first symptom and age at ECD diagnosis were also higher in patients with clonal hematopoiesis. Other clinical characteristics did not differ between ECD patients with and without clonal hematopoiesis (Table 1).
The total number of mutations among the 51 patients was 123. The median number of mutated genes per patient with clonal hematopoiesis was 1 (range, 1-6; mean, 1.9; SD, 1.33). The median number of mutations per patient with clonal hematopoiesis was 2 (range, 1-8; mean, 2.41; SD, 1.75) (Table 2). Thirty of 51 patients (59%) had multiple mutations (range, 2-7 mutations). The median VAF was variable among mutated genes (from 2% to 48%; Figure 2). The 123 mutations occurred in a limited number of genes (21 of 36; 58%). Six genes were mutated in at least 5 patients (TET2, n = 27; ASXL1, n = 11; DNMT3A, n = 9; NRAS, n = 7; CBL, n = 6; and JAK2, n = 5). Both the number of mutated genes and the total number of mutations positively correlated with age (R2 = 0.1025, P = .0004 and .1097, P = .0002, respectively). Of note, among 34 ECD patients without an overt hematologic malignancy, the 3 most recurrent mutated genes were TET2 (n = 19; 56%), DNMT3A (n = 8; 24%), and ASXL1 (n = 5; 15%). Mean duration of follow-up after BM evaluation was 13.4 months (SD, 4.46) (Figure 1). Survival was not different between patients with and without clonal hematopoiesis (hazard ratio, 1.378; 95% confidence interval, 0.3407 to 5.645; P = .65). The NGS analysis was repeated in 30 patients after 6 months, and the results (mutated genes, number of mutated genes, number of mutations) were consistent with the initial evaluation (data not shown). Eight deaths occurred during follow-up: 4 in the group with clonal hematopoiesis and 4 in the group without clonal hematopoiesis.

VAF of mutated genes. Box shows median, 25th, and 75th percentiles. Whiskers show minimal to maximal values. BM samples were used for genotyping. The full list of mutations with VAF is provided in supplemental Table 6.
VAF of mutated genes. Box shows median, 25th, and 75th percentiles. Whiskers show minimal to maximal values. BM samples were used for genotyping. The full list of mutations with VAF is provided in supplemental Table 6.
Myeloid malignancies in patients with ECD
Among the 120 ECD patients, 19 (16%) had an additional hematologic malignancy at the time of BM aspiration (Table 3): 1 with multiple myeloma and 18 with myeloid malignancies. Among these 18 patients, 6 had MPNs, 5 chronic myelomonocytic leukemia (CMML), 5 MDSs, and 2 AML. The clinical characteristics of ECD patients with hematological malignancy did not differ from those who had no hematological malignancy (supplemental Table 2). Notably, age at ECD diagnosis and age at BM aspiration were not different between these 2 populations. Four of 85 patients (5%) carried karyotype abnormalities, all 4 in patients with hematological malignancy. Additionally, 5 patients had a loss of the Y chromosome.
Clinical and biological characteristics of the 19 patients with ECD and hematologic neoplasm
| Diagnosis | Sex | BRAF | Age, y | Mutations | WHO classification | |
|---|---|---|---|---|---|---|
| 1 | ECD | M | N/A | 69 | ZRSR2 | MDS-EB, subtype MDS-EB-1 |
| 2 | ECD + LCH | F | VE | 66 | NRAS, TET2 | Multiple myeloma, smoldering |
| 3 | ECD | M | VE | 71 | ASXL1, TET2 | MDS–EB, subtype MDS-EB-1 |
| 4 | ECD | M | VE | 66 | ASXL1, CALR, U2AF1 | MPN/ET |
| 5 | ECD+ LCH | M | VE | 62 | JAK2, TET2 | MPN/ET with myelofibrosis |
| 6 | ECD | M | VE | 57 | ASXL1, PHF6, EZH2, TET2, ZRSR2 | AML NOS, subtype AML with maturation |
| 7 | ECD | M | VE | 72 | ASXL1, JAK2, NRAS, TET2, U2AF1 | MDS-multi |
| 8 | ECD | M | VE | 60 | KRAS, NRAS | MDS-EB, subtype MDS-EB-1 |
| 9 | ECD | M | VE | 73 | ASXL1, ZRSR2 | CMML, subtype CMML-1 |
| 10 | ECD + RDD | M | WT | 74 | IDH2, MPL, SRSF2 | CMML, subtype CMML-1 |
| 11 | ECD | M | WT | 65 | ASXL1, CALR | MPN/PMF, overt fibrotic stage |
| 12 | ECD | M | VE | 79 | TET2 | CMML, subtype CMML-1 |
| 13 | ECD | F | WT | 63 | KRAS | CMML, subtype CMML-1 |
| 14 | ECD + LCH | M | VE | 68 | KRAS, NPM1, NRAS, SMC1A, SRSF2, TET2 | AML |
| 15 | ECD | F | VE | 69 | CBL, NRAS, RIT1, SRSF2 TET2 | CMML, subtype CMML-1 |
| 16 | ECD | F | VE | 60 | JAK2, SF3B1 | MPN/ET |
| 17 | ECD | M | WT | 71 | CALR, DNMT3A, SF3B1 | MPN/PMF, overt fibrotic stage |
| 18 | ECD | M | VE | 58 | None | MPN/CML, BCR-ABL1+ |
| 19 | ECD | M | VE | 50 | None | MDS-MLD |
| Diagnosis | Sex | BRAF | Age, y | Mutations | WHO classification | |
|---|---|---|---|---|---|---|
| 1 | ECD | M | N/A | 69 | ZRSR2 | MDS-EB, subtype MDS-EB-1 |
| 2 | ECD + LCH | F | VE | 66 | NRAS, TET2 | Multiple myeloma, smoldering |
| 3 | ECD | M | VE | 71 | ASXL1, TET2 | MDS–EB, subtype MDS-EB-1 |
| 4 | ECD | M | VE | 66 | ASXL1, CALR, U2AF1 | MPN/ET |
| 5 | ECD+ LCH | M | VE | 62 | JAK2, TET2 | MPN/ET with myelofibrosis |
| 6 | ECD | M | VE | 57 | ASXL1, PHF6, EZH2, TET2, ZRSR2 | AML NOS, subtype AML with maturation |
| 7 | ECD | M | VE | 72 | ASXL1, JAK2, NRAS, TET2, U2AF1 | MDS-multi |
| 8 | ECD | M | VE | 60 | KRAS, NRAS | MDS-EB, subtype MDS-EB-1 |
| 9 | ECD | M | VE | 73 | ASXL1, ZRSR2 | CMML, subtype CMML-1 |
| 10 | ECD + RDD | M | WT | 74 | IDH2, MPL, SRSF2 | CMML, subtype CMML-1 |
| 11 | ECD | M | WT | 65 | ASXL1, CALR | MPN/PMF, overt fibrotic stage |
| 12 | ECD | M | VE | 79 | TET2 | CMML, subtype CMML-1 |
| 13 | ECD | F | WT | 63 | KRAS | CMML, subtype CMML-1 |
| 14 | ECD + LCH | M | VE | 68 | KRAS, NPM1, NRAS, SMC1A, SRSF2, TET2 | AML |
| 15 | ECD | F | VE | 69 | CBL, NRAS, RIT1, SRSF2 TET2 | CMML, subtype CMML-1 |
| 16 | ECD | F | VE | 60 | JAK2, SF3B1 | MPN/ET |
| 17 | ECD | M | WT | 71 | CALR, DNMT3A, SF3B1 | MPN/PMF, overt fibrotic stage |
| 18 | ECD | M | VE | 58 | None | MPN/CML, BCR-ABL1+ |
| 19 | ECD | M | VE | 50 | None | MDS-MLD |
CML, chronic myeloid leukemia; ET, essential thrombocythemia; MDS-1, myelodysplastic syndrome with single-lineage dysplasia; MDS-EB, myelodysplastic syndrome with excess blast; MDS-MLD, multi–myelodysplastic syndrome with multilineage dysplasia; N/A, not available; NOS, not otherwise specified; PMF, primary myelofibrosis; RDD, Destombes-Rosai-Dorfman disease; VE, BRAF with V600E mutation; WT, BRAF wild type (without V600E mutation).
ECD patients with TET2-mutant clonal hematopoiesis
Twenty-seven patients had at least 1 mutation in TET2. Vascular and retroperitoneal involvements were more frequent in patients with TET2 mutations (supplemental Table 3), and these individuals were older than patients without TET2 mutation. Other clinical characteristics were similar between ECD patients with and without TET2 mutation. The presence of at least 1 TET2 mutation was associated with the presence of the BRAFV600E mutation in tissues (P = .0006). TET2 mutations were closely linked to the presence of BRAF mutations (Figure 1).
Clonal origin of ECD and myeloid neoplasms
Given the high frequency of myeloid neoplasms and clonal hematopoiesis associated with ECD and recent evidence that the cell of origin of at least a proportion of patients with ECD resides in hematopoietic progenitor cells,12,15 we sought to evaluate whether the 2 diseases have a common clonal origin or arose independently. We first performed genotyping for known myeloid clonal hematopoiesis mutations in BM-sorted subpopulations (including monocytes [CD14+], B lymphocytes [CD19+], T lymphocytes [CD3+], and hematopoietic progenitors [CD34+38− and CD34+38+]) of ECD patients with or without an overt hematological malignancy.
In 12 of 36 ECD patients with available material, a total of 24 additional myeloid mutations were detected in the CD14+ fraction. The most frequently affected genes were TET2 (n = 11), ASXL1 (n = 2), DNMT3A (n = 2), and U2AF1 (n = 2) (supplemental Table 4). Among these 12 patients, 10 harbored at least 1 mutation with a significant VAF > 10% in the CD14+ fraction, allowing investigation for the presence of these variants in sorted B lymphocytes and CD34+ progenitors. All variants were found in the CD34+38− fractions (10 of 10 patients; mean VAF, 29%; range, 9% to 68%) and CD34+CD38+ fractions (7 of 7 patients; mean VAF, 24%; range, 10% to 51%) but were unevenly detected in CD19+ cells (mean VAF, 14%; range, <1% to 48%).
Of these 10 patients, to seek for the co-occurrence of the MAPK pathway and clonal hematopoiesis mutations at the level of hematopoietic progenitors, we next analyzed a total of 269 colonies (mean, 33 per patient; range, 13-58) derived from single CD34+CD38− cells of 8 patients known to have a MAPK pathway mutation in lesional histiocytes (6 BRAF V600E, 1 MAP2K1 P124L, 1 KRAS G12D). All tracked additional myeloid mutations were detected in single-cell CD34+38− derived colonies (269 of 269). A total of 36 of 96 colonies was mutated in 3 patients (1 of 45 for the BRAF V600E–mutated patient [Figure 3 patient #A], 6 of 22 for the MAP2K1-mutated patient [patient #C], and 29 of 29 for the KRAS-mutated patient [patient #B]). For each of these 3 patients, we detected colonies that harbored both MAPK pathway and clonal hematopoiesis (TET2, DNMT3A) mutations, suggesting a common clonal origin for both histiocytosis and hematologic disorder (Figure 3). Different subclones were therefore identified that allowed us to infer the order in which mutations were acquired. Interestingly, all potential orders of hierarchical acquisition of mutations could be observed as MAPK pathway mutations were acquired either before (patient #C), at the same time (patient #B), or after (patient #A) clonal hematopoiesis mutations (Figure 3). No MAPK pathway mutations were detected in the colonies of the 5 remaining patients (0 of 117 colonies) (Figure 3 patients #D, E, F, G, and H).

Mutational status of single-cell colonies derived from CD34+ 38−BM progenitor cells in 7 ECD patients. Mutational status of single-cell colonies derived from CD34+38− progenitor cells of 3 ECD patients (#A, B/13, and C) harboring both MAPK pathway and additional myeloid mutations, and of 5 ECD patients (#D/7, E, F/4, G/16, and H) harboring myeloid mutations. Of note, a larger number of colonies were sequenced for the MAPK pathway mutations and only the colonies sequenced for both MAPK pathway mutations and additional myeloid mutations are depicted. Patients who had hematological neoplasms are indicated by “/X”, the X number referring to the number on Table 3. The full details of the mutation list, and correspondence between BM and tissue mutations, are shown in supplemental Table 7.
Mutational status of single-cell colonies derived from CD34+ 38−BM progenitor cells in 7 ECD patients. Mutational status of single-cell colonies derived from CD34+38− progenitor cells of 3 ECD patients (#A, B/13, and C) harboring both MAPK pathway and additional myeloid mutations, and of 5 ECD patients (#D/7, E, F/4, G/16, and H) harboring myeloid mutations. Of note, a larger number of colonies were sequenced for the MAPK pathway mutations and only the colonies sequenced for both MAPK pathway mutations and additional myeloid mutations are depicted. Patients who had hematological neoplasms are indicated by “/X”, the X number referring to the number on Table 3. The full details of the mutation list, and correspondence between BM and tissue mutations, are shown in supplemental Table 7.
Comparison of mutations in tissue vs BM
In 28 patients, we performed NGS on both tissue and BM (supplemental Table 5), allowing the analysis of 91 mutations. The mutations were found in both BM and tissues in 60 of 91 cases (66%) and VAFs were higher in BM than in tissue (P < .0001). Mutations were found only in tissue in 10 cases (11%), and only in BM in 21 cases (23%). The VAFs for patients with analysis of hematopoietic progenitors are presented in supplemental Table 6, and those of non-sorted analyses in supplemental Table 7.
Discussion
In this study, ECD was characterized by a high frequency of myeloid mutations, with 42.5% of patients having genetic alterations in genes recurrently mutated in myeloid malignancies. Clonal hematopoiesis in ECD was manifested as somatic mutations in a limited number of driver genes (TET2, ASXL1, DNMT3A) for MDS, MPN, and AML. Both the number of mutated genes and the number of mutations positively correlated with age. Most of these mutations were found also in histiocytic tissue biopsies.
The frequency of clonal hematopoiesis in ECD was higher than reported in the general population,9,16 which is usually <10% in patients who are ≥70 years of age. Our results argue for a role of accumulated mutations in driver genes of myeloid malignancies in ECD. These findings argue for deeper investigations into the cause of this high frequency of clonal hematopoiesis in ECD. Patients with clonal hematopoiesis were older than patients without mutations, and this result was consistent with previous studies showing that CHIP mutations accumulate with age.7
TET2 was both the most frequently mutated gene, and the gene with the highest number of mutations per gene. TET2 mutations were closely linked with BRAF mutations. Previous evidence in ECD or mixed histiocytosis patients demonstrated a high frequency of mutations in MAPK-signaling intermediates in CD34+ cells, including detection of shared origin of LCH and AML driven by TET2-mutant CD34+ cell progenitors in 1 patient.4 Somatic mutations in TET2 are common in MDS, MPN, and MDS/MPN overlap syndromes, and have been associated with cardiovascular disease.17,18 Here, we found that vascular manifestations were more frequent in ECD patients with TET2 mutations. However, TET2 and BRAF mutations significantly coexisted, and the small number of patients with both mutations precluded multivariate analysis to evaluate a potential independent role of TET2 and BRAF mutations in cardiovascular disease.
Regarding the high prevalence of myeloid malignancies and clonal hematopoiesis in ECD patients, the relationship between clonal hematopoiesis and MAPK-mutant populations is a fundamental question. A striking result of this study is the co-occurrence of myeloid mutations or neoplasia, along with histiocytosis. Several different hypotheses may explain this co-occurrence, including both cell- as well as non–cell-autonomous mechanisms. In the cell-autonomous hypothesis, each disease could undergo sequential acquisition of different mutations as observed in MPNs, in which mutations in epigenetic regulators such as TET2 and DNMT3A occur with disease initiation and may precede the acquisition of JAK2V617F. In our cohort of ECD patients, the usually higher VAF of TET2 mutations compared with MAPK-ERK mutations in BM suggest the TET2 mutation as a possible disease initiation event and MAPK-ERK mutations as secondary events. This scenario possibly occurred with patient #A (Figure 3) as well as patients #D, #E (and to a lesser extent patient #H). In these latter cases, we did not detect CD34+38− MAPK-ERK–mutated colonies but this could be due in part to few available colonies as MAPK-ERK mutations are found at a very low frequency in BM progenitors.15 Another cell-autonomous scenario could be that both “myeloid” clonal hematopoiesis and MAPK-ERK–mutant cells emerge from a common progenitor ancestor cell that harbor mutations or others cellular transforming events that are not identified by targeted exon sequencing. For a non–cell-autonomous mechanism, TET2 mutations could favor clonal outgrowth of BRAF-mutant ECD cells in a paracrine manner but it could also be the other way around: the presence of malignant monocytes/macrophages/histiocytes could create a proinflammatory environment favoring the emergence of TET2-mutant hematopoiesis. Finally, the occurrence of clonal hematopoiesis and ECD disease could be both favored by abnormalities of BM niche/stroma microenvironment.
This study has several limitations: first, the lack of an age-matched control group studied in the same way as the patients here precludes a quantitative comparison of patients with ECD to normal subjects. Although we acknowledge that BM and blood cannot be validly compared, the frequency of clonal hematopoiesis in our ECD cohort of patients without overt hematological malignancy (101 patients) was higher (34 of 101 = 34%) than the frequencies reported in prior publications of clonal hematopoiesis in healthy individuals (5% to 10% for patients between 60 and 80 years old).8-10 Nevertheless, the frequency of TET2 mutations is striking and appears significant, although caution needs to be exercised as those with TET2 mutation were older at diagnosis that those without. The incidences of CALR, NRAS, KRAS, and splicing genes also appear to be higher than one might expect based on prior publications of clonal hematopoiesis in normal subjects. The lack of long follow-up is another limitation of this study, and we can suspect that some patients with clonal hematopoiesis could develop myeloid malignancies.
In conclusion, we demonstrated a high frequency of clonal hematopoiesis in ECD patients. Although we did not identify clonal hematopoiesis as a predictor of survival, the follow-up time was relatively short (13 months) and long-term outcomes should be addressed in future studies. The number of mutated genes and mutations positively correlated with age, but were observed at higher frequencies than in the general population. These results support the clonal myeloid origin of ECD. Potential implications for treatments (including targeted therapies) and follow-up of ECD patients should be considered. Further studies should be directed to establishing the potential of clonal hematopoiesis in ECD, its relationship with survival, and occurrence of myeloid neoplasia.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: F.C.A., Z.A., and J.H. designed the study; F.C.A., A.M.-R., D.R.-W., N.D., S.P., J.-F.E., F.C., and J.H. collected the data; A.M.-R., S.P., and N.D. provided the NGS on BM samples; J.-F.E. and F.C. centrally reviewed the histological samples; J.-F.E. determined the BRAF status and provided NGS data on biopsy samples; F.C.A., D.R.-W., and R.L. conducted the statistical analysis; D.R.-W., M.A., V.D.-V., and O.B. made experiments on BM precursors; F.C.A., D.R.-W., O.A.-W. and J.H. wrote the manuscript; and all authors analyzed and interpreted the data and critically reviewed and approved the final version of the manuscript.
Conflict-of-interest disclosure: F.C.A. and J.H. are investigators of an academic trial on the efficacy of cobimetinib on histiocytoses. The remaining authors declare no competing financial interests.
Correspondence: Fleur Cohen Aubart, Service de Médecine Interne 2, Institut e3m, Groupe Hospitalier Pitié-Salpêtrière, 47-83 Blvd de l’Hôpital, 75 651 Paris CEDEX 13, France; e-mail: fleur.cohen@aphp.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal