

Key Points
In the population aged ≥80 years, distinct mutational patterns define risk to develop myeloid neoplasms vs inflammatory-associated diseases.
In individuals with unexplained cytopenia, mutational status identifies subjects with presumptive evidence of myeloid neoplasms.

Abstract
Clonal hematopoiesis of indeterminate potential (CHIP) is associated with increased risk of cancers and inflammation-related diseases. This phenomenon becomes common in persons aged ≥80 years, in whom the implications of CHIP are not well defined. We performed a mutational screening in 1794 persons aged ≥80 years and investigated the relationships between CHIP and associated pathologies. Mutations were observed in one-third of persons aged ≥80 years and were associated with reduced survival. Mutations in JAK2 and splicing genes, multiple mutations (DNMT3A, TET2, and ASXL1 with additional genetic lesions), and variant allele frequency ≥0.096 had positive predictive value for myeloid neoplasms. Combining mutation profiles with abnormalities in red blood cell indices improved the ability of myeloid neoplasm prediction. On this basis, we defined a predictive model that identifies 3 risk groups with different probabilities of developing myeloid neoplasms. Mutations in DNMT3A, TET2, ASXL1, or JAK2 were associated with coronary heart disease and rheumatoid arthritis. Cytopenia was common in persons aged ≥80 years, with the underlying cause remaining unexplained in 30% of cases. Among individuals with unexplained cytopenia, the presence of highly specific mutation patterns was associated with myelodysplastic-like phenotype and a probability of survival comparable to that of myeloid neoplasms. Accordingly, 7.5% of subjects aged ≥80 years with cytopenia had presumptive evidence of myeloid neoplasm. In summary, specific mutational patterns define different risk of developing myeloid neoplasms vs inflammatory-associated diseases in persons aged ≥80 years. In individuals with unexplained cytopenia, mutational status may identify those subjects with presumptive evidence of myeloid neoplasms.
Introduction
Exome sequencing studies have identified the frequent age-dependent clonal expansion of somatic mutations in the hematopoietic system.1-5 Clonal hematopoiesis of indeterminate potential (CHIP) describes individuals with hematologic malignancy–associated mutations in blood or marrow but without other diagnostic criteria for a hematologic malignancy5 and is associated with increased risk of cancers (in particular myeloid neoplasms) and chronic inflammatory diseases (coronary heart disease).1-6
The phenomenon of CHIP becomes very common in the population of people aged ≥80 years,7-10 which represents the fastest growing age segment in developed countries.9,11 In these individuals, clinical implications of CHIP are expected to be relevant but are not well defined.7,8
The incidence of solid cancers and myeloid neoplasms increases with age, and mortality is higher after the age of 75 years.12 The risk of several chronic inflammatory diseases is age related as well, leading to large prevalence of frailty and disability among people aged ≥80 years.13 We hypothesized that the study of the population aged ≥80 years can contribute to define the relationship between specific mutational patterns in the hematopoietic system and the individual risk of developing cancers vs other adverse events (chronic inflammatory diseases).
Anemia is a common finding in the elderly and is associated with worse cognitive and functional outcomes and increased mortality.14-18 Underlying cause of anemia remained unexplained in 30% of cases, and a proportion of unexplained cytopenia may account for myeloid neoplasms.15,18 We hypothesized that the study of CHIP may contribute to determine specific causes of anemia in elderly people and define personalized treatment strategies to mitigate anemia-related negative sequela.
The primary objective of the present study was to evaluate the prevalence of CHIP and the relationships between CHIP and associated pathologies in the population aged ≥80 years. The secondary objective was to analyze clinical outcome of patients affected with unexplained cytopenia and define the clinical effect of clonal abnormalities among these individuals. The definitions of idiopathic cytopenia of unknown significance (ICUS) and clonal cytopenia of unknown significance (CCUS) were applied to identify individuals with nonclonal vs clonal unexplained cytopenia.5,19
Patients and methods
Study procedures are in accordance with the Declaration of Helsinki. Ethics Committees of Humanitas Research Hospital and Mario Negri Pharmacological Institute, Milan, Italy approved the study. Written informed consent was obtained prior to blood sampling. This trial was registered at www.clinicaltrials.gov as #NCT03907553.
Study population
Study procedures are described in supplemental File 1 (available on the Blood Web site).
We analyzed subjects included in 2 population-based studies enriched in individuals aged ≥80 years (ie, “Health_&_Anemia”19,20 and “Monzino_80+”21).
Health_&_Anemia is a prospective study (2003-2018) aimed to investigate clinical consequences of anemia in the elderly.15,17 We studied 1059 subjects aged ≥80 years (median age, 83 years; range, 80-105 years) in whom peripheral blood samples collected at study enrollment were available for mutational screening. At study enrollment (May 2003), clinical history was collected and complete blood count was performed. When a hemoglobin concentration was below the World Health Organization reference criteria for anemia (<12 g/dL in women and <13 g/dL in men), further investigations were made to define specific causes of anemia. Follow-up was updated to December 2018. A total of 344 565 laboratory tests were available during follow-up. Data on hospitalization and mortality were available for all subjects. Diagnoses of chronic inflammatory diseases and cancers were defined according to International Classification of Diseases for Oncology, 9th Revision, Clinical Modification codes and local cancer registry data. Diagnosis of myeloid neoplasms was based in addition on information provided by revision of bone marrow biopsy reports provided by the Hematology Unit of Biella Hospital. Three investigators (M.R., Emma Riva, and M.G.D.P.) reviewed this information independently, and a final diagnosis of myeloid neoplasms was provided after a consensus meeting.
As a second cohort, we analyzed 735 individuals (median age, 90 years; range, 80-104 years) enrolled in the Monzino_80+ prospective study (2002-2018) aimed at investigating relationships between age and cognitive decline and dementia.22
Because data collection on myeloid neoplasm diagnosis was less accurate in the Monzino_80+ population than in the Health_&_Anemia cohort (see supplemental File 1), we analyzed in addition 727 subjects aged ≥75 to <80 years from the Health_&_Anemia study, to specifically validate the predictive value of clinical and mutational features on the risk of developing myeloid neoplasms.
Finally, to compare clinical features and outcome of subjects aged ≥80 years with CHIP to those of patients with myeloid neoplasms, we analyzed a sex- and age-matched population of patients affected with myelodysplastic syndrome from the retrospective EuroMDS database. (2000-2018, www.clinicaltrials.gov # NCT04174547). Each subject with CHIP was matched with 5 patients with the same year of birth and sex; overall, 255 patients with myeloid neoplasms were included in this analysis.
Mutation screening
Using peripheral blood DNA, we looked for mutations in 47 genes related to myeloid neoplasms. The gene list is available in supplemental Table 1, and sequencing procedures and variant calling are available in supplemental File 2.5
CHIP was defined as the presence of a clonal blood cell population associated with a hematologic malignancy–related mutation at a variant allele frequency (VAF) ≥0.01. Median coverage was 3455×. Mutations with VAF <0.10 were resequenced on an independent platform. The effectiveness of resequencing in confirming genomic variants with VAF ≥0.01 and <0.10 was 96.5%, while DNA sequencing was significantly less performant and reproducible below the threshold of 0.01 (P = .02). The technique we used missed clonal skewing in the absence of mutations in putative myeloid neoplasm driver genes.23
Statistical analysis
Numerical variables were summarized by median and range; categorical variables were described with count and relative frequency (%) of subjects in each category.
Survival analyses were performed with the Kaplan-Meier method, and differences between groups were evaluated by log-rank test. Cox models were built to estimate hazard ratio (HR) with 95% confidence interval (CI) for probability of overall survival and risk of developing coronary heart disease and chronic inflammatory diseases (only incident cases were considered in the analyses).
The accuracy of mutational factors in predicting the risk of developing myeloid neoplasms was analyzed (only incident cases were considered in the analyses). The accuracy of categorical variables (presence vs absence of mutations in a specific gene) was estimated by calculating time-dependent positive predictive value (PPV) and negative predictive value (NPV).24 For variables measured on a continuous scale (VAF), time-dependent receiver-operating characteristic (ROC) curve, the corresponding area under the ROC curve (AUC), and the optimal cutoff point were calculated by using online available cenROC R package.21,25
To define a risk score for developing myeloid neoplasms, HRs from a multivariable Cox analysis on the Health_&_Anemia population including age, sex, mutational status, and nonmutational parameters as covariates were used. A diagnosis of myeloid neoplasm was considered as event; subjects were censored at the end of follow-up or at time of death. The goodness of concordance for the predictive score was measured by concordance index (C-index), internal fivefold cross-validation, and independent external validation.
Cumulative incidence of myeloid neoplasms was calculated by Kaplan-Meier method (death for any cause was considered as competing event in the estimation of cumulative incidence function).26 Left truncation was applied when calculating the cumulative incidence of myeloid neoplasms with age as the time scale.27
Results
Prevalence and clinical effect of CHIP in the population aged 80 years or older
We studied prevalence of CHIP and relationship between CHIP and probability of survival in the population aged ≥80 years. Analyses were performed on both the Health_&_Anemia and Monzino_80+ cohorts.
Mutations were observed in 32.6% (95% CI, 29.4-35.5) and 26.0% (22.9-29.8) of subjects enrolled in the Health_&_Anemia and Monzino_80+ cohorts, respectively. The majority of variants in both cohorts occurred in 3 genes: DNMT3A, TET2, and ASXL1 (prevalence of mutated genes is reported in Figure 1; supplemental Figure 1 and supplemental Table 2).
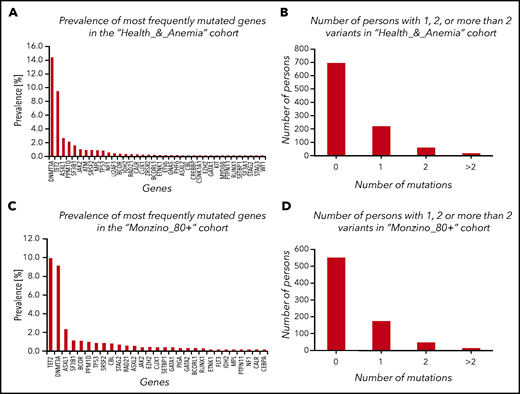
CHIP in subjects aged 80 years or older from the Health_&_Anemia and Monzino_80+ cohorts. (A,C) Prevalence of most frequently mutated genes in the 2 cohorts (considering mutated and unmutated persons). (B,D) Number of persons with 1, 2, or >2 variants.
CHIP in subjects aged 80 years or older from the Health_&_Anemia and Monzino_80+ cohorts. (A,C) Prevalence of most frequently mutated genes in the 2 cohorts (considering mutated and unmutated persons). (B,D) Number of persons with 1, 2, or >2 variants.
CHIP was more common in males than females (P = .02 and P = .005, respectively), and its prevalence increased with age (P = .001 and P = .03, respectively). Considering genes grouped according to functional patterns in both cohorts, we observed a significant increase of mutations with age in epigenetics and cohesin complex–related genes (P = .01 and P = .02, respectively). Considering single genes, we observed a significant increase in the prevalence of TET2 and ASXL1 mutations after the age of 90 years (P = .021 and P = .032, respectively). After testing for gender bias, we observed 2 genes significantly more mutated in males than in females: ZRSR2 and U2AF1.
We focused on centenarians in both cohorts (n = 44) stratified according to the presence of comorbidity (including heart disease, diabetes, stroke, cancer, osteoporosis, thyroid condition, Parkinson disease, and chronic obstructive pulmonary disease).28 Individuals with chronic age-related illness before the age of 100 were 33 vs 11 who remained disease-free at age 100 years. Prevalence of CHIP was higher in patients with vs without comorbidity (62% vs 20%, P = .015).
Subjects with CHIP were older than individuals without CHIP (median age of individuals without CHIP vs those with 1 mutation vs those with ≥2 mutations was 83 years vs 84 years vs 85 years, respectively, in the Health_&_Anemia cohort [P = .019] and 90 years vs 91 years vs 94 years, respectively, in the Monzino_80+ cohort (P < .001). The presence of CHIP was associated with a lower probability of survival (P < .001 in both cohorts), and prognosis was even poorer in subjects carrying ≥2 mutations (P < .001 in both cohorts; Figure 2). The independent association between CHIP and increased mortality was maintained in a multivariable analysis including age, sex, and cytopenia as covariates (HR, 1.28 [1.1-1.9], P = .009 in the Health_&_Anemia cohort and HR, 1.37 [1.2-1.71], P = .006 in the Monzino_80+ cohort) and when focusing on cancer-related death (HR, 1.91 [1.44-2.21], P = .001 and HR, 2.13 [1.94-2.39], P = .002) and non–cancer-related mortality (HR, 1.43 [1.29-1.77], P = .02 and HR, 1.56 [1.31-1.8], P = .01) as separate outcomes.
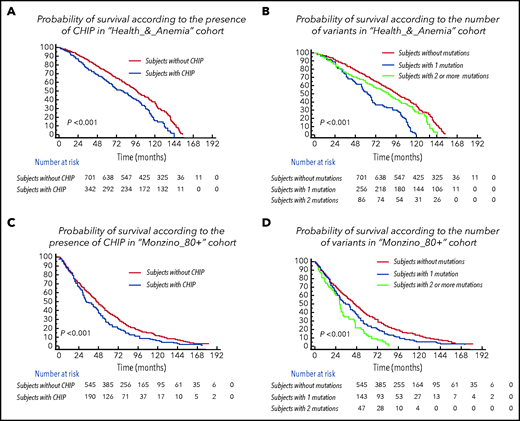
Clinical effect of CHIP in the population aged 80 years or older. (A,C) Cumulative probability of overall survival according to the presence of CHIP in subjects aged ≥80 years from the Health_&_Anemia and Monzino_80+ cohorts, respectively. (B,D) Cumulative probability of overall survival according to the number of variants (0 vs 1 vs ≥2) in subjects aged ≥80 years from the Health_&_Anemia and Monzino_80+ cohorts, respectively.
Clinical effect of CHIP in the population aged 80 years or older. (A,C) Cumulative probability of overall survival according to the presence of CHIP in subjects aged ≥80 years from the Health_&_Anemia and Monzino_80+ cohorts, respectively. (B,D) Cumulative probability of overall survival according to the number of variants (0 vs 1 vs ≥2) in subjects aged ≥80 years from the Health_&_Anemia and Monzino_80+ cohorts, respectively.
Focusing on subjects carrying ≥2 mutations in both cohorts, the independent effect of carrying ≥2 mutations on mortality was maintained in a multivariate analysis including age, sex and cytopenia as covariates (HR, 1.4 [1.19-2.31]; P = .008). Considering specific causes of death, a higher prevalence of cancer-related deaths was observed in individuals carrying ≥ 2 mutations vs both subjects carrying 1 mutation and those without CHIP (P = .028 and P = .009, respectively).
CHIP and risk of developing myeloid neoplasms in the population aged 80 years or older
We investigated the relationship between CHIP and risk of developing myeloid neoplasms. Analyses were performed on the Health_&_Anemia cohort. Prevalent and incident cases were 16 and 25, respectively. We calculated the time-dependent PPV and NPV for developing myeloid neoplasms at the age of 95 years for most relevant genomic features (supplemental Figure 2). Absence of mutations had high NPV (0.89 [0.86-0.92]), while the presence of CHIP per se had low PPV (0.11 [0.09-0.16]). Among investigated genes, splicing genes (SF3B1, SRSF2, U2AF1, and ZRSR2) and JAK2 had the highest PPV (0.58 [0.41; 0.63] and 0.70 [0.41-0.98], respectively). To evaluate the impact of multiple mutations in the same individual, we focused on the 3 most commonly mutated genes (DMNT3A, TET2, and ASXL1) comparing single mutations with comutation patterns. PPV of mutations in TET2, DNMT3A or ASXL1 with comutation patterns was higher than PPV of single lesions (0.28[0.14; 0.41] vs 0.08[0.02-0.23], P = .001). (supplemental Figure 2). Overall, mutations in splicing genes, mutations in JAK2 gene, and comutation patterns involving TET2, DNMT3A, and ASXL1 accounted for 75% of myeloid neoplasms diagnosed in subjects with prior CHIP.
We then explored the best cutoff value of VAF for developing myeloid neoplasms. VAF showed a significant accuracy as evaluated by time-dependent ROC curve (AUC was 0.88 [0.81-0.94], P < .001). At the age of 95 years, a VAF of 0.096 was found as optimal cutoff value (sensitivity, 0.84; specificity, 0.83).
In a multivariable analysis including the number of mutations per subject and the most frequently mutated genes, having comutation patterns involving TET2, DNMT3A, and ASXL1 (HR, 4.64 [2.11-12.23], P < .001), carrying splicing mutation (HR, 10.63 [5.23-17.68], P < .001), or having VAF > 0.096 (HR, 2.35 [1.22-5.48], P = .021) were independent predictors for developing myeloid neoplasms.
Finally, we aimed to study whether nonmutational factors may improve the capability to capture individual risk of developing myeloid neoplasms. We focused on changes in red blood cell (RBC) indices (mean corpuscular volume [MCV] and RBC distribution width [RDW]) that occur as early phenotypic abnormalities in subjects who later develop myeloid neoplasms.4
In the Health_&_Anemia cohort, we found that high MCV (>98 fL) and RDW (>14) values at study enrollment were associated with reduced survival (P = .01 and P = .008, respectively, supplemental Figure 3). In a multivariable analysis including splicing mutations; comutation patterns involving TET2, DNMT3A, and ASXL1; and VAF as covariates, abnormal RBC indices are associated with higher risk of developing myeloid neoplasms, independently of mutational features (HR, 2.02 [1.18-4.7], P < .001).
Definition of a risk score for developing myeloid neoplasms according to mutational status and RBC indices
We aimed to define a risk score for developing myeloid neoplasms according to mutational status and RBC indices. Subjects from the Health_&_Anemia cohort entered this analysis.
HRs from multivariable Cox analysis including age, sex, mutational status, and RBC indices as covariates were used to define a risk score (details are reported in Figure 3). A score of 1 was assigned for abnormalities in RBC indices and VAF >0.096; a score of 2 was assigned for comutation patterns involving TET2, DNMT3A, and ASXL1; and a score of 5 was assigned for splicing mutations. Risk groups were defined as low (score 0-1), intermediate (score 2-4), and high (score ≥5). A simplified risk classification was also provided. Cumulative incidence of myeloid neoplasms was significantly different among these 3 risk groups (P < .001). In particular, in high-risk individuals (3% of the whole general population aged ≥80 years), cumulative incidences of myeloid neoplasm was 14%, 34%, and 42% at the age of 85, 90, and 95 years, respectively. The accuracy of the predictive score was good (C-index, 0.851) and was confirmed by internal fivefold cross-validation (mean C-index in test sets, 0.849).
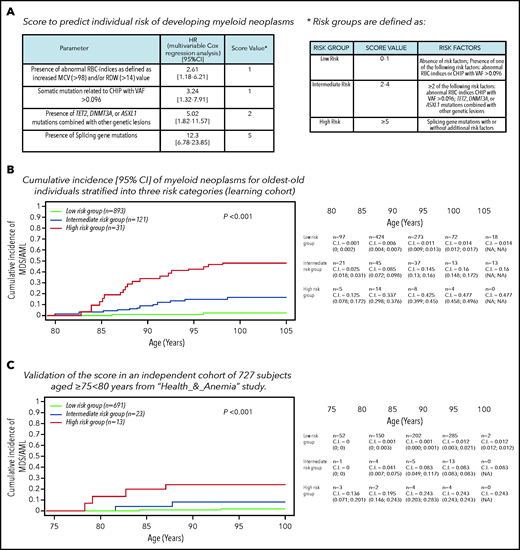
CHIP and risk of developing myeloid neoplasms. (A) Definition of a score based on specific mutational patterns (CHIP) and RBC indices to predict the risk of developing myeloid neoplasms. To define a risk score for developing myeloid neoplasms, we used HR from a multivariable Cox analysis on the Health_&_Anemia cohort (learning cohort) adjusted for age and sex, including mutational status (splicing mutations; comutation patterns involving TET2, DNMT3A and ASXL1; and VAF >0.096) and nonmutational parameters (RBC indices) as covariates. A diagnosis of myeloid neoplasm (including myelodysplastic syndrome and acute myeloid leukemia) was considered as event; subjects were censored at the end of follow-up or at time of death. (B) Cumulative incidence (C.I.) of myeloid neoplasms for individuals aged ≥80 years stratified into 3 risk categories (learning cohort). Cumulative incidence was calculated by Kaplan-Meier method (death for any cause was considered as competing-event in the estimation of cumulative incidence function). Left truncation was applied to calculate the cumulative incidence of myeloid neoplasms with age as the time scale. MDS/AML, myelodysplastic syndrome/acute myeloid leukemia. (C) Validation of the score on an independent cohort of 727 subjects aged ≥75 to <80 years from the Health_&_Anemia study.
CHIP and risk of developing myeloid neoplasms. (A) Definition of a score based on specific mutational patterns (CHIP) and RBC indices to predict the risk of developing myeloid neoplasms. To define a risk score for developing myeloid neoplasms, we used HR from a multivariable Cox analysis on the Health_&_Anemia cohort (learning cohort) adjusted for age and sex, including mutational status (splicing mutations; comutation patterns involving TET2, DNMT3A and ASXL1; and VAF >0.096) and nonmutational parameters (RBC indices) as covariates. A diagnosis of myeloid neoplasm (including myelodysplastic syndrome and acute myeloid leukemia) was considered as event; subjects were censored at the end of follow-up or at time of death. (B) Cumulative incidence (C.I.) of myeloid neoplasms for individuals aged ≥80 years stratified into 3 risk categories (learning cohort). Cumulative incidence was calculated by Kaplan-Meier method (death for any cause was considered as competing-event in the estimation of cumulative incidence function). Left truncation was applied to calculate the cumulative incidence of myeloid neoplasms with age as the time scale. MDS/AML, myelodysplastic syndrome/acute myeloid leukemia. (C) Validation of the score on an independent cohort of 727 subjects aged ≥75 to <80 years from the Health_&_Anemia study.
We performed in addition an external validation on an independent cohort of 727 subjects aged ≥75 to <80 years from the Health_&_Anemia study (Figure 3; supplemental Figure 4). The analyses performed on the validation cohort confirmed a high concordance of the model (C-index was 0.889), thus suggesting a high generalizability of the results.
Clonal evolution in the population aged 80 years or older with multiple samples available
We studied clonal evolution in 96 subjects from the Health_&_Anemia cohort, in which multiple samples were available over a period of 4 years.
CHIP was found at baseline in 22 cases (23%); during follow-up, 2 individuals acquired additional mutations, 10 displayed ≥0.05 VAF increase, and CHIP was lost in 3 cases. In 13 out of 74 subjects without mutations at baseline, CHIP was acquired during follow-up (17.5%). We identified 2 subjects in whom clonal evolution preceded a diagnosis of a myeloid neoplasm (myelodysplastic syndrome in both cases) (Figure 4; supplemental Table 3).
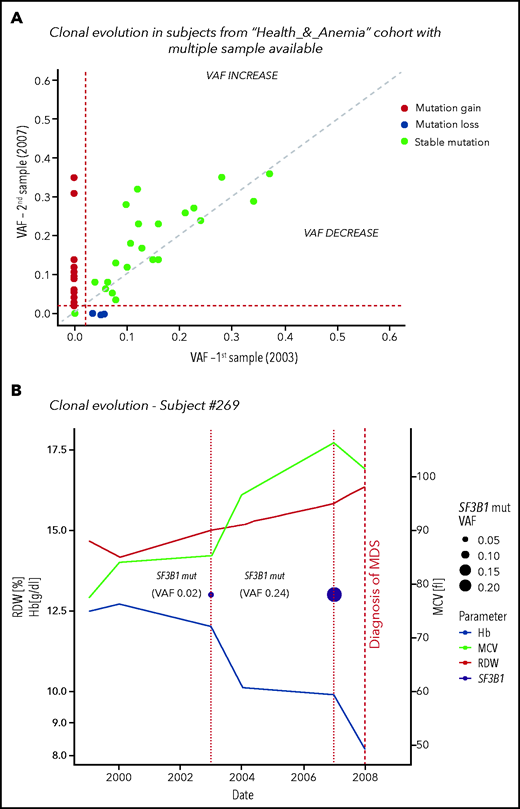
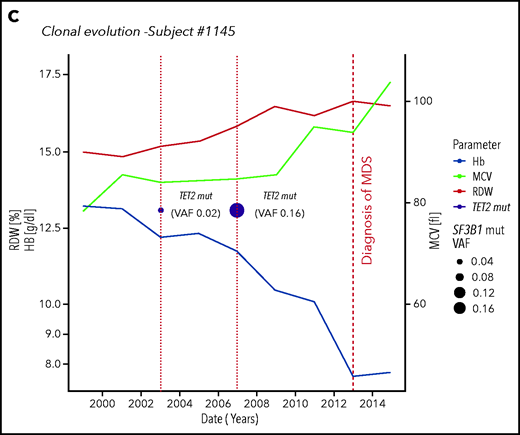
Clonal evolution in the population aged 80 years or older. (A) Clonal evolution in subjects from the Health_&_Anemia cohort with multiple samples available (n = 96). (B) Clonal evolution (subject 269). This female subject was born on 1921. In 1999, she displayed normal blood count. In 2003, a first mutational screening was performed with evidence of a mutation in SF3B1 gene with 0.02 VAF. At this time, the subject showed normal hemoglobin (Hb) level (12.1 g/dL), RDW (12), and MCV (87). Since 2003, this subject experienced increasing in RDW and MCV, and in 2007, a mild anemia was observed (10 g/dL). At this time, mutational screening showed increase in SF3B1 VAF (0.24). In 2008, a diagnosis of myelodysplastic syndrome with ring sideroblasts was performed. (C) Clonal evolution (subject 1145). This male subject was born in 1922. In 1999 blood count was normal. In 2003 a first mutational screening revealed a mutation in TET2 gene with 0.02 VAF. At this time, the subject showed a mild anemia (12.7 g/dL), with increased RDW (14.2) and normal MCV (89). In 2007, hemoglobin level decreased to 11.8 g/dL, and mutation analysis showed an increase in TET2 mutation VAF to 0.16. From 2007 to 2012, a further decrease in hemoglobin level, together with an increase in RDW and MCV, was noticed. In 2013, a diagnosis of myelodysplastic syndrome with unilineage dysplasia was made.
Clonal evolution in the population aged 80 years or older. (A) Clonal evolution in subjects from the Health_&_Anemia cohort with multiple samples available (n = 96). (B) Clonal evolution (subject 269). This female subject was born on 1921. In 1999, she displayed normal blood count. In 2003, a first mutational screening was performed with evidence of a mutation in SF3B1 gene with 0.02 VAF. At this time, the subject showed normal hemoglobin (Hb) level (12.1 g/dL), RDW (12), and MCV (87). Since 2003, this subject experienced increasing in RDW and MCV, and in 2007, a mild anemia was observed (10 g/dL). At this time, mutational screening showed increase in SF3B1 VAF (0.24). In 2008, a diagnosis of myelodysplastic syndrome with ring sideroblasts was performed. (C) Clonal evolution (subject 1145). This male subject was born in 1922. In 1999 blood count was normal. In 2003 a first mutational screening revealed a mutation in TET2 gene with 0.02 VAF. At this time, the subject showed a mild anemia (12.7 g/dL), with increased RDW (14.2) and normal MCV (89). In 2007, hemoglobin level decreased to 11.8 g/dL, and mutation analysis showed an increase in TET2 mutation VAF to 0.16. From 2007 to 2012, a further decrease in hemoglobin level, together with an increase in RDW and MCV, was noticed. In 2013, a diagnosis of myelodysplastic syndrome with unilineage dysplasia was made.
Relationship between CHIP and coronary heart disease or chronic inflammatory diseases in the population aged 80 years or older
We aimed to define the relationship between CHIP and risk of developing chronic inflammatory diseases. We analyzed the Health_&_Anemia cohort, in which the diagnosis of chronic inflammatory diseases was systematically recorded.
Coronary heart disease was defined as a history of myocardial infarction or coronary revascularization after the time of DNA collection. Prevalent and incident cases were 57 and 27, respectively. We defined ASXL1, TET2, DNMT3A, and JAK2 mutations as high risk for vascular events.6 Participants with CHIP had a significantly higher risk of coronary heart disease with respect to those without mutations (HR, 1.61 [1.28-3.21], P = .02). When considering patients with high-risk mutations, HR increased to 2.21 (1.45-4.01; P = .006), and the effect was maintained when adjusting for sex, age smoking, hypertension, and hyperlipidemia (not shown). Mutations in splicing genes were not associated with an increased risk of coronary heart disease (HR, 0.91 [0.79-1.65], P = .84).
As a further step, we investigated the possible role of CHIP in other chronic inflammatory diseases (stroke, diabetes, arthritis, and autoimmune diseases). We observed preliminary evidence of increased risk of developing rheumatoid arthritis (12 prevalent and 6 incident cases) in participants with vs without high-risk mutations6 (HR, 4.79 [1.9-17.62], P = .039). However, due to the low number of cases observed, this finding should be interpreted with caution.
Clinical relevance of CHIP in individuals aged 80 years or older with unexplained cytopenia
We aimed to specifically analyze clinical outcome of patients aged ≥80 years affected with unexplained cytopenia and to define the clinical effect of clonal abnormalities among these individuals. The definitions of ICUS and CCUS were applied to identify individuals with nonclonal vs clonal unexplained cytopenia.
Individuals from both the Health_&_Anemia and Monzino_80+ cohorts entered this analysis. The most common cytopenia reported at study enrollment was anemia (14.9% and 29.5%, respectively).15-18 The underlying cause of persistent (>6 months) cytopenia was unexplained in 29% and 34% of cases, respectively (Table 1). Prevalence of CHIP was not significantly different in subjects aged ≥80 years with vs without cytopenia (30.5% vs 29.6%, respectively; P = .24). Focusing on subjects affected with anemia stratified according to the underlying cause, no significant difference on the prevalence of CHIP was observed among different groups of patients. We noticed an enrichment of splicing gene mutations in patients with unexplained anemia with respect to subjects with anemia associated with specific underlying cause (P = .031).
Prevalence and leading causes of anemia and other cytopenias at study enrollment in subjects aged 80 years or older from the Health_&_Anemia and Monzino_80+ cohorts
| Health_&_Anemia study | Monzino_80+ study | P | |
|---|---|---|---|
| No. of subjects | 1059 | 735 | |
| Women (%) | 697 (65.8) | 546 (74.3) | <.001 |
| Men (%) | 362 (34.2) | 189 (25.7) | |
| Age (y), median (range) | 82.8 (80-105) | 90.5 (80.5-103.9) | <.001 |
| ≥80 to <85 y (%) | 725 (68.5) | 128 (17.4) | <.001 |
| ≥85 to <90 y (%) | 235 (22.2) | 215 (29.3) | |
| ≥90 to <95 y (%) | 71 (6.7) | 247 (33.6) | |
| ≥95 to <100 y (%) | 24 (2.3) | 105 (14.3) | |
| ≥100 y (%) | 4 (0.3) | 40 (5.4) | |
| Death (%) | 683/1059 (64.5) | 695/735 (94.5) | <.001 |
| Blood count at study enrollment | |||
| Hb (g/dL), median (range) | 13.6 (7.3-18) | 13.1 (6.7-16.8) | <.001 |
| HCT (%), median (range) | 42.4 (22.9-55.5) | 39.2 (22-50.6) | <.001 |
| MCV (fL), median (range) | 95.9 (61.9-123.3) | 92.2 (58.6-121.8) | <.001 |
| RDW (fL), median (range) | 14.4 (12.3-21.6) | 13.4 (11.3-22.2) | <.001 |
| WBCs (×109/L), median (range) | 6.4 (1.6-40.3) | 6.5 (2.3-78.9) | .664 |
| ANC (×109/L), median (range) | 3.8 (0.6-21.2) | 3.8 (1.2-15.2) | .059 |
| PLTs (×109/L), median (range) | 222 (52-1117) | 231 (25-915) | .044 |
| Anemia at study enrollment | |||
| All n (%) | 158/1059 (14.9) | 217/735 (29.5) | <.001 |
| Women (%) | 94/697 (13.5) | 142/546 (26) | <.001 |
| Men (%) | 64/362 (17.7) | 75/189 (39.7) | <.001 |
| Anemia by age (y) | |||
| ≥80 to <85 (%) | 81/725 (11.2) | 21/128 (16.4) | .142 |
| ≥85 to <90 (%) | 41/235 (17.4) | 49/215 (22.8) | .249 |
| ≥90 to <95 (%) | 25/71 (35.2) | 87/247 (35.2) | .930 |
| ≥95 to <100 (%) | 9/24 (37.5) | 40/105 (38.1) | .971 |
| ≥100 (%) | 2/4 (50) | 20/40 (50) | 1.000 |
| Severity of anemia | |||
| Mild (normal value to 10 g/dL) (%) | 141/158 (89.3) | 197/217 (90.7) | .910 |
| Moderate (9.9-8 g/dL) (%) | 16/158 (10.1) | 16/217 (7.3) | .389 |
| Severe (<8 g/dL) (%) | 1/158 (0.6) | 4/217 (2) | .320 |
| Causes of anemia | |||
| Iron deficiency (%) | 9/158 (5.7) | 14/217 (6.4) | .777 |
| β thalassemia (%) | 12/158 (7.6) | 5/217 (2.3) | .021 |
| B12/folate deficiency (%) | 18/158 (11.4) | 41/217 (18.8) | .091 |
| Renal (%) | 29/158 (18.4) | 49/217 (22.6) | .419 |
| Chronic disease (%) | 16/158 (10.1) | 16/217 (7.3) | .389 |
| Multifactorial (%) | 23/158 (14.6) | 18/217 (8.2) | .087 |
| Unexplained (%) | 41/158 (25.9) | 74/217 (34.1) | .216 |
| Neutropenia at study enrollment | |||
| All (%) | 10/1059 (1) | 26/735 (3.5) | <.001 |
| Unexplained (%) | 4/10 (40) | 11/26 (42.3) | .936 |
| Thrombocytopenia at study enrollment | |||
| All (%) | 51/1059 (4.8) | 56/735 (7.6) | 0.021 |
| Unexplained (%) | 9/51 (17.6) | 16/56 (28.5) | .294 |
| Unexplained cytopenia | 48/1059 (4.5) | 85/735 (11.5) | <.001 |
| Health_&_Anemia study | Monzino_80+ study | P | |
|---|---|---|---|
| No. of subjects | 1059 | 735 | |
| Women (%) | 697 (65.8) | 546 (74.3) | <.001 |
| Men (%) | 362 (34.2) | 189 (25.7) | |
| Age (y), median (range) | 82.8 (80-105) | 90.5 (80.5-103.9) | <.001 |
| ≥80 to <85 y (%) | 725 (68.5) | 128 (17.4) | <.001 |
| ≥85 to <90 y (%) | 235 (22.2) | 215 (29.3) | |
| ≥90 to <95 y (%) | 71 (6.7) | 247 (33.6) | |
| ≥95 to <100 y (%) | 24 (2.3) | 105 (14.3) | |
| ≥100 y (%) | 4 (0.3) | 40 (5.4) | |
| Death (%) | 683/1059 (64.5) | 695/735 (94.5) | <.001 |
| Blood count at study enrollment | |||
| Hb (g/dL), median (range) | 13.6 (7.3-18) | 13.1 (6.7-16.8) | <.001 |
| HCT (%), median (range) | 42.4 (22.9-55.5) | 39.2 (22-50.6) | <.001 |
| MCV (fL), median (range) | 95.9 (61.9-123.3) | 92.2 (58.6-121.8) | <.001 |
| RDW (fL), median (range) | 14.4 (12.3-21.6) | 13.4 (11.3-22.2) | <.001 |
| WBCs (×109/L), median (range) | 6.4 (1.6-40.3) | 6.5 (2.3-78.9) | .664 |
| ANC (×109/L), median (range) | 3.8 (0.6-21.2) | 3.8 (1.2-15.2) | .059 |
| PLTs (×109/L), median (range) | 222 (52-1117) | 231 (25-915) | .044 |
| Anemia at study enrollment | |||
| All n (%) | 158/1059 (14.9) | 217/735 (29.5) | <.001 |
| Women (%) | 94/697 (13.5) | 142/546 (26) | <.001 |
| Men (%) | 64/362 (17.7) | 75/189 (39.7) | <.001 |
| Anemia by age (y) | |||
| ≥80 to <85 (%) | 81/725 (11.2) | 21/128 (16.4) | .142 |
| ≥85 to <90 (%) | 41/235 (17.4) | 49/215 (22.8) | .249 |
| ≥90 to <95 (%) | 25/71 (35.2) | 87/247 (35.2) | .930 |
| ≥95 to <100 (%) | 9/24 (37.5) | 40/105 (38.1) | .971 |
| ≥100 (%) | 2/4 (50) | 20/40 (50) | 1.000 |
| Severity of anemia | |||
| Mild (normal value to 10 g/dL) (%) | 141/158 (89.3) | 197/217 (90.7) | .910 |
| Moderate (9.9-8 g/dL) (%) | 16/158 (10.1) | 16/217 (7.3) | .389 |
| Severe (<8 g/dL) (%) | 1/158 (0.6) | 4/217 (2) | .320 |
| Causes of anemia | |||
| Iron deficiency (%) | 9/158 (5.7) | 14/217 (6.4) | .777 |
| β thalassemia (%) | 12/158 (7.6) | 5/217 (2.3) | .021 |
| B12/folate deficiency (%) | 18/158 (11.4) | 41/217 (18.8) | .091 |
| Renal (%) | 29/158 (18.4) | 49/217 (22.6) | .419 |
| Chronic disease (%) | 16/158 (10.1) | 16/217 (7.3) | .389 |
| Multifactorial (%) | 23/158 (14.6) | 18/217 (8.2) | .087 |
| Unexplained (%) | 41/158 (25.9) | 74/217 (34.1) | .216 |
| Neutropenia at study enrollment | |||
| All (%) | 10/1059 (1) | 26/735 (3.5) | <.001 |
| Unexplained (%) | 4/10 (40) | 11/26 (42.3) | .936 |
| Thrombocytopenia at study enrollment | |||
| All (%) | 51/1059 (4.8) | 56/735 (7.6) | 0.021 |
| Unexplained (%) | 9/51 (17.6) | 16/56 (28.5) | .294 |
| Unexplained cytopenia | 48/1059 (4.5) | 85/735 (11.5) | <.001 |
The classification of anemia and other cytopenias (based on the hematologic findings) was supported by the clinical conditions and pharmacological therapies of older patients (criteria are defined in supplemental File 1). Anemias and other cytopenias that could not be classified into any of the previously defined categories were considered to be of unexplained origin. A panel 3 physicians reviewed, discussed, and reached a final consensus for each case of cytopenia with discrepant classification.
ANC, absolute neutrophil count; Hb, hemoglobin; HCT, hematocrit; MCV, mean corpuscular volume; PLTs, platelets; RDW, red cell distribution width; WBCs, white blood cells.
We considered subjects with unexplained cytopenia from both studies (n = 133), stratified according to the presence of mutations as ICUS (n = 82) vs CCUS (n = 51,38%).19 Subjects with CCUS showed a significantly lower probability of survival compared with ICUS (P = .002), while no significantly different probability of survival was noticed between ICUS and individual without cytopenia (Figure 5).
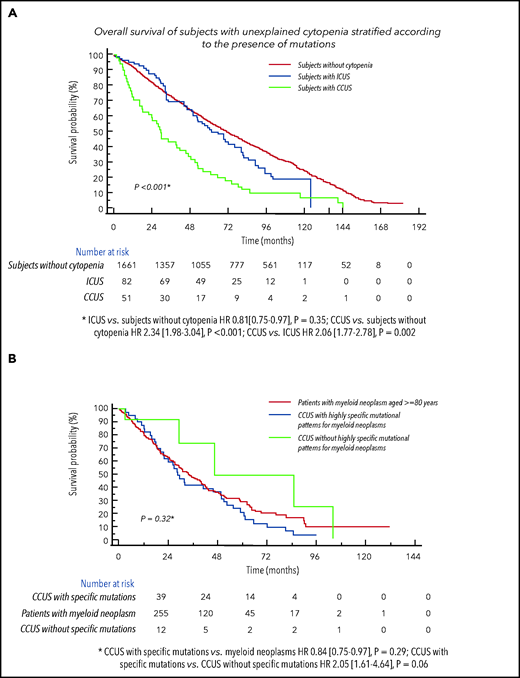
Clinical relevance of CHIP in individuals aged 80 years or older with unexplained cytopenia.(A) Overall survival of subjects with unexplained cytopenia form both the Health_&_Anemia and Monzino_80+ cohorts stratified according to the presence of mutations as ICUS vs CCUS. Probability of survival of individuals without cytopenia was also reported. (B) Overall survival of subjects with CCUS with highly specific mutational patterns for myeloid neoplasms vs CCUS without specific mutational patterns and vs age- and sex-matched patients affected with myeloid neoplasm (myelodysplastic syndromes, from the EuroMDS database).
Clinical relevance of CHIP in individuals aged 80 years or older with unexplained cytopenia.(A) Overall survival of subjects with unexplained cytopenia form both the Health_&_Anemia and Monzino_80+ cohorts stratified according to the presence of mutations as ICUS vs CCUS. Probability of survival of individuals without cytopenia was also reported. (B) Overall survival of subjects with CCUS with highly specific mutational patterns for myeloid neoplasms vs CCUS without specific mutational patterns and vs age- and sex-matched patients affected with myeloid neoplasm (myelodysplastic syndromes, from the EuroMDS database).
As we observed a difference in survival among CCUS vs ICUS, we tested the hypothesis that highly specific mutation patterns for myeloid neoplasms may provide presumptive evidence of hematological malignancy in patients with unexplained cytopenia even in absence of definitive morphological criteria. According to our findings, highly specific mutation patterns for myeloid neoplasms were defined as the presence of mutations of splicing factors; comutation patterns involving TET2, ASXL1, or DNMT3A; and/or mutations with VAF > 0.096.
CCUS with highly specific mutation patterns frequently showed macrocytic anemia and/or multilineage cytopenia (30 of 39 cases) consisting with a myelodysplastic syndrome phenotype. Two subjects died because of acute myeloid leukemia, while the most frequent cause of death was cardiac disease (25 subjects).
We then compared clinical features and outcomes of subjects with CCUS with those of an age- and sex-matched population affected with myeloid neoplasms (myelodysplastic syndromes) reported to retrospective EuroMDS database (n = 255; supplemental Table 4). No significant differences were observed in probability of survival between CCUS with highly specific mutation patterns with respect to patients with myeloid neoplasms, while CCUS without highly specific mutation patterns showed higher probability of survival with respect to CCUS with highly specific mutation patterns (HR, 2.05 [1.61-4.64], P = .06; Figure 5).
Considering CCUS with highly specific mutation patterns as “potential myeloid neoplasms,” 7.5% of subjects aged ≥80 years with cytopenia had presumptive evidence of myeloid neoplasm.
Discussion
We observed that CHIP is a common finding in the population aged ≥80 years (30% of individuals) and that specific mutational profiles are associated with distinct clinical outcomes.
Mutations in splicing genes (mostly occurring as single genetic lesion)29 showed the highest predictive value for myeloid neoplasms,1-4,30 while they are not significantly associated with increased risk of developing chronic inflammatory diseases.
By contrast, the PPV for myeloid neoplasms of isolated mutations in TET2, DNMT3A, and ASXL1 genes was lower, and additional genetic events are required to give rise to a myeloid neoplasm.1-4,30 These mutations are mostly linked to increased risk of myocardial infarction and arthritis.1-4,6,31
Focusing on prediction of myeloid neoplasms, both number of mutations per subject and size of the mutant clone (VAF) had significant PPV.30 We observed in addition that abnormalities in RBC indices significantly improve the capability of molecular features to capture individual risk of developing myeloid neoplasms.4 By combining specific mutational patterns, size of mutant clone, and abnormalities in RBC indices, we defined 3 groups of individuals with different risk of developing myeloid neoplasms. In details, we identified a small population (3% of individuals aged ≥80 years) in which cumulative incidence of myeloid neoplasm was 14%, 34%, and 42% at the age of 85, 90, and 95 years, respectively. Importantly, the predictive power of the score was confirmed by an external, independent validation.
Clonal evolution was a frequently observed event in subjects aged ≥80 years with sequential samples available and in some cases preceded the occurrence of an overt myeloid neoplasm, suggesting that serial analysis may improve clinical monitoring.30
We had the opportunity to study CHIP in centenarian according to different morbidity profiles. CHIP is common in centenarians with age-associated diseases while rarely observed in those who reach their 100th birthday without comorbidity. The possible relationship between absence of CHIP and exceptional longevity should be addressed by specific investigations.32
Finally, we hypothesized that myeloid neoplasms could be underdiagnosed in the population subjects ≥80 years, especially in cases with unexplained cytopenia.14,32 We observed that among individuals with unexplained cytopenia, the presence of a highly specific mutation pattern for myeloid neoplasms30,33 is associated with reduced survival. According to mutational features, 7.5% of subjects aged ≥80 years with cytopenia may have a presumptive evidence of myeloid neoplasm. The study of CHIP is therefore expected to contribute to determine specific causes of cytopenia in elderly people and define personalized treatment strategies.
Since CHIP was described, caution is suggested against adopting mutational testing in clinical practice. In fact, the presence of mutations per se in a given individual has only limited predictive power, as conversion to overt diseases is rare regardless of mutation status.1-3,34 Our findings improve the capability to capture clinical information at individual patient level with respect to the presence of specific mutation patterns. These data support the rationale for prospective studies including CHIP as a part of conventional laboratory investigations to evaluate the health general status of elderly people and drive strategies to prevent adverse events.
Acknowledgments
This work was supported by the Cariplo Foundation (Milan, Italy; project 2016-0860 [M.G.D._P., U.L.]), the AIRC Foundation (Associazione Italiana per la Ricerca contro il Cancro, Milan, Italy; project 22053 [M.G.D.P.]), Ricerca Finalizzata 2016 (Italian Ministry of Health, Italy; project RF2016-02364918 [M.G.D.P., U.L.]), the Beat Leukemia Foundation (Milan, Italy [M.G.D.P.]), PRIN 2017 (Ministry of University & Research, Italy; Project 2017WXR7ZT [M.G.D.P.]), and the European Union (Transcan 7, Horizon 2020, EuroMDS project 20180424 [M.G.D.P., F.S., U.P., P.F.]; and Horizon 2020, GenoMed4All project 101017549 [M.G.D.P., G.C., T.H., U.P., P.F.]). The Health_&_Anemia study was supported by a research grant from Amgen Italy. The Monzino_80+ study was supported by a research grant from Italo Monzino Foundation, Milan, Italy.
Authorship
Contribution: M.R., U.L., and M.G.D.P. designed the study; M.R., M.M., M.Z., M.T., Emma Riva, E.T., E.S., C.C., N.M., C.M., M.U., L.M., C.P., C. Selmi, M.D.S., K.M., G.M., M. Bicchieri, A.C., C.A.T., A.R., E.C., P.A., P.D., A.M., L.S., S.R., R.Z., C. Saitta, Elena Riva, G. Condorelli, F.P., A.S., W.K., T.H., and U.L. collected data; M.R., M.M., M.Z., M.T., Emma Riva, E.T., M. Bersanelli, S.M., A.A.G., E.M., G.S., R.A., S.D., R.P., G. Corrao, C. Sala, A.T., L.G., N.B., G. Castellani, W.K., T.H., U.L., and M.G.D.P. analyzed data; M.R., M.T., Emma Riva, E.T., M. Bersanelli, F.S., U.P., P.F., N.B., G. Castellani, W.K., G.S.V., T.H., U.L., and M.G.D.P. interpreted data; and all authors wrote and revised the manuscript.
Conflict -of-interest disclosure: M.R. has a consulting or advisory role with Pfizer, Celgene, IQvia, and Janssen; M.M. is employed by the MLL Munich Leukemia Laboratory. F.P. is a member of the speakers’ bureau of Novartis and AOP Orphan Pharmaceuticals. A.S. has a consulting or advisory role with Bristol-Myers Squibb, Servier, Gilead Sciences, Pfizer, Eisai, Bayer AG, MSD, Sanofi, and ArQule and is a member of the speakers’ bureau of Takeda, Roche, AbbVie, Amgen, Celgene, AstraZeneca, ArQule, Lilly, Sandoz, Novartis, Bristol-Myers Squibb, Servier, Gilead Sciences, Pfizer, Eisai, Bayer AG, and MSD. U.P. received honoraria from Celgene/Jazz; has a consulting or advisory role with Celgene/Jazz; received research funding from Amgen, Janssen, Novartis, BerGenBio, and Celgene; and received travel and accommodation expense reimbursement from Celgene. P.F. received honoraria from Celgene and research funding from Celgene. N.B. has a consulting or advisory role with Janssen and is a member of the speakers’ bureau at Celgene and Amgen. W.K. is employed by and has a leadership role at the MLL Munich Leukemia Laboratory and has stock and other ownership interests there. T.H. is employed by and has a leadership role at the MLL Munich Leukemia Laboratory and has a consulting or advisory role at Illumina. The remaining authors declare no competing financial interests.
Correspondence: Matteo G. Della Porta, Center for Accelerating Leukemia/Lymphoma Research IRCCS Humanitas Research Hospital, via Manzoni 56, 20089 Rozzano, Milan, Italy; e-mail: matteo.della_porta@hunimed.eu.
Access is provided to DNA sequence obtained from peripheral blood samples of the present study through the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) public repository. Data can be found under accession number PRJNA736552.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal