


Abstract
BCL2 and MCL1 are commonly expressed prosurvival (antiapoptotic) proteins in hematologic cancers and play important roles in their biology either through dysregulation or by virtue of intrinsic importance to the cell-of-origin of the malignancy. A new class of small-molecule anticancer drugs, BH3 mimetics, now enable specific targeting of these proteins in patients. BH3 mimetics act by inhibiting the prosurvival BCL2 proteins to enable the activation of BAX and BAK, apoptosis effectors that permeabilize the outer mitochondrial membrane, triggering apoptosis directly in many cells and sensitizing others to cell death when combined with other antineoplastic drugs. Venetoclax, a specific inhibitor of BCL2, is the first approved in class, demonstrating striking single agent activity in chronic lymphocytic leukemia and in other lymphoid neoplasms, as well as activity against acute myeloid leukemia (AML), especially when used in combination. Key insights from the venetoclax experience include that responses occur rapidly, with major activity as monotherapy proving to be the best indicator for success in combination regimens. This emphasizes the importance of adequate single-agent studies for drugs in this class. Furthermore, secondary resistance is common with long-term exposure and often mediated by genetic or adaptive changes in the apoptotic pathway, suggesting that BH3 mimetics are better suited to limited duration, rather than continuous, therapy. The success of venetoclax has inspired development of BH3 mimetics targeting MCL1. Despite promising preclinical activity against MYC-driven lymphomas, myeloma, and AML, their success may particularly depend on their tolerability profile given physiological roles for MCL1 in several nonhematologic tissues.
Introduction
The regulatory approvals for venetoclax herald the arrival of a new class of small-molecule cancer therapeutics that target prosurvival proteins that protect against apoptosis. Venetoclax, a potent and selective inhibitor of the intracellular protein BCL2, has received US Food and Drug Administration (FDA) approval for the treatment of chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML). Fast on its heels in development are similarly acting drugs that target either BCL2 or proteins with comparable prosurvival function, particularly MCL1. This review focuses on key areas relevant to clinical use and development of these BH3 mimetic drugs. Underpinning preclinical data will be only selectively touched on, given previous comprehensive reviews. Neither have we attempted to summarize all early-phase trial data in hematologic diseases given the currently rapidly changing trial landscape. Rather, we distil principles that have been established and identify major questions that the field needs to address to enable BCL2 and MCL1 inhibitors to achieve maximum clinical impact.
Biology of the BCL2 family of proteins regulating apoptosis
The development this new class of antineoplastics required a deep understanding of the intrinsic (mitochondrial) pathway to apoptosis.1-5 The pathway is orchestrated by the BCL2 network, a family of partially homologous cytoplasmic proteins, best considered as 3 functional subfamilies. Pivotal in this are the effectors of mitochondrial apoptosis: BAX and BAK.
Within a cell, BAX/BAK are kept inert by the direct and indirect actions of prosurvival family members. These proteins (BCL2, MCL1, BCLxL, BCLw, and BCL2A1) act to prevent BAX/BAK-driven apoptosis, thereby maintaining cellular viability. Physiologic stresses (eg, lack of growth signals, oncogenic stresses, DNA damage) trigger increased expression of the BH3-only proteins, the third subfamily in the network. These BH3-only proteins (eg, BIM, BAD, NOXA) act to initiate apoptosis by binding tightly to prosurvival BCL2 proteins. If present in sufficient amounts, the BH3-only proteins overwhelm the capacity of prosurvival proteins to hold BAX/BAK in check, thus allowing these effectors to oligomerize and form activated pores that damage the mitochondrial outer membrane. Certain BH3-only proteins have also been reported to activate BAX/BAK directly; however, BAX/BAK-driven apoptosis can proceed even in the complete absence of any BH3-only protein.6
BAX/BAK-driven mitochondria outer membrane permeabilization (MOMP) commits a cell toward apoptosis undermining normal mitochondrial function, including energy production, and allowing the leakage of mitochondrial contents, including cytochrome c, which trigger proteolytic enzymes (caspases) causing cellular demolition.
Most normal cells rely on multiple prosurvival BCL2 proteins to maintain their viability although in some cell types particular prosurvival proteins predominate. For example, mature B lymphocytes are highly reliant on BCL2, whereas MCL1 plays a central role in plasma cells.7 This is exaggerated by overexpression of specific prosurvival proteins in certain neoplasms, such as BCL2 in CLL. Regardless, once the action of the prosurvival proteins is blocked by the BH3-only proteins, BAX and BAK are freed to drive apoptosis. Conversely, mitochondrial apoptosis cannot proceed in the absence of BAX and BAK.
Small-molecule BH3 mimetic inhibitors of BCL2 and MCL1 and their mechanism of action
A cardinal feature of this cellular life-death switch is that it is underpinned by protein–protein interactions, distinct from enzymatic reactions such as phosphorylation by kinases (eg, BCR-ABL, FLT3, BTK). To initiate apoptosis, the killer BH3 domain of the BH3-only proteins engages a large, poorly defined groove on the prosurvival proteins. Unlike enzymes such as protein kinases that bind their substrates via small binding interfaces (akin to a “lock-and-key”), the BH3:prosurvival groove interactions use an “induced-fit” mechanism mediated largely by hydrophobic interactions.8 Moreover, multiple molecular interactions are required for tight binding across the large protein-protein interfaces.
Small molecules have been developed to mimic the action of the BH3-only proteins, hence BH3 mimetics. Such small-molecule entities have been generated to preferentially bind ≥1 prosurvival proteins: cells reliant on BCL2 will be highly sensitive to a BCL2-selective inhibitor. By mimicking the action of the BH3-only proteins, a selective chemical inhibitor will induce BAX/BAK-mediated mitochondrial apoptosis unless other prosurvival proteins are present in sufficient amounts to restrain endogenous apoptosis effectors (Figure 1).
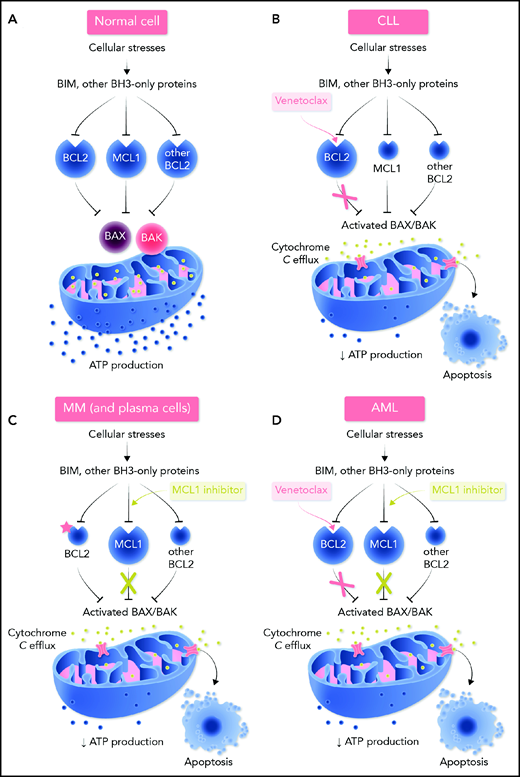
Regulation of the intrinsic (mitochondrial) pathway to apoptosis by the BCL2 protein family and mechanism of action of BH3 mimetics in normal cells and selected hematologic neoplasms. (A) In most normal cells, multiple prosurvival BCL2 proteins act to maintain their survival by preventing the activation of the cell death effectors BAX and BAK. Cellular stresses upregulate the BH3-only proteins (eg, BIM, BAD, PUMA, NOXA) that act by binding to BCL2 or its prosurvival relatives (MCL1, BCLxL, BCLw, BCL2A1). These interactions between BH3-only proteins and prosurvival proteins can be specific (eg, BAD only binds BCL2, BCLxL, and BCLW with high affinity; NOXA only binds MCL1 and BCL2A1) or more encompassing (eg, BIM and PUMA will bind all prosurvival proteins with high affinity).121 Tight binding of the BH3-only proteins blocks the ability of the prosurvival BCL2 proteins to hold BAX/BAK in check. The threshold for apoptosis is determined largely by the relative abundance of the key players and this varies according to cell lineage and microenvironmental cues. In many hematologic cancers, oncogenic stress-induced BH3-only protein activity means that the threshold for inducing apoptosis is low with the malignant cell being “primed for death” in some instances.2 (B) BCL2 expression is often dysregulated in CLL with high levels of expression and the leukemic cells are heavily reliant on BCL2.17,30,122 Hence, CLL cells are susceptible to the selective inhibition of BCL2 with venetoclax (in red). Inhibition of BCL2 by venetoclax allows activated BAX/BAK oligomers to form on the mitochondrial outer membrane, permeabilizing it and thereby compromising normal mitochondrial function (eg, reduced ATP production), as well as triggering the leakage of intramitochondrial molecules (eg, cytochrome c; green). These trigger the activation of proteolytic caspases that lead to cellular demolition. Because BAX/BAK are essential to drive MOMP, they are required for the action of venetoclax and all true BH3 mimetics. (C) In normal plasma cells and in many cases of multiple myeloma, especially relapsed disease, the predominant survival protein expressed is MCL1.7,117,123 Hence, MCL1 inhibition (in green) could well prove to be effective for this plasma cell malignancy. However, as indicated by the red asterisk, some subtypes of multiple myeloma are also highly susceptible to BCL2 inhibition (see Figure 2). (D) In AML, the degree of dependence on BCL2 or on MCL1 varies, with some subtypes being more BCL2 reliant than others (see Figure 2). Overall, both BCL2 and MCL1 appear to play prominent prosurvival roles in most AML cases. Loss of oxidative phosphorylation and energy production after MOMP is a prominent effect of venetoclax in AML cells.91,124 Professional illustration by Somersault18:24.
Regulation of the intrinsic (mitochondrial) pathway to apoptosis by the BCL2 protein family and mechanism of action of BH3 mimetics in normal cells and selected hematologic neoplasms. (A) In most normal cells, multiple prosurvival BCL2 proteins act to maintain their survival by preventing the activation of the cell death effectors BAX and BAK. Cellular stresses upregulate the BH3-only proteins (eg, BIM, BAD, PUMA, NOXA) that act by binding to BCL2 or its prosurvival relatives (MCL1, BCLxL, BCLw, BCL2A1). These interactions between BH3-only proteins and prosurvival proteins can be specific (eg, BAD only binds BCL2, BCLxL, and BCLW with high affinity; NOXA only binds MCL1 and BCL2A1) or more encompassing (eg, BIM and PUMA will bind all prosurvival proteins with high affinity).121 Tight binding of the BH3-only proteins blocks the ability of the prosurvival BCL2 proteins to hold BAX/BAK in check. The threshold for apoptosis is determined largely by the relative abundance of the key players and this varies according to cell lineage and microenvironmental cues. In many hematologic cancers, oncogenic stress-induced BH3-only protein activity means that the threshold for inducing apoptosis is low with the malignant cell being “primed for death” in some instances.2 (B) BCL2 expression is often dysregulated in CLL with high levels of expression and the leukemic cells are heavily reliant on BCL2.17,30,122 Hence, CLL cells are susceptible to the selective inhibition of BCL2 with venetoclax (in red). Inhibition of BCL2 by venetoclax allows activated BAX/BAK oligomers to form on the mitochondrial outer membrane, permeabilizing it and thereby compromising normal mitochondrial function (eg, reduced ATP production), as well as triggering the leakage of intramitochondrial molecules (eg, cytochrome c; green). These trigger the activation of proteolytic caspases that lead to cellular demolition. Because BAX/BAK are essential to drive MOMP, they are required for the action of venetoclax and all true BH3 mimetics. (C) In normal plasma cells and in many cases of multiple myeloma, especially relapsed disease, the predominant survival protein expressed is MCL1.7,117,123 Hence, MCL1 inhibition (in green) could well prove to be effective for this plasma cell malignancy. However, as indicated by the red asterisk, some subtypes of multiple myeloma are also highly susceptible to BCL2 inhibition (see Figure 2). (D) In AML, the degree of dependence on BCL2 or on MCL1 varies, with some subtypes being more BCL2 reliant than others (see Figure 2). Overall, both BCL2 and MCL1 appear to play prominent prosurvival roles in most AML cases. Loss of oxidative phosphorylation and energy production after MOMP is a prominent effect of venetoclax in AML cells.91,124 Professional illustration by Somersault18:24.
BH3 mimetics are therefore defined by their ability to kill cells by apoptosis in a strictly BAX/BAK-dependent manner.3 BH3 mimetics targeting BCL2 (±BCLxL) or MCL1 that have progressed into clinical trials are listed in Table 1. Other than venetoclax and navitoclax, it can be appreciated that the clinical development of other BH3 mimetics is only in the earliest stages. Navitoclax9 was the first potent BCL2 inhibitor to enter clinical trials and demonstrated moderate single-agent activity in patients with relapsed CLL and indolent B-cell lymphomas.10-13 However, acute reduction in platelet count is a direct legacy of its cotargeting of BCLxL, as survival of platelets is highly dependent on BCLxL.14 Consequently, thrombocytopenia proved to be the dose-limiting toxicity, precluding full exploration of BCL2 inhibition. Navitoclax remains in clinical development only for hematologic diseases where targeting of BCLxL or both BCL2 and BCLxL, is anticipated to be beneficial, such as myelofibrosis and acute lymphoblastic leukemia (ALL), respectively.15,16
BCL2-targeting or MCL1-targeting bone fide BH3 mimetics currently in clinical trials
| State of development | Characteristics | ||||||
|---|---|---|---|---|---|---|---|
| Target molecule | Developer | FDA approved | Developmental phase | Heme malignancy | Target Ki* (nM) | BAX/BAK dependent | Route |
| BCL2 | |||||||
| Venetoclax (ABT-199)17 | Abbvie /Genentech | CLL, AML | 3 | MM, NHL, AML, CLL | <<1 | ✓ | Oral |
| S655487 S55746125 | Servier | 1 1 | Multiple CLL, AML | NR ∼1 | NR ✓ | IV Oral | |
| BGB-11417126 | Beigene | 1 | Multiple | <<1 | NR | Oral | |
| APG-2575127 | Ascentage | 1 | CLL, AML, NHL, T-PLL | <<1 | ✓ | Oral | |
| FCN-338128 | Fochon | 1 | CLL, SLL | NR | NR | Oral | |
| BCL2/BCLxL | |||||||
| Navitoclax (ABT-263)9 | Abbvie | 3 | Myelofibrosis | <1/<1 | ✓ | Oral | |
| AZD0466129,130 | Astra Zeneca | 1 | Multiple | NR | ✓ | IV | |
| Pelcitoclax (APG-1252)131,132 | Ascentage | 1/2 | Myelofibrosis | <1/<1 | ✓ | IV | |
| MCL1 | |||||||
| AMG 17619 AMG 39720 | Amgen | 1 | AML, MM Multiple | <<1 <<1 | ✓ | IV Oral | |
| S64315 (MIK665)21 | Servier/Novartis | 1 | AML, MM, NHL | <<1 | ✓ | IV | |
| AZD599122 | Astra Zeneca | 1 | Multiple | <1 | ✓ | IV | |
| ABBV-467 | Abbvie | 1 | MM | NR | NR | IV | |
| PRT1419 | Prelude | 1 | MDS, AML, NHL, MM | NR | NR | Oral | |
| State of development | Characteristics | ||||||
|---|---|---|---|---|---|---|---|
| Target molecule | Developer | FDA approved | Developmental phase | Heme malignancy | Target Ki* (nM) | BAX/BAK dependent | Route |
| BCL2 | |||||||
| Venetoclax (ABT-199)17 | Abbvie /Genentech | CLL, AML | 3 | MM, NHL, AML, CLL | <<1 | ✓ | Oral |
| S655487 S55746125 | Servier | 1 1 | Multiple CLL, AML | NR ∼1 | NR ✓ | IV Oral | |
| BGB-11417126 | Beigene | 1 | Multiple | <<1 | NR | Oral | |
| APG-2575127 | Ascentage | 1 | CLL, AML, NHL, T-PLL | <<1 | ✓ | Oral | |
| FCN-338128 | Fochon | 1 | CLL, SLL | NR | NR | Oral | |
| BCL2/BCLxL | |||||||
| Navitoclax (ABT-263)9 | Abbvie | 3 | Myelofibrosis | <1/<1 | ✓ | Oral | |
| AZD0466129,130 | Astra Zeneca | 1 | Multiple | NR | ✓ | IV | |
| Pelcitoclax (APG-1252)131,132 | Ascentage | 1/2 | Myelofibrosis | <1/<1 | ✓ | IV | |
| MCL1 | |||||||
| AMG 17619 AMG 39720 | Amgen | 1 | AML, MM Multiple | <<1 <<1 | ✓ | IV Oral | |
| S64315 (MIK665)21 | Servier/Novartis | 1 | AML, MM, NHL | <<1 | ✓ | IV | |
| AZD599122 | Astra Zeneca | 1 | Multiple | <1 | ✓ | IV | |
| ABBV-467 | Abbvie | 1 | MM | NR | NR | IV | |
| PRT1419 | Prelude | 1 | MDS, AML, NHL, MM | NR | NR | Oral | |
Trial data from ClinicalTrials.gov (accessed 2 January 2021). The most advanced trial phase is listed. References are provided where publication has disclosed the chemical structure and function of the compound. NR, not reported.
Ki measures the affinity for the target in a competition assay. As Ki is measured relative to a competitor and the competitors and assay formats are not uniform across drugs, direct comparison of Ki values between the drugs listed is precluded. Therefore, approximations are provided based on cited specific published data. A Ki of 1 nM or less reflects tight binding to the target and potency in competing with BH3-containing proteins.
To avoid the on-target thrombocytopenia associated with navitoclax, venetoclax was designed to be a BCL2-specific inhibitor. No naturally occurring BH3-only proteins inhibit BCL2 exclusively. Venetoclax binds and inhibits BCL2 with picomolar affinity and displays greater than 100-fold selectivity for BCL2 over BCLxL and >1000-fold for MCL1.17 The demonstration that a single prosurvival protein can be inhibited and have important clinical activity turbo-charged drug discovery and development of this class. Similarly, MCL1 can now be selectively targeted by potent BH3 mimetics.18-22
Hematologic malignancies where BCL2 inhibition has an established indication
CLL
Dependence on BCL2 is high and relatively uniformly so in CLL. BCL2 is required but not essential for normal B-cell development, and mature B cells normally express high levels of BCL2.23 CLL cells in all patients express high levels of BCL2 and typically much lower levels of MCL1 or BCLxL.23-27 CLL bearing del(13q) have loss of mir15/16, negative regulators of BCL2 expression, with enhanced levels of BCL2 expression.28,29 In vitro, CLL cells are highly sensitive to BCL2-selective inhibition (Figures 1 and 2).17,30,31

Summary of key preclinical and clinical characteristics of hematologic cancers where BCL2 inhibitors demonstrate clinical activity. At the single cell level, high vulnerability to BCL2 inhibition (left) is typically observed when BCL2 expression is constitutively high, and there is relatively minor expression of other prosurvival proteins.17,133 These BCL2-dependent cells reflect the near totality of cells among populations where CRs can readily be induced with monotherapy (eg, CLL,11,17,27,30,31,41 some MCL,103,105 t(11;14) myeloma and myeloma with heightened BCL2:BCLxl expression,96,97,123IDH-mutant AML69,80). For these cell populations, BH3 mimetics like venetoclax strike the bullseye. More commonly (right), cells are not BCL2 dependent, and BCL2 is not the bullseye. Rather these cells are vulnerable to BCL2 inhibition when a wave (depicted as green lines) of secondary inhibition of other prosurvival proteins by displaced BH3-only proteins134 is initiated, resulting in antagonism of other prosurvival proteins and BCL2. In these partially BCL2-dependent cells, BCL2 inhibitor monotherapy may be insufficient to kill cells with these characteristics (moderate and variable BCL2 expression, relatively higher levels of MCL1 or BCLxL), but some cells will die, whereas others suffer sublethal injury consequent on BAX/BAK-dependent mitochondrial depolarization and reduced energy production.53,91 Additional stressors will be required to maximize apoptosis and achieve high response rates in diseases in which the majority of cells are affected in this manner. This includes most myeloma,97 AML,67,69,135 ALL,16 and many B-cell lymphomas.17,105,136 Adapted137 with permission. Professional illustration by Somersault18:24.
Summary of key preclinical and clinical characteristics of hematologic cancers where BCL2 inhibitors demonstrate clinical activity. At the single cell level, high vulnerability to BCL2 inhibition (left) is typically observed when BCL2 expression is constitutively high, and there is relatively minor expression of other prosurvival proteins.17,133 These BCL2-dependent cells reflect the near totality of cells among populations where CRs can readily be induced with monotherapy (eg, CLL,11,17,27,30,31,41 some MCL,103,105 t(11;14) myeloma and myeloma with heightened BCL2:BCLxl expression,96,97,123IDH-mutant AML69,80). For these cell populations, BH3 mimetics like venetoclax strike the bullseye. More commonly (right), cells are not BCL2 dependent, and BCL2 is not the bullseye. Rather these cells are vulnerable to BCL2 inhibition when a wave (depicted as green lines) of secondary inhibition of other prosurvival proteins by displaced BH3-only proteins134 is initiated, resulting in antagonism of other prosurvival proteins and BCL2. In these partially BCL2-dependent cells, BCL2 inhibitor monotherapy may be insufficient to kill cells with these characteristics (moderate and variable BCL2 expression, relatively higher levels of MCL1 or BCLxL), but some cells will die, whereas others suffer sublethal injury consequent on BAX/BAK-dependent mitochondrial depolarization and reduced energy production.53,91 Additional stressors will be required to maximize apoptosis and achieve high response rates in diseases in which the majority of cells are affected in this manner. This includes most myeloma,97 AML,67,69,135 ALL,16 and many B-cell lymphomas.17,105,136 Adapted137 with permission. Professional illustration by Somersault18:24.
Efficacy
Venetoclax was first approved by the FDA as monotherapy for relapsed or refractory del(17p) CLL in April 2016. It now has multiple listings internationally for relapsed/refractory disease in combination with rituximab and for unfit patients with newly diagnosed CLL in combination with obinutuzumab. Monotherapy studies in relapsed/refractory disease indicated an immediate reduction in CLL burden in almost all patients. Objective responses occurred in 79% of patients, complete remissions (CRs) in 20% and undetectable measurable residual disease (uMRD) in up to 30%32-34 with acceptable toxicity.35 Indeed, the dose-limiting toxicity (DLT) was biochemical tumor lysis, with several patients experiencing severe or fatal clinical tumor lysis syndrome (TLS).32 Patients with high burden disease or reduced renal function were at highest risk. This necessitated introduction of a standard 4-week ramp-up phase commencing at 20 mg/day and rising incrementally each week to the target dose of 400 mg/day. In patients, apoptosis of CLL cells was readily detectable within 6 to 8 hours of dosing,31 consistent with the rapid induction observed in vitro and the timing of peak plasma venetoclax concentrations (6 hours).32 Importantly, venetoclax is highly active after prior therapy with chemo-immunotherapy, BTK inhibitors, or phosphatidylinositol 3-kinase inhibitors.32,34,36,37
Apparently higher CR (51%) and uMRD (57%) rates were observed in early-phase trials combining rituximab with venetoclax,38 prompting this combination to be compared in a randomized trial against bendamustine-rituximab in relapsed/refractory CLL. Venetoclax-rituximab demonstrated superior efficacy and safety; respective hazard ratios (HRs) for progression-free survival (PFS) and overall survival (OS) were 0.17 and 0.48, with a number-needed-to treat (NNT) of 2 for PFS at 2 years.39,40 Despite its now uniform adoption as the standard venetoclax regimen for relapsed/refractory CLL, in the absence of a randomized comparison vs venetoclax monotherapy, the evidence for additive benefit from rituximab is modest and confined to improvement in CR rate in a multivariable analysis of pooled trial data.41 Uncontrolled real-world data analyses of PFS do not suggest additive benefit,42 and it seems most probable that venetoclax is delivering the majority of the observed clinical efficacy.
In first-line treatment of patients unfit for fludarabine-based regimens, venetoclax-obinutuzumab proved superior to chlorambucil-obinutuzumab with respect to PFS (HR, 0.31; NNT of 3 at 3 years), with similar toxicity and OS.43 As previously observed for fludarabine-cyclophosphamide-rituximab chemo-immunotherapy, the durability of benefit with venetoclax-based regimens correlated with achievement of deeper responses (measured either by CR or uMRD rate).38,40,41,44 In the salvage setting, the probability of achieving uMRD with continuing venetoclax plateaus around 2 years34,40,41,44 and earlier in the first-line setting.44 This has provided a rational basis for the introduction of regimens with fixed-duration venetoclax (2 years for relapsed/refractory, 1 year for first-line) in registration studies rather than continuous use which may not provide additional benefit.45
Biomarkers
However, long-term follow-up data indicate an ongoing risk of relapse with these venetoclax-based regimens, and it seems improbable that these treatments are curative. Accumulated trial data now enable analyses of which factors either modify the response to venetoclax and/or predict for particular benefit from the use of venetoclax-based regimens over chemo-immunotherapy and/or are prognostic with venetoclax-based regimens. In relapsed CLL, the validated genetic biomarkers most relevant to clinical practice are del(17p), TP53 mutation, and immunoglobulin heavy chain variable region (IGHV) mutational status.46Table 2 summarizes the key findings from randomized trials. Other factors have been associated with relatively reduced response rates or inferior duration of response compared with their absence (bulky lymphadenopathy >10 cm; refractoriness to ibrutinib; NOTCH1 mutations) in multivariable analyses of pooled data from single arm trials,41 but these have not yet been confirmed in prospective trials.
Treatment effect modifiers for venetoclax in CLL, myeloma, and AML
| Disease line, regimen | Biomarker | Improvements in treatment outcomes vs control arm (in comparison with absence of marker) | Predict benefit vs control arm? | Prognostic with Ven? | |||||
|---|---|---|---|---|---|---|---|---|---|
| Chemo-Ab (Chl-Obin or Benda-R) → Ven-Ab | |||||||||
| CLL | Name | Status | CR | uMRD (<10–4) | 2-y PFS | 3-y PFS | CR and uMRD | PFS | |
| First line43,44,47 Ven-Obin | del(17p) | Present Absent | 7% → 44% (24% → 50%) | 7% → 71% 38% → 76% | 23% → 65% (∼71% → 91%) | 12% → 49% (>50% → ∼84%) | Yes ↑↑ | Yes ↑ | Yes ↓ |
| Relapsed39,48 Ven-R | Present Absent | NR | 5%* → 60% (20%* → 76%) | 28% → 82% (41% → 86%) | 0% → 40% (∼30% → 80%) | Yes ↑↑ | Yes ↑↑ | Yes ↓ | |
| First line | TP53 mut | Present Absent | 5% → 48% (25% → 50%) | 21 → 65 (37% → 77%) | 37% → 73% (∼73% → 93%) | 35% → 60% (53% → 85%) | Yes ↑↑ | Yes ↑ | Yes ↓ |
| Relapsed | Present Absent | NR | 5% → 57%† (20% → 66%) | HR 0.19† (HR 0.15) | ∼10% → 63% (∼25% → 80%) | Yes ↑↑ | Yes ↑ | Yes ↓ | |
| First line | Unmutated IGHV | Unmutated Mutated | 15% → 61% (16% → 64%) | 28% → 79% (43% → 74%) | 51% → 89% (86% → 90%) | 34% → 81% (75% → 87%) | Yes ↑↑ | Yes ↑↑ | Probably‡ ↓ |
| Relapsed | Unmutated Mutated | NR | 15% → 61% (16% →64%) | HR 0.16 HR (0.11) | NR | Yes ↑↑ | Yes ↑ | NR | |
| Pbo-BortDex → Ven-BortDex | |||||||||
| Myeloma | Name | Status | ≥vGPR | uMRD (<10–5) | PFS | Survival HR | ≥vGPR & PFS | OS | |
| Relapsed99 Ven-BD | t(11;14) | Present Absent | 27% → 75% (38% → 58%) | 0% → 25% (1% → 12%) | HR 0.11 (HR 0.67) | 0.34 (2.5) | Yes ↑↑ | No | Yes ↑ |
| High BCL2§ | First quartile Fourth quartile | 28% → 71% (40% → 52%) | 0% → 18% (2% →11%) | HR 0.24 (HR 0.76) | 1.0 (>2.0) | Yes ↑↑ | No | Yes ↑ | |
| Pbo-Aza → Ven-Aza | |||||||||
| AML | Name | Status | CR/Cri | ΔCR/Cri | 2-y OS | HR OS | CR | OS | |
| First line74 Ven-Aza | IDH1/2 mut | Present Absent¶ | 11% → 75% 32% → 66% | +64% +34% | 14% → 52% (14% → 41%) | 0.34 | Yes ↑↑ | Yes ↑↑ | Yes ↑ |
| TP53 mut | Present Absent¶ | 0% → 55% 31% → 68% | +55% +37% | 7% → 11% 27% → 49% | 0.76 | Yes ↑↑ | No | Yes ↓ | |
| Disease line, regimen | Biomarker | Improvements in treatment outcomes vs control arm (in comparison with absence of marker) | Predict benefit vs control arm? | Prognostic with Ven? | |||||
|---|---|---|---|---|---|---|---|---|---|
| Chemo-Ab (Chl-Obin or Benda-R) → Ven-Ab | |||||||||
| CLL | Name | Status | CR | uMRD (<10–4) | 2-y PFS | 3-y PFS | CR and uMRD | PFS | |
| First line43,44,47 Ven-Obin | del(17p) | Present Absent | 7% → 44% (24% → 50%) | 7% → 71% 38% → 76% | 23% → 65% (∼71% → 91%) | 12% → 49% (>50% → ∼84%) | Yes ↑↑ | Yes ↑ | Yes ↓ |
| Relapsed39,48 Ven-R | Present Absent | NR | 5%* → 60% (20%* → 76%) | 28% → 82% (41% → 86%) | 0% → 40% (∼30% → 80%) | Yes ↑↑ | Yes ↑↑ | Yes ↓ | |
| First line | TP53 mut | Present Absent | 5% → 48% (25% → 50%) | 21 → 65 (37% → 77%) | 37% → 73% (∼73% → 93%) | 35% → 60% (53% → 85%) | Yes ↑↑ | Yes ↑ | Yes ↓ |
| Relapsed | Present Absent | NR | 5% → 57%† (20% → 66%) | HR 0.19† (HR 0.15) | ∼10% → 63% (∼25% → 80%) | Yes ↑↑ | Yes ↑ | Yes ↓ | |
| First line | Unmutated IGHV | Unmutated Mutated | 15% → 61% (16% → 64%) | 28% → 79% (43% → 74%) | 51% → 89% (86% → 90%) | 34% → 81% (75% → 87%) | Yes ↑↑ | Yes ↑↑ | Probably‡ ↓ |
| Relapsed | Unmutated Mutated | NR | 15% → 61% (16% →64%) | HR 0.16 HR (0.11) | NR | Yes ↑↑ | Yes ↑ | NR | |
| Pbo-BortDex → Ven-BortDex | |||||||||
| Myeloma | Name | Status | ≥vGPR | uMRD (<10–5) | PFS | Survival HR | ≥vGPR & PFS | OS | |
| Relapsed99 Ven-BD | t(11;14) | Present Absent | 27% → 75% (38% → 58%) | 0% → 25% (1% → 12%) | HR 0.11 (HR 0.67) | 0.34 (2.5) | Yes ↑↑ | No | Yes ↑ |
| High BCL2§ | First quartile Fourth quartile | 28% → 71% (40% → 52%) | 0% → 18% (2% →11%) | HR 0.24 (HR 0.76) | 1.0 (>2.0) | Yes ↑↑ | No | Yes ↑ | |
| Pbo-Aza → Ven-Aza | |||||||||
| AML | Name | Status | CR/Cri | ΔCR/Cri | 2-y OS | HR OS | CR | OS | |
| First line74 Ven-Aza | IDH1/2 mut | Present Absent¶ | 11% → 75% 32% → 66% | +64% +34% | 14% → 52% (14% → 41%) | 0.34 | Yes ↑↑ | Yes ↑↑ | Yes ↑ |
| TP53 mut | Present Absent¶ | 0% → 55% 31% → 68% | +55% +37% | 7% → 11% 27% → 49% | 0.76 | Yes ↑↑ | No | Yes ↓ | |
The table summarizes the randomized trial data for genetic markers with the most robust evidence informing treatment effect modification: either abrogation of previously established negative treatment effect modification by venetoclax or the presence of novel positive treatment effect modification with venetoclax therapy. In CLL, the established negative treatment effect modifiers for chemo-immunotherapy (del(17p), TP53 mutations and unmutated IGHV status) do not affect responsiveness to venetoclax, and their presence predicts major benefit from use of venetoclax over chemo-immunotherapy, but they remain negatively prognostic. In multiple myeloma, the presence of either t(11;14) or high BCL2 RNA expression is a strong positive treatment effect modifier, with the magnitude of response and PFS benefits for venetoclax significantly exceeding that seen in marker-negative myeloma. In AML, the presence of mutations in IDH1 or IDH2 is a strong positive treatment effect modifier with the magnitude of benefits for venetoclax significantly exceeding that seen in marker-negative AML. Although TP53 mutations in AML are associated with magnified benefit with respect CR/CRi rate, improved survival has not been shown; the presence of TP53 mutations is associated with a lower CR/CRi rate with venetoclax and a negative prognosis compared with AML with normal TP53. ∼, estimated from Kaplan-Meier plots;
Aza, azacytidine; BD, bortezomib + dexamethasone; Benda, bendamustine; Chl, chlorambucil; CRi, CR, including with hematologic incomplete recovery; HR, hazard ratio for progression vs control, included when actual rates were not provided, and italicized when not statistically significantly different from control therapy; NR, not reported; Obin, obinutuzumab; Pbo, placebo; R, rituximab; Ven, venetoclax.
Includes data for either del(17p) or TP53 mutated cases for control treatment arm, as del(17p) data alone were not reported.
All data in these comparisons include either del(17p) or TP53 mutations.
PFS: HR, 1.96; 95% CI, 0.92-4.17.
High BCL2 RNA expression.
Absence of a marker includes patients with unknown status.
Abnormalities of TP53 function are known negative response-effect modifiers for DNA-damaging therapy in CLL (and other hematologic malignancies), characterized by consistent associations with inferior CR, uMRD, PFS, and OS rates in trials and mechanistic studies in model systems.46 With venetoclax ± anti-CD20 antibodies, no negative effect is observed for overall response, CR, or MRD clearance rates or with CLL killing in vitro.31,39,43,47,48 However, the presence of a TP53 abnormality before therapy is associated with an increased rate of relapse, compared with CLL with normal TP53, and remains prognostic.41,47,48 The magnitude of benefit for venetoclax-based regimens over chemo-immunotherapy is greater for patients with TP53-aberrant CLL than observed for those with TP53 wild-type CLL.47,48 Similarly, patients with unmutated IGHV CLL have inferior outcomes with chemo-immunotherapy compared with those whose CLL has mutated IGHV. Although unmutated IGHV is a negative treatment effect modifier for chemo-immunotherapy, it is not for venetoclax-based regimens, and its presence predicts for major benefit from the use of venetoclax.41,47 Both mutated and unmutated IGHV CLL have similar rates and depths of response. However, unmutated IGHV may remain a negative prognostic factor (compared with mutated IGHV; Table 2), in long-term follow-up of approved venetoclax-based regimens.47
Resistance
Robust data are also emerging about the molecular basis for treatment failure in CLL (Table 3). Early progression (within the first year) is uncommon and closely associated with complex karyotype and Richter transformation in heavily pretreated patients.49,50 Secondary resistance in patients receiving continuous venetoclax can be caused by several independently occurring mechanisms (Table 3; Figure 3). These include mutations in BCL2 (eg, Gly101Val), overexpression of MCL1, and overexpression of BCLxL.51-54 All have been validated functionally, with each reducing the intrinsic sensitivity of CLL cells to venetoclax by 1.5 to 3 logs in both patient cells and model systems. BCL2 mutations reduce binding of the drug but retain pro-survival function of the protein.51,55 Heightened expression of MCL1 or BCLxL provides cells with sufficient alternative prosurvival function to avoid apoptosis in the presence of venetoclax. It is common for relapse to be polyclonal, with independent mechanisms evident in different subclones. For example, as well as the 2 more common BCL2 mutations (Gly101Val and Asp104Tyr), 6 other BCL2 mutations have been described, with many patients demonstrating several independent subclones, each characterized by a different mutation.56 How the BCL2 mutations are generated remains unknown. The great majority have never been observed outside the context of secondary venetoclax resistance. MCL1 overexpression can be the consequence of amplification of the MCL1 locus on chromosome 1q21 but can also occur in the absence of gene amplification.53 The presumed epigenetic mechanisms leading to MCL1 overexpression in those circumstances and BCLxL overexpression in other patients’ cells remain to be elucidated. With most patients treated with venetoclax now completing time-limited therapy in objective response and early data suggesting that re-exposure to the drug induces secondary responses in 12 of 17 cases reported in 2 prospective studies to date,45,48 further studies are required to understand whether the landscape of resistance will be different with repeat exposure.
Clinical, genetic, and epigenetic alterations associated with venetoclax resistance in patients, classified by class and tier of supportive evidence
| Class | Gene or gene product | CLL | Mantle cell lymphoma | AML | |
|---|---|---|---|---|---|
| Clinical | High bulk41 | Tier 3 | |||
| BTKi clinical resistance41 | Tier 3 | ||||
| Prior azacitidine75 | Tier 3 | ||||
| Monocytic lineage86 | Tier 2 | ||||
| Cytogenetic | Complex karyotype48,74 | Tier 1* | Tier 2 | ||
| del9p138 | Tier 1† | ||||
| Mutations/CNV | Apoptosis | TP53 mut/loss (incl del17p)41,47,81,82 | Tier 2 | Tier 2 | |
| BCL2 mut51,52,54,56 | Tier 1 | ||||
| MCL1 amp53 | Tier 1 | ||||
| BAX mut84,85 | Tier 1 | Tier 2 | |||
| Signaling | Notch1 mut (activating)41 | Tier 2 | |||
| Activated growth factor49,81 | Braf; Tier 2 | FLT3; Tier 1 Ras; Tier 2 | |||
| Cell cycle | CDKN2A/B mut/loss49 | Tier 1* | |||
| BTG1 mut49 | Tier 2 | ||||
| Epigenetic regulator | SWI/SNF family mut/loss138 | Tier1† | |||
| Expression | Apoptosis | ↓BCL2138 | Tier 1 | ||
| ↑BCLxL51,78,82,86,138 | Tier 1 | Tier 1 | Tier 1 | ||
| ↑MCL153,86 | Tier 1 | Tier 1 | |||
| ↑BCL2A1139,140 | Tier 1 | Tier 1 | |||
| ↓PUMA78 | Tier 2 | ||||
| ↓NOXA82,88 | Tier 2 | ||||
| Mitochondrial metabolism | AMPK53 | Tier 1 | Tier 1 |
| Class | Gene or gene product | CLL | Mantle cell lymphoma | AML | |
|---|---|---|---|---|---|
| Clinical | High bulk41 | Tier 3 | |||
| BTKi clinical resistance41 | Tier 3 | ||||
| Prior azacitidine75 | Tier 3 | ||||
| Monocytic lineage86 | Tier 2 | ||||
| Cytogenetic | Complex karyotype48,74 | Tier 1* | Tier 2 | ||
| del9p138 | Tier 1† | ||||
| Mutations/CNV | Apoptosis | TP53 mut/loss (incl del17p)41,47,81,82 | Tier 2 | Tier 2 | |
| BCL2 mut51,52,54,56 | Tier 1 | ||||
| MCL1 amp53 | Tier 1 | ||||
| BAX mut84,85 | Tier 1 | Tier 2 | |||
| Signaling | Notch1 mut (activating)41 | Tier 2 | |||
| Activated growth factor49,81 | Braf; Tier 2 | FLT3; Tier 1 Ras; Tier 2 | |||
| Cell cycle | CDKN2A/B mut/loss49 | Tier 1* | |||
| BTG1 mut49 | Tier 2 | ||||
| Epigenetic regulator | SWI/SNF family mut/loss138 | Tier1† | |||
| Expression | Apoptosis | ↓BCL2138 | Tier 1 | ||
| ↑BCLxL51,78,82,86,138 | Tier 1 | Tier 1 | Tier 1 | ||
| ↑MCL153,86 | Tier 1 | Tier 1 | |||
| ↑BCL2A1139,140 | Tier 1 | Tier 1 | |||
| ↓PUMA78 | Tier 2 | ||||
| ↓NOXA82,88 | Tier 2 | ||||
| Mitochondrial metabolism | AMPK53 | Tier 1 | Tier 1 |
The strength of evidence for a clinically important role of an associated marker in venetoclax resistance is categorized into tiers: tier 1, proven mechanism and associated with resistance in multiple patients; tier 2, plausible mechanism and observed in some patients; tier 3, associated with resistance in multiple patients, but no mechanism established. Where a molecular marker is associated with primary resistance to venetoclax, the tier is in bold. Markers associated with secondary resistance are recorded in normal font. Consistent with expectations arising from our knowledge of the regulation of apoptosis, genetic or epigenetic alterations in BCL2 family expression are common causes of secondary resistance to venetoclax.
BTKi, Bruton tyrosine kinase inhibitor; CK, complex karyotype; CNV, copy number variation.
Associated with Richter transformation.
del(9p) and SWI/SNF family mutations are causative of resistance when they co-occur.
![Direct and indirect alterations to apoptosis regulators explain many of the proven mechanisms of clinical resistance to venetoclax. The graphic highlights the interrelations between validated genetic and epigenetic mechanisms of venetoclax resistance observed in patients (tier 1 mechanisms listed in Table 3 with references) and their convergence on the expression or function of BCL2 family proteins and/or on mitochondria. Genetic aberrations in BCL2 family members such as amplifications affecting MCL1, coding mutations in BCL2 (*such as encode BCL2 Gly101Val) that alter venetoclax binding, or nonsense mutations (eg, BAX) may occur. Alternatively, genetic changes in their regulators (eg, TP53, del9p, members of the SWI/SNF complex) may alter their expression indirectly (epigenetic changes [Δ]). TP53 loss or mutation (in green) can indirectly reduce sensitivity to venetoclax by reducing expression of BH3-only proteins (eg, PUMA, NOXA) and/or by increasing the threshold for BAX/BAK-induced permeabilization of the outer mitochondrial membrane.83 Activated signaling in various growth factor pathways (in blue; eg, via kinase mutations) can indirectly increase expression of other prosurvival proteins or increase mitochondrial metabolism.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/13/10.1182_blood.2020006785/10/m_bloodbld2020006785cf3.png?Expires=1765884115&Signature=Ev0uUx6o958s5AQ3qYvVDeYYNcEsxx3Cs2~00875Bb3i0eM8abLZL61Ol~E9GmrTu5vfK2k6JVyzZYBsHcgkB7933i9GUIlOWMkLp9feSIU3G-6x9GGQxJqT~H3efPtHHdZuOjqZp116RH0VNRFuetUUdX6DDMnW2C4Dt8lS32NLN62SjCENvQx8Ayxxg~VubTT1WeRHk5td2csIj4kFmHqbW~a59I8n~XvRz7YtbUfEW79uT1G6it1FZMKsjJj94r0OcYEgrt5ylEiXub0sg34Pnzw0JMmfcPhA-7VTupnq8-Xx98qjsaFuilC5RDxlQ3sYyL-cZngg2ABzRF600g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Direct and indirect alterations to apoptosis regulators explain many of the proven mechanisms of clinical resistance to venetoclax. The graphic highlights the interrelations between validated genetic and epigenetic mechanisms of venetoclax resistance observed in patients (tier 1 mechanisms listed in Table 3 with references) and their convergence on the expression or function of BCL2 family proteins and/or on mitochondria. Genetic aberrations in BCL2 family members such as amplifications affecting MCL1, coding mutations in BCL2 (*such as encode BCL2 Gly101Val) that alter venetoclax binding, or nonsense mutations (eg, BAX) may occur. Alternatively, genetic changes in their regulators (eg, TP53, del9p, members of the SWI/SNF complex) may alter their expression indirectly (epigenetic changes [Δ]). TP53 loss or mutation (in green) can indirectly reduce sensitivity to venetoclax by reducing expression of BH3-only proteins (eg, PUMA, NOXA) and/or by increasing the threshold for BAX/BAK-induced permeabilization of the outer mitochondrial membrane.83 Activated signaling in various growth factor pathways (in blue; eg, via kinase mutations) can indirectly increase expression of other prosurvival proteins or increase mitochondrial metabolism.
Direct and indirect alterations to apoptosis regulators explain many of the proven mechanisms of clinical resistance to venetoclax. The graphic highlights the interrelations between validated genetic and epigenetic mechanisms of venetoclax resistance observed in patients (tier 1 mechanisms listed in Table 3 with references) and their convergence on the expression or function of BCL2 family proteins and/or on mitochondria. Genetic aberrations in BCL2 family members such as amplifications affecting MCL1, coding mutations in BCL2 (*such as encode BCL2 Gly101Val) that alter venetoclax binding, or nonsense mutations (eg, BAX) may occur. Alternatively, genetic changes in their regulators (eg, TP53, del9p, members of the SWI/SNF complex) may alter their expression indirectly (epigenetic changes [Δ]). TP53 loss or mutation (in green) can indirectly reduce sensitivity to venetoclax by reducing expression of BH3-only proteins (eg, PUMA, NOXA) and/or by increasing the threshold for BAX/BAK-induced permeabilization of the outer mitochondrial membrane.83 Activated signaling in various growth factor pathways (in blue; eg, via kinase mutations) can indirectly increase expression of other prosurvival proteins or increase mitochondrial metabolism.
BTK inhibitors (BTKi) are the other class of targeted novel drugs that have revolutionized care in CLL. Like venetoclax, they are highly effective as monotherapy and against TP53-aberrant disease.57,58 Unlike venetoclax, they are not rapidly cytotoxic; rather CLL cells die of neglect because of blockade of B-cell receptor and chemokine receptor signaling.58 Indefinite continuous rather than fixed-duration therapy appears required. Preclinical data indicate synergy between BTKi and BCL2 inhibition, and circulating CLL cells from patients taking ibrutinib are sensitized to in vitro killing by venetoclax.59-61 Together with the largely nonoverlapping toxicity profiles, these findings made exploration of the combination of BTKi and venetoclax logical in CLL (and mantle cell lymphoma [MCL]). Phase 1/2 trials indicate 90% to 100% rates of response and very high levels of CRs (50% to 88%) and uMRD (60% to 70%) in both the first-line and relapsed settings.62,63 Phase 3 trials comparing various combinations of venetoclax with a BTKi ± anti-CD20 antibody are ongoing, including randomized studies vs chemoimmunotherapy in fit patients.
AML
AML is a more heterogeneous disease than CLL, representing neoplastic transformation of either hematopoietic stem cells (HSCs) or myeloid progenitor cells. BCL2 expression varies across its spectrum and can be heterogeneous even within a patient’s leukemic cell population. Genetic evidence indicates that BCL2 is not essential for HSC or progenitor cell function, whereas MCL1 is required for HSC function64 and for expansion of AML in model systems.65 Nevertheless, ABT-737 had in vitro activity against primary patient samples.66 Subsequent studies revealed venetoclax and ABT-737 to be equipotent in killing AML cells, supporting the dominant prosurvival role played by BCL2.67 Venetoclax was also shown to induce apoptosis more effectively in leukemic compared with normal CD34+ HSCs and progenitor cells, suggesting a therapeutic window.68 In the first study of venetoclax monotherapy in relapsed/refractory AML, CRs were observed in 19%.69 These responses, however, were not durable, in contrast to those seen in CLL.
Combination therapy is therefore necessary, and the first regimens trialed were those incorporating conventional treatments for patients unfit for intensive induction therapy. Azacitidine can enhance venetoclax activity against AML through TP53-independent NOXA induction after activation of the integrated stress response pathway,70 whereas genotoxic drugs do so through TP53-mediated upregulation of NOXA and PUMA.67,71
Efficacy
In November 2018, venetoclax was granted accelerated approval for use in adults with newly diagnosed AML ≥ 75 years of age or with comorbidities precluding intensive chemotherapy. Phase 1b/2 studies had shown that venetoclax in combination with either low-dose cytarabine or hypomethylating agents produced high rates of CR (54%-67%) in patients with a median age of 74 years.72,73 These remissions were achieved rapidly and with low early (30-day) mortality (3% to 6%), consistent with the profile expected for a low-intensity therapy. In October 2020, venetoclax was granted full approval after 2 confirmatory phase 3 trials, conducted in the same unfit patient population, demonstrated a significant survival benefit over placebo for the addition of venetoclax to azacitidine (VIALE-A; HR 0.66, NNT of ∼5 at 2 years) or to low-dose cytarabine (VIALE-C).74,75 In VIALE-C, improvement in survival was only significant after a post hoc longer-term follow-up analysis. Therefore, venetoclax-azacitidine has been widely adopted as the preferred first line therapy for older and unfit AML patients. In these patients, the major toxicities relate to myelosuppression which is increased and can be prolonged.
Biomarkers
With the graduation of venetoclax into the routine management of AML, a current priority is the identification of clinically relevant correlates of response and resistance to guide patient selection and inform design of new drug combinations aimed at subverting drug resistance or salvaging patients with disease progression.
However, biomarkers of primary response to venetoclax in AML are not straightforward to delineate, as AML has diverse genetics, and the drug has not been widely used as monotherapy. Two clinical studies have examined bone marrow blast responses to venetoclax monotherapy. In relapsed/refractory AML, blast reductions were greatest in AML bearing IDH, SRSF2, or ZRSR2 mutations.76 In previously untreated AML, a 7-day prephase of venetoclax monotherapy suppressed marrow blasts most effectively in NPM1, IDH, and SRSF2 mutant AML.77 Preclinical studies have suggested the presence of a HOX gene expression signature,78MLL-X fusion, RAD21, GATA2, or WT1 mutations to correspond with venetoclax sensitivity.78,79 It has yet to be determined, however, how these entities perturb AML biology in favor of venetoclax responsiveness, and they have not been studied prospectively in trials. Importantly, the presence of either IDH1 or IDH2 canonical mutations is a bone fide treatment effect modifier in the context of venetoclax-azacitidine treatment. Consistent with preclinical discoveries and early signals from the phase 1 study, IDH-mutant AML is relatively highly susceptible to BCL2 inhibition compared with other AMLs and compared with azacitidine therapy alone (Table 2).69,74,80
Resistance
In terms of secondary venetoclax resistance, longitudinal studies have identified some factors associated with adaptive resistance (Table 3). Selection of clones enriched for FLT3-ITD or mutant TP53 has been found at the time of AML relapse.81 The addition of exogenous growth factors reduces sensitivity to venetoclax in vitro and activating mutations in growth factor signaling pathways (eg, FLT3-ITD) appear to drive expression of prosurvival proteins, including MCL1, which can mediate resistance to venetoclax and other cytotoxic drugs.81 Unlike in CLL, BCL2 mutations impairing venetoclax affinity have not been reported in patients with relapsed AML. Loss-of-function CRISPR (clustered regularly interspaced short palindromic repeats) screens have identified TP53 and BAX as candidate loss-of-function routes to venetoclax resistance, but exactly how TP53 dysfunction achieves this remains uncertain.82 In part, it is explained by recent data demonstrating that TP53 loss impairs BAX/BAK activation, conferring a competitive advantage for subclones with aberrant TP53 during ongoing BCL2 (or MCL1) inhibition.83 This appears particularly important when BH3 mimetic concentrations are suboptimal and resolves the apparent disconnect between similarly high response rates in TP53-aberrant and TP53-sufficient AML and CLL but inferior long-term benefit in TP53-aberrant leukemias. Emergent BAX mutations have been identified recently in patients with AML relapsing after venetoclax and also in the myeloid compartment of patients with clonal hematopoiesis receiving venetoclax for treatment of CLL.84,85 Higher constitutive expression of MCL1 in monocytes may also result in lineage-associated adaptive resistance during venetoclax therapy (Table 3).86 Other reported associations with venetoclax resistance in AML in vitro include increased expression of alternative prosurvival proteins, such as BCLxL, MCL1, BCL2A1, downregulation of proapoptotic BH3-only members PUMA or NOXA, or activating mutations affecting other kinases in the MAPK pathway, such as RAS or PTPN11.78,82,87,88 However, whether these lesions do cause resistance in patients in vivo is currently unknown.
Previous studies have identified MCL1 to be amplified or overexpressed in AML and models conditionally targeting MCL1 have confirmed its prosurvival role in certain subsets of AML, including MLL-X–driven disease.65 Xenograft model systems demonstrated synergistic antileukemic activity in vivo when venetoclax was combined with an inducible vector targeting MCL1.67 Although a subset of primary AML samples also appear sensitive to MCL1 inhibition with BH3 mimetics, monotherapy activity in vitro against primary AML is generally modest.18,89 However, combined targeting of BCL2 and MCL1 (using S63845,18 AMG176,19 AZD5991,22 or VU66101390) has been reported by 4 independent groups to be synergistic against AML, including TP53-aberrant AML,83 confirming the importance of these 2 prosurvival targets in sustaining survival of a diverse spectrum of leukemic blasts (Figures 1 and 2). Although toxicity from dual inhibition may be a barrier, to date, lethal damage to normal tissues in these models has not been observed, perhaps suggesting that clinical development of such combinations may be feasible.19,89
Other opportunities to improve venetoclax efficacy in AML relate to the apparent reliance of leukemic stem and progenitor cells (LSPCs) on glycolytic metabolism as an energy source.91 Following BAX/BAK-induced MOMP, the oxidative phosphorylation pathway is disrupted, including in cells incompletely committed to apoptosis (Figure 1). Adding drugs that compromise amino acid metabolism92 or transportation of fatty acids into the cell can deprive AML of alternative energy sources to compensate for the effect of venetoclax-azacitidine on oxidative phosphorylation in LSPCs.93 In fit patients with poor prognosis AML, venetoclax is now being combined with more intensive cytotoxic regimens. Preliminary data indicate higher initial response rates but also more pronounced hematopoietic toxicity, particularly with repetitive cycles of therapy.77
BCL2 inhibition as treatment of other hematologic malignancies: potential indications
Multiple myeloma
Neoplastic plasma cells not only typically express high levels of BCL2 but commonly express BCLxL and MCL1.94 Indeed, MCL1 is required for normal plasma cell development.7 In vitro testing of primary cells indicate broad activity of BCL2 inhibitors against myeloma, with marked sensitivity observed for myeloma bearing t(11;14).94-96 BCL2 inhibition as treatment of patients with myeloma remains experimental. Although durable remissions in the t(11;14) subset have been observed with venetoclax monotherapy,97 combination therapy appears necessary in most patients.98 The most advanced combination, venetoclax with bortezomib-dexamethasone, has been investigated in a placebo-controlled randomized trial in relapsed/refractory disease.99 Venetoclax substantially improved PFS but not OS, at a cost of increased morbidity and some excess mortality in patients with myeloma progression (Table 4). Increased risk of infections was an important contributor to this excess toxicity and current trials in multiple myeloma include specific recommendations on how this risk can be mitigated. t(11;14) is clearly a positive response-effect modifier (Table 2). Ongoing trials are required to identify how this drug can be incorporated into well tolerated regimens with enhanced efficacy.
Published phase 2 or 3 data in other hematologic malignancies
| Disease phase (n) | Regimen (dose) | Comparator | Efficacy* | Safety | Comment |
|---|---|---|---|---|---|
| Relapsed multiple myeloma | |||||
| Phase 399 (n = 291) | Ven-BortD 800 mg daily | Pbo-BortD | Median PFS: 22m vs 11m; HR 0.63 (0·44–0·90) | ↑ Neutropenia; ↑ Mortality related to infection | ↑ Efficacy without excess mortality in t(11;14) subgroup |
| Relapsed follicular lymphoma | |||||
| Phase 2141 (n = 163) | Ven-R; Ven-BR 800 mg daily | BR | CMR/CR rate VR: 17% VBR: 75% BR: 69% | ↑ Toxicity with VBR; and ↓ BR dose intensity when combined with Ven | No benefit from adding Ven to BR |
| First-line diffuse large B-cell lymphoma | |||||
| Phase 2142 (N = 206) | Ven-R-CHOP 800 mg 10d | R-CHOP† | CR rate: 69% vs 63%; NS 2-y PFS: 80% vs 67%; HR 0.61 (0.43-0.87) | ↑ Grade 3/4 AEs (86% vs 66%), principally neutropenia | Scheduling needs optimization; ↑ Incremental efficacy if BCL2 IHC+Ven (exploratory) |
| Relapsed mantle-cell lymphoma | |||||
| Phase 2143 (n = 24‡) | Ven-Ibr 400 mg daily | Nil | CR at 16 wk 42% CMR rate 71% | 33% G3/4 neutropenia 17% G3/4 tcp | Dose adjustments for either Ven or Ibr common |
| Phase 1/2144 (n = 24) | Obin-Ven-Ibr 400 mg daily§ | Nil | CMR rate 67% | 33% G4 neutropenia 12% G4 tcp | Dose adjustments for either Ven or Ibr common |
| Disease phase (n) | Regimen (dose) | Comparator | Efficacy* | Safety | Comment |
|---|---|---|---|---|---|
| Relapsed multiple myeloma | |||||
| Phase 399 (n = 291) | Ven-BortD 800 mg daily | Pbo-BortD | Median PFS: 22m vs 11m; HR 0.63 (0·44–0·90) | ↑ Neutropenia; ↑ Mortality related to infection | ↑ Efficacy without excess mortality in t(11;14) subgroup |
| Relapsed follicular lymphoma | |||||
| Phase 2141 (n = 163) | Ven-R; Ven-BR 800 mg daily | BR | CMR/CR rate VR: 17% VBR: 75% BR: 69% | ↑ Toxicity with VBR; and ↓ BR dose intensity when combined with Ven | No benefit from adding Ven to BR |
| First-line diffuse large B-cell lymphoma | |||||
| Phase 2142 (N = 206) | Ven-R-CHOP 800 mg 10d | R-CHOP† | CR rate: 69% vs 63%; NS 2-y PFS: 80% vs 67%; HR 0.61 (0.43-0.87) | ↑ Grade 3/4 AEs (86% vs 66%), principally neutropenia | Scheduling needs optimization; ↑ Incremental efficacy if BCL2 IHC+Ven (exploratory) |
| Relapsed mantle-cell lymphoma | |||||
| Phase 2143 (n = 24‡) | Ven-Ibr 400 mg daily | Nil | CR at 16 wk 42% CMR rate 71% | 33% G3/4 neutropenia 17% G3/4 tcp | Dose adjustments for either Ven or Ibr common |
| Phase 1/2144 (n = 24) | Obin-Ven-Ibr 400 mg daily§ | Nil | CMR rate 67% | 33% G4 neutropenia 12% G4 tcp | Dose adjustments for either Ven or Ibr common |
AE, adverse events; B, bendamustine; Bort, bortezomib; CMR, complete metabolic response (no PET evidence of active lymphoma, but may have residual structural abnormalities); D, dexamethasone; G, grade; Ibr, ibrutinib; IHC+, positive immunohistochemistry for BCL2; Obin, obinutuzumab; Pbo, placebo; R, rituximab; tcp, thrombocytopenia; Ven, venetoclax.
Primary end point of the trial.
Indirect comparison with covariate-adjusted controls from GOYA trial.
Includes 1 first-line patient with TP53 aberration.
Six of 24 patients received either 600 or 800 mg daily of venetoclax.
Lymphoma
B-cell lymphomas by virtue of their lineage commonly express BCL2.100 Overexpression is driven in some through translocation (eg, t[14;18]),101 in follicular lymphoma and some double-hit lymphomas, gene amplification (eg, some diffuse large B-cell lymphomas [DLBCL]),102 or epigenetic dysregulation (eg, MCL).103 High-level MCL1 expression is common in MYC-driven lymphomas such as Burkitt lymphoma.104
BCL2 inhibition as treatment of patients with lymphoma remains experimental. Early-phase trials established modest single agent activity in MCL, follicular lymphoma, and Waldenström macroglobulinemia, but minimal activity in DLBCL.105 Various combination regimens, rational for each disease, are being tested in ongoing trials. Published results for completed phase 2 trials are summarized in Table 4. The results of randomized trials of R-CHOP (rituximab-cyclophosphamide-doxorubicin hydrochloride-vincristine sulphate-prednisone), with or without venetoclax for first-line high risk DLBCL and ibrutinib, with or without venetoclax, in relapsed MCL are awaited with interest.
Other diseases
Single-agent activity has also been observed in small series of patients with diseases with high levels of BCL2 expression: blastic plasmacytoid dendritic cell neoplasm,106 prolymphocytic leukemia,107 and t(11;14) light chain amyloidosis.108 In relapsed acute lymphoblastic leukemia, venetoclax in combination with navitoclax (to inhibit BCLxL) and nonmyelotoxic chemotherapy is being explored, with a phase 1b study reporting a high response rate.109
MCL1 inhibitors in clinical development
What should we expect from MCL1 inhibitors?
As listed in Table 1, there are multiple specific MCL1i in early-stage clinical development. Based on preclinical data and lessons learned from selective BCL2 inhibition with venetoclax, several predictions can be made for selective MCL1i undergoing clinical trials. First, apoptosis of susceptible cells is likely to be rapid, with biological activity evident within hours after exposure. TLS can be anticipated to occur occasionally, especially in patients with bulky or proliferative forms of myeloma, lymphoma, or acute leukemia. Second, "on target" hematopoietic toxicities, including neutropenia, monocytopenia, and depletion of plasma cells (with consequent hypogammaglobulinemia), are likely to occur in patients receiving antitumoral doses.19,110 Gene ablation models have also highlighted the biologically important role played by MCL1 in cardiomyocyte,111,112 neuronal,113 hepatocyte,114,115 and intestinal cell survival.116 Consequently, collateral toxicities that should be anticipated in clinical trials include biochemical derangement of liver and cardiac enzymes and gastrointestinal symptoms. Patients should be screened for pretreatment hepatic and/or cardiac abnormalities and monitored closely for evidence of biochemical damage after commencing therapy. As MCL1i are likely to be less well tolerated than BCL2i, intermittent schedules (eg, weekly or bi weekly) are being explored in initial first-in-human trials. Third, objective responses with monotherapy should be observed in myeloma,18,19,117 AML,18,19,22,65,90 and MYC-driven lymphomas,18,104 but these may not be durable. Although some exceptional responders are likely to be seen, at this point, no putative biomarkers have been validated. Candidate biomarkers of response include amplification of MCL1 on chromosome 1q21118 and monocytic differentiation in AML.86,119
Analogous to venetoclax in various blood cancers (CLL, AML), aberrant TP53 is unlikely to diminish response to MCL1i but durability of benefit is likely to be reduced.83 Consequently, combination therapy will be required.18,19,22,90 As with venetoclax, potential partners should be tailored to specific diseases and can be anticipated to include monoclonal antibodies , tyrosine kinase inhibitors (TKIs), hypomethylating agents, and conventional cytotoxics. Furthermore, once tolerable monotherapy regimens demonstrating biological activity without DLTs are established, it seems appropriate to commence assessment of novel combinations. Based on a narrower therapeutic window than venetoclax, it is probable that MCL1i will most likely succeed if efficacy only requires brief bursts of exposure to maximal tolerated doses.
To date, the only clinical trial data reported for MCL1 inhibition are preliminary results from the first 26 patients with relapsed refractory myeloma enrolled to the first-in-human trial of intravenous AMG-176. Emergent side effects were mostly hematologic (neutropenia and anemia) and gastrointestinal (nausea and diarrhea). An antitumor effect was reported in 3 patients (1 CR and 2 partial responses).120 Enrollment to another trial investigating the orally administered MCL1 inhibitor AMG-397 in various blood malignancies was placed on clinical hold after a cardiac toxicity signal was seen. Clinical trial data from all MCL1i are awaited to determine how appropriate patient selection and dosing schedules can result in identification of a safe and efficacious clinical pathway for these new BH3 mimetic drugs.
Key clinical research questions and future expectations
For highly susceptible diseases (eg, CLL with BCL2 inhibition), the outstanding questions relate to (1) the optimum duration of treatment: is this for a fixed duration or adapted to the tempo and depth of MRD clearance, and (2) whether triplet regimens combining venetoclax with BTKi and monoclonal antibodies add substantially to durability of disease clearance? The latter question also applies for mantle cell lymphoma. Long term, it is conceivable that combinations of BCL2i and BTKi may be curative for some patients with CLL. For less susceptible diseases (eg, BCL2-expressing DLBCL), the critical question is whether routine addition of venetoclax adds to the cure fraction currently achievable with chemo-immunotherapy alone. This question is also of great interest in patients with AML suitable for standard intensive chemotherapy. BH3 mimetics have long promised to augment efficacy when combined with DNA-damaging cytotoxics in preclinical models, and it seems these 2 clinical settings will inform us as to whether the inevitable increase in hematologic toxicity is outweighed by substantially higher proportions of patients with durable responses. In AML, another important question is whether treatment should continue until disease progression or whether limited-duration therapy should be considered, similar to CLL. This is especially pertinent in subgroups such as IDH-mutated leukemia and where MRD is absent.
As outlined above, the immediate research imperative for MCL1 inhibitors is definition of safe and biologically active regimens with preliminary evidence of efficacy in relapsed and refractory disease settings. The field is expecting that some clinically important dose-limiting toxicities will emerge necessitating use of submaximal doses in combination regimens in order to limit exposure at peak concentrations while maximizing clinical efficacy. Priorities for phase 2 exploration should be guided by where the preliminary clinical data suggest the greatest impact may be, with likely candidate areas including relapsed AML, first-line AML bearing highly unfavorable genomic features, multiply relapsed MYC-driven lymphomas, and relapsed myeloma bearing amplifications of chromosome 1q21 (containing the MCL1 locus).
The potential of BH3 mimetics as anticancer drugs is only beginning to emerge. These rapidly acting, non-DNA, damaging cytotoxics may fill key gaps in our current armamentaria against hematologic malignancies. Lessons learned thus far form sound principles for future therapeutic endeavors. The small, potential risk of tumor lysis syndrome, as well as bone marrow and other organ toxicity, mandates that new combinations are carefully tested in clinical trials. As long as therapeutic safety margins can be maintained, BH3 mimetic agents have the potential to further improve clinical outcomes in multiple hematologic malignancies.
Acknowledgments
The authors thank their many long-term collaborators for advice and important contributions to the generation of knowledge included in this review.
Academic research in the A.W.R., A.H.W., and D.C.S.H. laboratories has been supported over the last 20 years by grants from the National Health and Medical Research Council of Australia , the Leukemia and Lymphoma Society, Cancer Council of Victoria, Victorian Cancer Agency, Australian Cancer Research Foundation, Leukaemia Foundation of Australia, the Snowdome Foundation, and the Medical Research Future Fund.
Authorship
Contribution: A.W.R., A.H.W., and D.C.S.H. conceived, reviewed the literature, and wrote the manuscript.
Conflict-of-interest disclosure: A.W.R. and D.C.S.H. are employees and A.H.W. is a former employee of the Walter and Eliza Hall Institute, which receives milestone and royalty payments related to venetoclax; each receives a share of these royalties from the Institute. A.W.R. has received research funding to his institutions from Abbvie, Janssen, and Servier for investigator-initiated clinical trials or laboratory research. A.H.W. has served on advisory boards for Novartis, Janssen, Amgen, Roche, Pfizer, Abbvie, Servier, Celgene-BMS, Macrogenics, Agios, and Gilead; receives research funding to the institution from Novartis, Abbvie, Servier, Celgene-BMS, Astra Zeneca, and Amgen; and serves on speaker bureaus for Abbvie, Novartis, and Celgene. D.C.S.H. has received research funding from Genentech and Servier.
Correspondence: Andrew Roberts, The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, VIC 3052, Australia; e-mail: roberts@wehi.edu.au.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal