


Abstract
Cohesin is a multisubunit protein complex that forms a ring-like structure around DNA. It is essential for sister chromatid cohesion, chromatin organization, transcriptional regulation, and DNA damage repair and plays a major role in dynamically shaping the genome architecture and maintaining DNA integrity. The core complex subunits STAG2, RAD21, SMC1, and SMC3, as well as its modulators PDS5A/B, WAPL, and NIPBL, have been found to be recurrently mutated in hematologic and solid malignancies. These mutations are found across the full spectrum of myeloid neoplasia, including pediatric Down syndrome–associated acute megakaryoblastic leukemia, myelodysplastic syndromes, chronic myelomonocytic leukemia, and de novo and secondary acute myeloid leukemias. The mechanisms by which cohesin mutations act as drivers of clonal expansion and disease progression are still poorly understood. Recent studies have described the impact of cohesin alterations on self-renewal and differentiation of hematopoietic stem and progenitor cells, which are associated with changes in chromatin and epigenetic state directing lineage commitment, as well as genomic integrity. Herein, we review the role of the cohesin complex in healthy and malignant hematopoiesis. We discuss clinical implications of cohesin mutations in myeloid malignancies and discuss opportunities for therapeutic targeting.
Introduction
The eukaryotic genome is hierarchically and spatially organized from the level of the DNA double helix wrapped around histone proteins in nucleosomes to the 3-dimensional conformation of chromatin loops, topologically associating domains (TADs), compartments, and chromosome territories. This higher order genome organization is essential for long-distance gene regulation, which controls cell fate commitment and differentiation. The cohesin complex is an essential protein complex that wraps around the DNA and plays a critical role in sister chromatid cohesion and segregation, dynamic structural genome organization, transcriptional activation, and DNA replication and damage repair.
Cohesin is a multimeric protein complex that consists of 4 core subunits forming a ring-shaped structure encircling the DNA double helix (Figure 1A). In human cells, proteins encoded by the structural maintenance of the chromosome genes SMC1 and SMC3 form a V-shaped heterodimer creating a hinge at the dimerization domain. The RAD21 protein links the ATPase heads of SMC1 and SMC3, closes the loop, and is bound to the helical repeat proteins STAG1, STAG2, or the meiosis-specific paralogue STAG3. The relative abundance of the paralogue proteins STAG1 and STAG2 varies among cell types and developmental stages, with specific functions that are only beginning to be elucidated.1-3 Cohesin is loaded onto chromosomes by the NIPBL-MAU2 complex and released by WAPL and PDS5.4,5 Sororin constitutes an additional regulatory subunit competing with WAPL to stabilize cohesin (reviewed by Losada6 ).

Cohesin complex mutations in myeloid malignancies. The core members of the cohesin complex ring and its loader complex (A) and the frequency of mutations (B) according to diagnostic subgroup: de novo AML (n = 2170)8,15,128 ; NPM1-mutant AML (n = 418)15 ; FLT3-ITD-mutant AML (n = 341)15 ; t(8;21) AML (n = 254)15,32,49 ; inv(16) AML (n = 189)32,49,129 ; MDS (n = 1596)35,38 ; sAML (n = 93)130 ; AML-MRC (n = 106)8,18 ; CMML (n = 224)8,131 ; pediatric MDS (n = 38)39 ; pediatric AML (n = 993)129 ; and DS-AMKL (n = 190).41,42 Mutation frequency was calculated as frequency of positive reported cases within the total tested cohort in a single study or averaged across multiple available cohorts. AML-MRC, AML with myelodysplasia-related changes; CMML, chronic myelomonocytic leukemia; DS-AMKL, Down syndrome–associated acute megakaryoblastic leukemia.
Cohesin complex mutations in myeloid malignancies. The core members of the cohesin complex ring and its loader complex (A) and the frequency of mutations (B) according to diagnostic subgroup: de novo AML (n = 2170)8,15,128 ; NPM1-mutant AML (n = 418)15 ; FLT3-ITD-mutant AML (n = 341)15 ; t(8;21) AML (n = 254)15,32,49 ; inv(16) AML (n = 189)32,49,129 ; MDS (n = 1596)35,38 ; sAML (n = 93)130 ; AML-MRC (n = 106)8,18 ; CMML (n = 224)8,131 ; pediatric MDS (n = 38)39 ; pediatric AML (n = 993)129 ; and DS-AMKL (n = 190).41,42 Mutation frequency was calculated as frequency of positive reported cases within the total tested cohort in a single study or averaged across multiple available cohorts. AML-MRC, AML with myelodysplasia-related changes; CMML, chronic myelomonocytic leukemia; DS-AMKL, Down syndrome–associated acute megakaryoblastic leukemia.
Recurrent somatic mutations of the cohesin ring subunits and modulators have been identified across a wide spectrum of human malignancies, including myeloid neoplasms, glioblastoma, breast cancer, bladder cancer, melanoma, and Ewing sarcoma, among others. STAG2 is 1 of only 12 human genes to be significantly mutated in 4 or more distinct cancer types.7 In this review, we focus on the spectrum of cohesin mutations in myeloid neoplasms and their association with specific clinical entities. We discuss the general principles of cohesin biology and review the current understanding of how altered cohesin function affects hematopoietic homeostasis and contributes to disease development.
Incidence of cohesin mutations in human disease
Four of the core components of the cohesin complex (SMC1A, SMC3, RAD21, and STAG2) have been found to be collectively mutated in 10% to 20% of myeloid malignancies. Mutations in the regulatory subunits (NIPBL, WAPL, PDS5, and ESCO2) have been reported in less than 0.5% of cases, although these are prevalent in certain rare germline disorders, including Cornelia de Lange (CLS) and Roberts syndromes. Cohesin mutations generally occur as heterozygous lesions, with predominance of missense (∼50%), nonsense (22%), frameshift (17%), and splice-site (11%) mutations. They are distributed throughout the coding sequence without any obvious hotspots, and, at the protein level, they lead to haploinsufficiency or production of a dominant negative form.8,9
The overall frequency of cohesin mutations in myeloid malignancies is between 12% (in de novo acute myeloid leukemia [AML]) and 20% (in high-risk myelodysplastic syndrome [MDS] and secondary AML [sAML])8,10 (Figure 1B). Furthermore, cohesin mutations have been identified in chronic myeloid leukemia, chronic myelomonocytic leukemia (CMML), myeloproliferative neoplasms, and in rare cases of therapy-related clonal hematopoiesis of indeterminate potential (CHIP).8,11 Notably, cohesin mutations are rarely found in CHIP without prior exposure to cytotoxic agents.12-14 In the largest genetically annotated cohort of AML to date, cohesin mutations are classified as secondary-type mutations that belong to a distinct chromatin-spliceosome AML genomic subgroup associated with poor prognosis, independent of an antecedent diagnosis of MDS.15 This has been observed in additional cohorts,16-18 although an independent prognostic value for any mutant cohesin gene has not been established.
STAG2 is the predominant mutant cohesin subunit in solid malignancies, including Ewing sarcoma,19 melanoma,20 glioblastoma,21 and bladder,22 colon,23,24 and endometrial25 cancers. Of note, the spectrum of mutant cohesin genes in solid tumors is different from that observed in myeloid malignancies, where other cohesin subunits are recurrently mutated in addition to STAG2, especially in the context of de novo AML.20,26-28 RAD21 mutations, the most prevalent cohesin mutations in de novo AML, have not been described in solid tumors, although altered expression may be associated with poor clinical outcome29,30 in breast cancer. In contrast, STAG1 is almost never mutated in myeloid malignancies, but such mutations are relatively common in bladder and breast cancers31 and are frequently amplified in various carcinomas.
The distribution of mutant cohesin subunits across myeloid malignancies is also not uniform (Figure 1B; Table 1). RAD21 mutations dominate the de novo AML cohorts: they are present in 3% of combined de novo AML cases and in up to 8% and 11% to 20% of NPM1- and t(8;21)-mutant de novo AML subsets, respectively.15 This enrichment in core binding factor leukemia is most striking in RUNX1-RUNXT1+ cases, however, cohesin mutations are rarely found in AML with inv(16) CBF-MHY1 rearrangements.32-34 STAG2 mutations dominate the MDS and sAML cohorts. In MDS without clear leukemic drivers, STAG2 mutations co-occur with SRSF2, RUNX1, ASXL1, EZH2, CEBPA, TET2, and BCOR mutations35,36 and are associated with increased blast counts. In addition, gene expression patterns of STAG2-, SRSF2-, and EZH2-mutated MDSs are significantly correlated, suggesting, at least in part, some shared downstream transcriptional changes.37 In contrast to SRSF2 and U2AF1, mutations in the splicing factor SF3B1 and STAG2 tend not to co-occur.38
Cohesin mutation frequency in myeloid neoplasms
| Frequency (%)* | |||||||
|---|---|---|---|---|---|---|---|
| Entity | Total, n | SMC1A | SMC3 | STAG2 | RAD21 | NIPBL | CTCF |
| De novo AML | 21708,15,128 | 2.7 | 2.7 | 4.4 | 2.8 | 0.57 | N/A |
| NPM1+ AML | 41815 | 1 | 3 | 3 | 7 | N/A | 1 |
| FLT3+ AML | 34115 | 3 | 6 | 3 | 3 | N/A | N/A |
| t(8;21) AML | 25415,32-34,49 | 6.3 | 5.1 | 1.5 | 9.9 | N/A | N/A |
| inv(16) AML | 18915,32–34 | 0.6 | 0.9 | 0 | 0.5 | N/A | N/A |
| MDS | 159635,38 | N/A | 1.6 | 6.3 | 1.2 | 0.04 | N/A |
| sAML | 93130 | 3 | 2 | 14 | 2 | N/A | N/A |
| AML-MRC | 1068,18 | N/A | 1 | 6 | 3 | N/A | N/A |
| CMML | 2248,131 | N/A | N/A | 7.4 | 1.5 | N/A | N/A |
| Pediatric MDS | 3839 | 0 | 0 | 0 | 0 | N/A | N/A |
| Pediatric AML | 993129 | 0 | 1.6 | 1.2 | 1.8 | 1 | N/A |
| DS-AMKL | 19041,42 | 4.2 | 2.5 | 15.7 | 16.7 | 4.6 | 11.7 |
| Frequency (%)* | |||||||
|---|---|---|---|---|---|---|---|
| Entity | Total, n | SMC1A | SMC3 | STAG2 | RAD21 | NIPBL | CTCF |
| De novo AML | 21708,15,128 | 2.7 | 2.7 | 4.4 | 2.8 | 0.57 | N/A |
| NPM1+ AML | 41815 | 1 | 3 | 3 | 7 | N/A | 1 |
| FLT3+ AML | 34115 | 3 | 6 | 3 | 3 | N/A | N/A |
| t(8;21) AML | 25415,32-34,49 | 6.3 | 5.1 | 1.5 | 9.9 | N/A | N/A |
| inv(16) AML | 18915,32–34 | 0.6 | 0.9 | 0 | 0.5 | N/A | N/A |
| MDS | 159635,38 | N/A | 1.6 | 6.3 | 1.2 | 0.04 | N/A |
| sAML | 93130 | 3 | 2 | 14 | 2 | N/A | N/A |
| AML-MRC | 1068,18 | N/A | 1 | 6 | 3 | N/A | N/A |
| CMML | 2248,131 | N/A | N/A | 7.4 | 1.5 | N/A | N/A |
| Pediatric MDS | 3839 | 0 | 0 | 0 | 0 | N/A | N/A |
| Pediatric AML | 993129 | 0 | 1.6 | 1.2 | 1.8 | 1 | N/A |
| DS-AMKL | 19041,42 | 4.2 | 2.5 | 15.7 | 16.7 | 4.6 | 11.7 |
N/A, data not available. AML-MRC, AML-myelodysplasia-related changes.
Aggregated for multiple cohorts as listed.
Unlike mutations in splicing factors or epigenetic regulators, which are rare in pediatric MDS and AML,39,40 cohesin mutations are prevalent in a few specific childhood myeloid neoplasms (Figure 1B). More than 50% of cases of Down syndrome–associated acute megakaryoblastic leukemia (DS-AMKL)41-43 contain cohesin mutations, which are acquired as progression lesions from transient abnormal myelopoiesis (TAM) to AML. Similarly, cohesin mutations represent important progression lesions in myeloid neoplasms associated with MDS/AML germline predisposition syndromes such as GATA2 deficiency or RUNX1-familial platelet disorder.44-49 This is in accordance with our view of cohesin mutations as progression but not initiating lesions in adult myeloid neoplasms where they are usually a part of the dominant clone8,17 and are acquired as secondary hits after the acquisition of a CHIP-like (DNMT3A, TET2, or ASXL1) or SRSF2 mutation.17,36,50,51 Furthermore, single-cell studies of FLT3-internal tandem duplication (FLT3-ITD) mutant AML52 support the hypothesis that cohesin mutations are part of a preleukemic clone in which acquisition of mutations in NPM1 and/or FLT3-ITD lead to a full leukemic transformation.
Lastly, mutations in the cohesin complex have been identified in several germline syndromes termed cohesinopathies, including CLS and Roberts syndrome.53 NIPBL mutations account for ∼50% of cases of CLS,54 and mutations or microdeletion of RAD21,55 SMC1/SMC3,56 STAG1, and STAG257,58 have also been identified. The most frequently reported hematological phenotype associated with CLS is a mild thrombocytopenia in a subset of cases,59,60 and there have been case reports of rare association with lymphoid and myeloid malignancies.61,62 Notably, the megakaryocytic lineage is significantly affected in all germline syndromes (DS-AMKL , CLS, and RUNX1-familial platelet disorder) with associated cohesin mutations. Collectively, human genetics data strongly support the role of cohesin mutations as genetic drivers of progression in development of myelodysplasia and leukemia.
Cohesin as a gatekeeper of genomic integrity
The cohesin complex was initially described in cycling cells because of its role in the fundamental process of sister chromatid cohesion, also referred to as its cohesion-dependent function. During the G1 phase of the cell cycle, the opening of the cohesin ring is facilitated by the ATPase activity of the SMC1-SMC3 heterodimer, and the ring is subsequently loaded onto chromatin with the help of the NIPBL-MAU2 loader complex. Sister chromatid cohesion is maintained during DNA replication, and the cohesin complex prevents replication fork collapse and helps restart stalled replication forks63-66 (Figure 2A). During anaphase, removal of the cohesin complex is facilitated by HDAC8-mediated deacetylation67 and sister chromatids are released by WAPL-PDS5 after an orchestrated signaling cascade involving PLK1, AURKB, and CDK1.68 Centromeric cohesin remains loaded onto chromatin until RAD21 gets cleaved by separase. Proper loading and release of cohesin from DNA is essential for proper chromosome segregation.
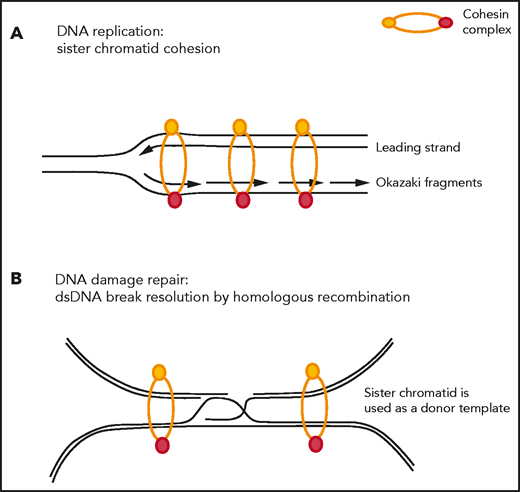
Sister chromatid cohesion–dependent functions of the cohesin complex. (A) Cohesin stabilizes replication forks during DNA replication and restarts stalled replication forks. (B) Cohesin is actively recruited to sites of DNA double-strand breaks to bring the damaged sister chromatid in close proximity of its nondamaged sister chromatid to be used as a donor template during the process of homologous recombination-mediated repair.
Sister chromatid cohesion–dependent functions of the cohesin complex. (A) Cohesin stabilizes replication forks during DNA replication and restarts stalled replication forks. (B) Cohesin is actively recruited to sites of DNA double-strand breaks to bring the damaged sister chromatid in close proximity of its nondamaged sister chromatid to be used as a donor template during the process of homologous recombination-mediated repair.
Furthermore, cohesin has been shown to facilitate DNA repair at sites of double-strand DNA (dsDNA) breaks by homologous recombination,69-71 (Figure 2B) and SMC1 is a substrate of the checkpoint protein kinase ATM.72 In rapidly dividing cells, such as embryonic stem cells, cohesin depletion leads to increased DNA damage, checkpoint activation, and cell cycle arrest with induction of TP53.73,74 In a mutant KRAS-driven oncogenic system, WAPL and RAD21 have been shown to be recruited to promote DNA repair and restart stalled replication forks,75 and WAPL deficiency leads to low DNA replication speed, reduced origin firing, and frequent DNA breakage. Models of cohesin-mutant MDS and AML similarly display evidence of DNA damage, with accumulation of γH2AX and activation of DNA damage checkpoints, slowing and stalling of replication forks, and increased sensitivity to PARP inhibition.76
Notably, cohesin gene mutations can induce genomic instability without creating overt aneuploidy.76-78 Cohesin mutations have not been found to be associated with complex karyotype or aneuploidy,22,79 except in an isolated report in bladder cancer.19 In myeloid malignancies, cohesin complex mutations are similarly not associated with increased cytogenetic aberrations, and they have been found to correlate inversely with aberrations in chromosome 5 and 7.8,17,80,81 Taken together, cohesin is an important gatekeeper of genomic integrity, and it remains to be seen whether genomic instability acts as a driver or a consequence of cohesin dysfunction in human cancers.
Role of cohesin in eukaryotic genome organization
Beyond this fundamental role of the cohesin complex in the maintenance of genomic integrity, cohesin plays a critical role in organizing the 3-dimensional architecture of eukaryotic genomes (Figure 3). During interphase, DNA is organized into chromatin loops, TADs, and compartments. Cohesin folds DNA into loops in a process called DNA loop extrusion, which allows for dynamic changes in specific arrangement of regulatory elements, including promoters and enhancers, and allows for orderly execution of cellular programs such as lineage commitment or self-renewal.82-86 This genomic architecture is maintained and dynamically shaped by anchors in CCCTC-binding factor (CTCF) proteins, which largely colocalize with cohesin and help form boundaries and insulate TADs.87,88

Sister chromatid cohesion–independent functions of the cohesin complex. (A) Cohesin mutations lead to increased intermixing of transcriptionally active A compartments and transcriptionally inactive B compartments. (B) Cohesin-mutant cells preserve TAD boundaries, albeit with weaker insulation, extrude longer loops and lose a subset of shorter E-P loops, which drives transcriptional changes. STAG1-cohesin is recruited to maintain chromatin organization in STAG2-mutant cells. (C) Cohesin is recruited along with the PRC complex to mediate epigenetic silencing of the HOX gene cluster at the level of nucleosomes.
Sister chromatid cohesion–independent functions of the cohesin complex. (A) Cohesin mutations lead to increased intermixing of transcriptionally active A compartments and transcriptionally inactive B compartments. (B) Cohesin-mutant cells preserve TAD boundaries, albeit with weaker insulation, extrude longer loops and lose a subset of shorter E-P loops, which drives transcriptional changes. STAG1-cohesin is recruited to maintain chromatin organization in STAG2-mutant cells. (C) Cohesin is recruited along with the PRC complex to mediate epigenetic silencing of the HOX gene cluster at the level of nucleosomes.
The development of chromosome conformation capture methods has enabled investigation of the role of epigenetic regulators, including cohesin proteins, in genomic organization and has linked them to transcriptional control. Cohesin, CTCF, and the transcriptional coactivator mediator have been shown to link gene expression with chromatin structure by looping enhancers and promoters of active genes in a cell-type–specific manner.89 Depletion of the cohesin release factor WAPL leads to processive loop enlargement and increased interaction between neighboring TADs,90 as well as an increase in TAD size and weakening of compartments.91 In contrast, deletion of the cohesin-loading factor NIPBL results in loss of TADs but reinforcement of compartment structure, suggesting an interplay of a cohesin-dependent formation of TADs with cohesin-independent segregation of the genome into compartments.92 Depletion of the cohesin subunit RAD21 has similarly been shown to lead to elimination of TADs and strengthening of compartment structure,91,93 whereas other studies of cohesin depletion have shown preservation of the TAD architecture.94,95 Evaluation of the distinct roles of STAG1-cohesin and STAG2-cohesin in chromatin organization revealed that STAG1-cohesin preferentially stabilizes TAD boundaries, whereas STAG2-cohesin facilitates enhancer-promoter (E-P) loop formation.1,77 Finally, both CTCF depletion and genetic manipulation of CTCF binding sites disrupt genome conformation and impair enhancer-dependent gene expression.96-98 Of note, and somewhat unexpectedly, there was no or a very modest change in gene expression after depletion of a cohesin subunit or its regulators in any of these studies, which is consistent with what has been previously observed with different cohesinopathies.
Modeling cohesin dysfunction in hematopoiesis and leukemia
The first piece of evidence supporting the role of cohesin in normal hematopoiesis came from genetic screens in zebrafish which were conducted to identify positive regulators of hematopoietic runx1 expression. These studies showed that biallelic inactivation of either rad21 or smc3 resulted in a complete loss of differentiated blood cells and monoallelic loss–affected gene expression.99 The identification of recurrent somatic mutations in cohesin subunits as drivers in myeloid malignancies have subsequently prompted several groups to further study the role of the cohesin complex in hematopoietic stem and progenitor cells (HSPCs), in both primary mouse and human cell models (Table 2).
In vivo models to study cohesin function in hematopoiesis and leukemia
| Model system | Reference | Hematopoietic phenotype | Chromatin changes | Transcriptional changes |
|---|---|---|---|---|
| Mx1-Cre+; Smc3fl/fl Mx1-Cre+; Smc3fl/+ | 100 | BM aplasia, lethal -↑LSK, ↓MP, ↑ST-HSC, ↓LT-HSC, ↑MPP -Competitive advantage in transplant assays -AML in cooperation with FLT3-ITD | — ↓Chromatin accessibility at enhancers of downregulated genes | — -Global reduction in transcription -↓Expression of TFs associated with lineage commitment |
| Vav1-Cre+; Smc3fl/fl ERT2-Cre+; Smc3fl/fl ERT2-Cre+; Smc3fl/+ Vav1-Cre+; Smc3fl/+ | 101 | Embryonic lethal BM aplasia, lethal -No change in number of HSPCs -Competitive disadvantage in transplant assays, partial rescue in cooperation with DNMT3A+/− No change in number of HSPCs | — — — Minimal changes in chromatin accessibility | — — — Minimal global transcriptional changes |
| Mx1-Cre+; Stag2fl/fl Mx1-Cre+; Stag1fl/fl Mx1-Cre+; Stag1fl/fl Stag2fl/fl | 102 | -↑LSK, ↑MPP, ↑ST-HSC, ↑LT-HSC, ↑GMP -Competitive advantage in transplant assays -PB with progressive leukopenia, thrombocytopenia, ↑myeloid, ↓B cells -Myelodysplasia in the BM -No change in number of HSPCs -No competitive advantage in transplant assays -No morphologic changes BM failure, lethal | -↓ Chromatin accessibility at enhancers of downregulated genes -↓Chromatin insulation — — | -↓Cell commitment (B cell > myeloid, erythroid) -↑Self-renewal signature — — |
| Transplant models of Rad21, Smc1a, and Stag2 TRE-shRNA; ROSA26(M2rtTA/+) | 103 | -↓ST- and LT-HSC, ↑GMP, ↓MEP, ↑MPP -Extramedullary hematopoiesis and MPN in aged Smc1ashRNA mice, exaggerated in compound Stag2shRNA Smc1ashRNA | ↑Chromatin accessibility of myeloid lineage genes | -↑Myeloid differentiation -↓Lymphoid-specific gene expression |
| Transplant models of human cord blood CD34+ cells expressing RAD21 shRNA in NSG Culture of RAD21 E212*, RAD21 Q592* and SMC1A R711G-expressing human CD34+ cord blood cells | 9 | -↑CD34+ cell frequency and engraftment in BM of NSG mice -Myeloid differentiation skewing -↓Erythroid and myeloid differentiation -↑Serial replating | — -Global↓ in chromatin accessibility at transcriptional regulators -↑Chromatin accessibility for ERG, GATA2, and RUNX1 consensus binding sites | — -↑HSC gene expression (HOX genes, MEIS1) -↓Myeloid differentiation genes (MPO, CSF1R) |
| Transplant models of human cord blood CD34+CD38−CD90+ CD45RA− cells expressing STAG2 or SMC3 shRNA in NSG | 105 | -↑Engraftment in the BM in primary and secondary transplants -Myeloid skewing (with STAG2 shRNA, not SMC3 shRNA) -↑Frequency of CD34+CD38-cells; | ↑HSC gene expression | |
| Transplant model of LSK transduced with Rad21 shRNA | 104 | -↓HSPC differentiation in animals on dietary restriction > ad libitum diet -↑HSC self-renewal with serial transplantation with AL diet, aging and inflammation -↓LPS-induced myeloid differentiation of LSK cells | ↓Chromatin accessibility, accentuated with LPS treatment | ↓NF-κB dependent signaling in transplanted HSCs |
| Mx1-Cre+; Stag2fl/− Mx1-Cre+; Stag2fl/− Runx1fl/fl | 36 | -Mild ↓WBC, ↓B cells, ↑RDW -Mild trilineage dysplasia -↑LSK,ST- and LT-HSC, MPP -Myeloid skewing, ↑CMP, ↑GMP, ↓CLP, ↓MEP, ↓erythroid program -Pancytopenia, ↑RDW, ↑MCV -↑LSK, ↑MPP, ↓ST-and LT-HSC -Myeloid skewing -MDS, severe trilineage dysplasia | -↑Chromatin accessibility at RUNX1, GATA2 sites -↓Chromatin accessibility at IRF sites -Slight ↑TAD boundary insulation -↑Chromatin accessibility changes compared with Mx1-Cre+; Stag2fl/− -Slight ↑TAD boundary insulation -↓E-P loop formation | -↑Myeloid program -↓Lymphoid program -↑Gene expression changes compared with Mx1-Cre+; Stag2fl/− -↓HoxA9, ↑Gata2, ↑Fos -↓Expression of high pausing genes |
| Sequential transplant model of Cas9+ c-kit+ cells expressing Tet2 and Stag2 sgRNA Culture of isogenic STAG2 knockout U937 cells | 76 | -Leukocytosis, anemia, thrombocytopenia, lymphopenia -MDS with evidence of dysplasia, ↓megakaryocytes, ↑erythrophagocytosis>-↑LSK,ST- and LT-HSC, MPP — | — -↑Intermixing of A and B compartments -↓TAD boundary insulation -↑Size extruded loops | — -↑Myeloid program -↓DNA damage repair -↑Type 1 interferon response |
| Model system | Reference | Hematopoietic phenotype | Chromatin changes | Transcriptional changes |
|---|---|---|---|---|
| Mx1-Cre+; Smc3fl/fl Mx1-Cre+; Smc3fl/+ | 100 | BM aplasia, lethal -↑LSK, ↓MP, ↑ST-HSC, ↓LT-HSC, ↑MPP -Competitive advantage in transplant assays -AML in cooperation with FLT3-ITD | — ↓Chromatin accessibility at enhancers of downregulated genes | — -Global reduction in transcription -↓Expression of TFs associated with lineage commitment |
| Vav1-Cre+; Smc3fl/fl ERT2-Cre+; Smc3fl/fl ERT2-Cre+; Smc3fl/+ Vav1-Cre+; Smc3fl/+ | 101 | Embryonic lethal BM aplasia, lethal -No change in number of HSPCs -Competitive disadvantage in transplant assays, partial rescue in cooperation with DNMT3A+/− No change in number of HSPCs | — — — Minimal changes in chromatin accessibility | — — — Minimal global transcriptional changes |
| Mx1-Cre+; Stag2fl/fl Mx1-Cre+; Stag1fl/fl Mx1-Cre+; Stag1fl/fl Stag2fl/fl | 102 | -↑LSK, ↑MPP, ↑ST-HSC, ↑LT-HSC, ↑GMP -Competitive advantage in transplant assays -PB with progressive leukopenia, thrombocytopenia, ↑myeloid, ↓B cells -Myelodysplasia in the BM -No change in number of HSPCs -No competitive advantage in transplant assays -No morphologic changes BM failure, lethal | -↓ Chromatin accessibility at enhancers of downregulated genes -↓Chromatin insulation — — | -↓Cell commitment (B cell > myeloid, erythroid) -↑Self-renewal signature — — |
| Transplant models of Rad21, Smc1a, and Stag2 TRE-shRNA; ROSA26(M2rtTA/+) | 103 | -↓ST- and LT-HSC, ↑GMP, ↓MEP, ↑MPP -Extramedullary hematopoiesis and MPN in aged Smc1ashRNA mice, exaggerated in compound Stag2shRNA Smc1ashRNA | ↑Chromatin accessibility of myeloid lineage genes | -↑Myeloid differentiation -↓Lymphoid-specific gene expression |
| Transplant models of human cord blood CD34+ cells expressing RAD21 shRNA in NSG Culture of RAD21 E212*, RAD21 Q592* and SMC1A R711G-expressing human CD34+ cord blood cells | 9 | -↑CD34+ cell frequency and engraftment in BM of NSG mice -Myeloid differentiation skewing -↓Erythroid and myeloid differentiation -↑Serial replating | — -Global↓ in chromatin accessibility at transcriptional regulators -↑Chromatin accessibility for ERG, GATA2, and RUNX1 consensus binding sites | — -↑HSC gene expression (HOX genes, MEIS1) -↓Myeloid differentiation genes (MPO, CSF1R) |
| Transplant models of human cord blood CD34+CD38−CD90+ CD45RA− cells expressing STAG2 or SMC3 shRNA in NSG | 105 | -↑Engraftment in the BM in primary and secondary transplants -Myeloid skewing (with STAG2 shRNA, not SMC3 shRNA) -↑Frequency of CD34+CD38-cells; | ↑HSC gene expression | |
| Transplant model of LSK transduced with Rad21 shRNA | 104 | -↓HSPC differentiation in animals on dietary restriction > ad libitum diet -↑HSC self-renewal with serial transplantation with AL diet, aging and inflammation -↓LPS-induced myeloid differentiation of LSK cells | ↓Chromatin accessibility, accentuated with LPS treatment | ↓NF-κB dependent signaling in transplanted HSCs |
| Mx1-Cre+; Stag2fl/− Mx1-Cre+; Stag2fl/− Runx1fl/fl | 36 | -Mild ↓WBC, ↓B cells, ↑RDW -Mild trilineage dysplasia -↑LSK,ST- and LT-HSC, MPP -Myeloid skewing, ↑CMP, ↑GMP, ↓CLP, ↓MEP, ↓erythroid program -Pancytopenia, ↑RDW, ↑MCV -↑LSK, ↑MPP, ↓ST-and LT-HSC -Myeloid skewing -MDS, severe trilineage dysplasia | -↑Chromatin accessibility at RUNX1, GATA2 sites -↓Chromatin accessibility at IRF sites -Slight ↑TAD boundary insulation -↑Chromatin accessibility changes compared with Mx1-Cre+; Stag2fl/− -Slight ↑TAD boundary insulation -↓E-P loop formation | -↑Myeloid program -↓Lymphoid program -↑Gene expression changes compared with Mx1-Cre+; Stag2fl/− -↓HoxA9, ↑Gata2, ↑Fos -↓Expression of high pausing genes |
| Sequential transplant model of Cas9+ c-kit+ cells expressing Tet2 and Stag2 sgRNA Culture of isogenic STAG2 knockout U937 cells | 76 | -Leukocytosis, anemia, thrombocytopenia, lymphopenia -MDS with evidence of dysplasia, ↓megakaryocytes, ↑erythrophagocytosis>-↑LSK,ST- and LT-HSC, MPP — | — -↑Intermixing of A and B compartments -↓TAD boundary insulation -↑Size extruded loops | — -↑Myeloid program -↓DNA damage repair -↑Type 1 interferon response |
GMP, Lin− cKit+ Sca1− Cd34+ Fcg+; LSK, Lin− Sca-1+ c-Kit+; LT-HSC, LSK+ Cd150+ Cd48−); MP myeloid progenitors (Lin− Sca-1− c-Kit+); MPP, LSK+ Cd150− Cd48+ Cd127−; NSG NOD/SCID/IL2R-gamma null mice; PB peripheral blood; ST-HSC, LSK+ Cd150− Cd48−; RDW red cell distribution width; WBC, white blood cell.
Several models have been used to examine an isolated loss or downregulation of a single cohesin subunit in the adult bone marrow (BM) compartment. For example, whereas a complete loss of Smc3 in the hematopoietic compartment leads to BM aplasia, consistent with Smc3 being an essential gene, conditional Smc3 haploinsufficiency drives an expansion of short-term hemapoietic stem cells (ST-HSCs) and multipotent progenitors (MPPs), accompanied by a decrease in myeloid progenitors and long-term (LT)-HSCs. Smc3 haploinsufficient BM cells demonstrated a clonal advantage in a transplant setting, but overt leukemic transformation was seen only when combined with FLT3-ITD.100 Additional models of Smc3 haploinsufficiency had few effects on steady-state hematopoiesis and yielded a competitive disadvantage over time, suggesting a need for cooperating genetic events.101
A similar genetic approach to model Stag2 loss in mice showed that Stag2 deficiency in the adult hematopoietic compartment leads to an expansion of ST- and LT-HSCs, MPPs, and granulocyte macrophage progenitors (GMPs) and is also associated with a competitive advantage in transplant assays. In addition, unlike the Smc3-haploinsufficient models, Stag2 conditional knockout mice showed evidence of myelodysplasia in the BM and impaired lymphoid differentiation.102 A parallel approach of doxycycline-inducible knockdown of Stag2, Rad21, and Smc1a led to the development of an age-dependent myeloproliferative phenotype and increased hematopoietic stem cell (HSC) self-renewal,103,104 without an overt leukemic transformation. Similar results were obtained in the human CD34+ cell context, where knockdown or overexpression of truncated or mutant cohesin subunits led to increased engraftment with myeloid skewing.9,105
One caveat of the single cohesin subunit depletion models to study cohesin dysfunction in myeloid disease is that cohesin mutations are rarely, if ever, initiating events in myeloid neoplasia and are generally not seen in CHIP. Human genetics data support a model in which cohesin mutations are acquired after a preexisting mutation, usually in an epigenetic regulator, such as ASXL1, TET2, or SRSF2, has clonally expanded.17,36,50,51 Therefore, the specific genetic background and subsequent epigenetic rewiring of cells that acquire cohesin mutations likely significantly affect the resulting phenotype of the cohesin-mutant clone. In return, this may also enable the mutant cells to tolerate some of the phenotypic consequences resulting from cohesin deficiency that may otherwise be selected against. Modeling of loss-of-function mutations in STAG2 or SMC3 in human CD34+ cells using CRISPR followed by transplantation into immunodeficient mice leads to significantly less clonal expansion than what has been observed with the 3 most recurrently mutated genes in CHIP, ASXL1, TET2, and DNMT3A.106 Combinatorial models of cohesin deficiency have recently been described. Mice deficient in Stag2 and Runx1 develop MDS with severe trilineage dysplasia, expansion of LSK and MPP pools and pancytopenia with myeloid skewing.36 Sequential acquisition of Tet2 and Stag2 loss-of-function mutations leads to development of MDS with leukocytosis, anemia, and significant dysmegakaryopoiesis.76
Taken together, both human genetic data and experimental evidence argue that cohesin mutations are early, but not initiating, genetic lesions during myeloid disease development. We hypothesize that they rely on a primed state (which could be driven by a CHIP-like mutation, germline RUNX1, or GATA2 mutation, TAM in the context of trisomy 21, an inflammatory microenvironment, and/or genomic stress), to give a clonal advantage and facilitate a positive mutational selection. This process precedes and likely predetermines the types of additional secondary mutations that result in overt leukemic transformation (eg, NPM1 or RUNX1:RUNXT1 and RUNX1). Additional experimental models that recapitulate human genetics and allow for temporal control of mutation acquisition are needed to fully understand the function that cohesin mutations play both in the development of MDS arising in a setting of CHIP, TAM, or other preexisting BM abnormalities, and in the progression to sAML.
Impact of cohesin mutations on chromatin architecture and transcriptional control in hematopoiesis
A large number of studies have linked phenotypic changes in hematopoietic differentiation and self-renewal to changes in chromatin accessibility in distinct hematopoietic populations,9,102,103,107 as well as in AML cell lines76,108,109 (Table 2). The common message emerging from these data sets suggests that loss of cohesin in the hematopoietic compartment leads to global changes in chromatin accessibility, including increased intermixing of transcriptionally active A compartments and transcriptionally inactive B compartments, decreased insulation at TAD boundaries, and preferential loss of shorter E-P loops (Figure 3A-B). These effects have been in turn linked to changes in chromatin accessibility of regulatory elements that drive expression of differentially expressed genes. For example, increased chromatin accessibility at binding sites of lineage-defining transcription factors, such as ERG, GATA2, and RUNX1, in CD34+ cells overexpressing different cohesin mutations, was associated with increased self-renewal.9 Similarly, Stag2 deficiency in adult mouse BM interferes with B-cell lineage–specific differentiation programs, as exemplified by Ebf1/Pax5 loci where local interactions mediated by Stag2-cohesin are not fully compensated for by Stag1-cohesin binding.107 In addition, cohesin has been shown to play a dynamic role in inducing erythroid differentiation by interacting with tissue-specific transcription factors, such as Etv6. Depletion of cohesin leads to failure of activation of genes repressed by Etv6 and proper erythroid differentiation110 (Figure 3B). Conversely, Rad21 was found to interact with the polycomb-repressive complex111 and repress Hox gene expression. Cohesin-depleted HSPCs have also been shown to lose H3K27me3 marks in the Hox cluster of genes and lead to Hoxa9 upregulation, which has been linked to increased self-renewal (Figure 3C).112
The loci that control stem cell identity tend to be highly complex regulatory elements, with several promoter-enhancer and enhancer-enhancer contacts clustered together as “superenhancers.”113 Why these complex regulatory regions in cohesin-mutant cells are preferentially protected over those driving lineage commitment and differentiation has not been fully elucidated. In a model of complete cohesin depletion, only a very small set of genes in proximity with the superenhancers has been shown to be strongly downregulated.93 In Stag2/Runx1–deficient cells, superenhancer-associated genes with low transcriptional pausing were less affected than genes with high basal transcriptional pausing, such as interferon response or DNA repair response genes, which were more downregulated.36 A possible explanation for these observations is that mutant cohesin or low levels of residual wild-type cohesin complexes remain preferentially bound to superenhancer elements, as compared with typical enhancers with less complexity. This would be consistent with previous observations made in yeast, where quantitative titration of cohesin complex levels led to preferential binding to specific genomic loci and a striking difference in the resulting effect on sister chromatid cohesion–dependent vs independent functions of the complex.69 The low residual level of wild-type cohesin complex or functionally altered mutant cohesin complexes may be sufficient to maintain most regulatory elements required for HSPC proliferation and self-renewal, while reducing the ability to restructure the chromatin for induction of lineage determination and differentiation.
The critical role that cohesin complexes play in dynamic facilitation of specific E-P contacts is also evident in studies of inducible gene expression in response to inflammatory stimuli. Isolated Rad21 depletion in otherwise healthy mature macrophages has been shown to lead to impaired induction of inflammatory gene expression programs.114 Similarly, isolated depletion of Stag2, Smc3, or Smc1a in healthy mouse HSPCs reduced the NF-κB–dependent inflammatory gene expression programs and increased resistance to differentiation-inducing inflammatory stimuli.114 Taken together, DNA looping interactions mediated by cohesin complexes are critical for HSPC function, and cohesin dysfunction can lead to impairment of differentiation and enforcement of stem cell programs.
Distinct roles of STAG1- vs STAG2-cohesin complexes
Multiple models of cohesin dysfunction in hematopoietic cells have demonstrated that although certain aspects of chromatin organization may be perturbed, the 3-dimensional structure is still mostly preserved. This observation was not unexpected, given that cohesin-mutant cells retain a wild-type copy of the affected cohesin subunit, and, in studies of complete X-linked STAG2 deficiency, increased expression of the paralogue protein STAG1 was shown to rescue a significant portion of STAG2-binding sites.77,107 Analysis of the composition of cohesin complexes in cohesin-mutant AML cell lines identified a switch from STAG2- to STAG1-containing cohesin complexes, which has been suggested to be a common downstream effect across different cohesin mutations that are not limited to STAG2.76 Such a switch may in turn help explain some of the shared features of cohesin-mutant phenotypes and directly link them to functional differences between the STAG1- and STAG2-containing complexes.
The STAG1 and STAG2 complexes have been long appreciated to play distinct roles during development and cellular differentiation. For example, whereas STAG1 cohesin is enriched in the egg extracts of the xenopus oocyte, STAG2 cohesin is the predominant form in xenopus somatic tissues, suggesting a developmental switch.2,3 In addition, the 2 versions of the complex have been linked to differential regulation of telomere and centromere cohesion.115 STAG1- and STAG2-cohesin complexes have also been shown to differentially affect the choice of dsDNA repair pathway, with STAG2-cohesin complex being preferentially involved in homologous recombination repair70 and replication fork progression. This may in turn explain why cohesin-mutant cells, including STAG2-, RAD21-, and SMC3-mutant MDS and AML cells, all of which display a switch from STAG2 to STAG1 complexes, show high levels of dsDNA breaks, stalled replication forks, and sensitivity to PARP inhibition76 and other inhibitors of DNA damage repair.116
More recent studies highlighted additional nonoverlapping functions of the 2 complexes through investigation of the association of STAG1 and STAG2 with chromatin and its impact on chromatin organization and transcriptional control.117,118 In one such study, STAG1 cohesin was found to bind preferentially to and stabilize TAD boundaries, whereas STAG2 cohesin was more involved in facilitating shorter E-P loops independent of CTCF.77 Furthermore, STAG1 and STAG2 cohesin seem to have opposite effects in their contribution to the polycomb-mediated network of long-range chromosome interactions.119 Although STAG2 preferentially facilitates polycomb domain compaction through PRC recruitment and is necessary for organization of local transcriptional hubs that are essential for cell identity,120 STAG1 contributes to TAD boundary strength by loop extrusion119 and counteracts compartmentalization more than STAG2 does.77 Some of these phenotypes have now been recapitulated in mouse models of hematopoietic compartment–specific Stag2 deficiency,102 as well as Stag2-mutant MDS and AML.36,76 We hypothesize that additional models of cohesin-mutant myeloid malignancies, not just those restricted to STAG2 mutations, would similarly highlight distinct features of STAG1 and STAG2 complexes, given the switch from STAG2 to STAG1 cohesin that has been observed in SMC3- and RAD21-mutant AML cells.76 Targeting of STAG1 across cohesin-mutant myeloid malignancies and cancer in general may therefore be a particularly attractive therapeutic approach.
Clinical implications
As outlined in this review, multiple lines of evidence demonstrate the critical role that cohesin plays in normal hematopoiesis and development of leukemia. Current evidence suggests that cohesin mutations observed in myeloid malignancies lead to development of a stem cell-like phenotype, impaired hematopoietic differentiation and leukemic progression. There is currently no clear evidence that the presence of a cohesin mutation has any impact on response to intensive chemotherapy alone or in combination with more targeted agents, although increased response to hypomethylating agents has been reported in some cohesin-mutant patients17 and mouse xenograft models106 and recent data suggest that STAG2 mutations may be associated with poor response to the FLT3 inhibitor crenolanib.121
Given the dependency of STAG2-mutant cells on STAG1,76,107,122,123 targeting STAG1- or STAG1 cohesin–dependent functions may be a promising therapeutic approach in this context. STAG1 has been successfully degraded using the auxin-inducible degradation system and resulted in a mitotic arrest of STAG2-mutant but not wild-type cells.124 Therefore, a STAG1-directed, small-molecule degrader could provide an approach to targeting this structural protein. Furthermore, crystal structures of STAG1 segments bound to RAD21 peptides led to identification of the D797 residue of STAG1 as being unique and critical for interaction with RAD21, chromatin association, and sister chromatid cohesion. Therefore, an inhibitor blocking STAG1 interaction with RAD21 around this residue represents yet another strategy to tackle the complex.
Several groups have shown that targeting of other members of the cohesin complex could be exploited therapeutically in a broader context. For example, NIPBL overexpression has been shown to correlate with poor outcomes and treatment resistance in non–small cell lung cancer, and knockdown of NIPBL in non–small cell lung cancer cell lines leads to impaired proliferation, migration, and invasion in vitro, as well as increased response to chemotherapy.125,126 Therefore, modulation of levels of expression, stability, or activity of different cohesin subunits and modulators should be explored further, particularly in the context of nonessential genes, such as STAG1, NIPBL, or HDAC8, which is essential for deacetylation and recycling of cohesin in interphase.67
Finally, although successful targeting of the STAG1-cohesin complex may be inherently challenging given the similarities between STAG1 and STAG2 proteins, some of the phenotypes associated with STAG1-cohesin complexes, such as replication fork stalling and increased dsDNA breaks, are more readily targetable. PARP inhibitors, such as talazoparib, have been shown to deplete STAG2-mutant MDS, AML, glioblastoma, Ewing sarcoma, and bladder cancer cells, and AML models of mutations in additional cohesin ring components, including SMC3 and RAD21, have also demonstrated sensitivity to PARP inhibition.21,76,116,127 A phase 1B clinical trial evaluating the effect of talazoparib in cohesin-mutant MDS and AML is currently ongoing (registered on www.clinicaltrials.gov as NCT03974217).
Conclusions
Remarkable progress in our understanding of the role of the cohesin complex in shaping genomic architecture, regulation of gene expression, and maintenance of genomic integrity has been made since the first genetic screens in yeast that identified its different components. More recent discovery of cohesin mutations as genetic drivers of transformation across multiple cancer types, including myeloid malignancies, has fueled deeper mechanistic investigations into the role of this complex in development of human disease. Whereas we understand some aspects of the DNA looping, gene expression, DNA replication, and DNA damage repair processes that are disrupted in models of cohesin-mutant disease, we still do not know which of the many downstream effects of cohesin mutations are causative of the process of transformation. Development of models of cohesin-mutant disease that faithfully recapitulate the right cellular context, timing, and genetic complexity observed in patients will be critical as we push forward to understand the mechanisms of transformation in cohesin-mutant diseases and develop more effective therapies .
Acknowledgments
Z.T. was supported by National Institutes of Health National Heart, Lung, and Blood Institute grant K08HL140138-01, the Burroughs Wellcome Fund, the American Society of Hematology, the Doris Duke Charitable Foundation, the Alex’s Lemonade Stand Foundation, the RUNX1 Research Foundation, the Edward P. Evans Foundation, the V Foundation, the HMS/Ludwig Center, the KI-DF/HCC Bridge Project and Novartis. J.-C.J. was supported by German Research Foundation (DFG) grant JA 3193/1-1.
Authorship
Contribution: J-C.J. and Z.T. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Zuzana Tothova, Department of Medical Oncology, Dana Farber Cancer Institute, 450 Brookline Ave, Dana 540A, Boston, MA 02115; e-mail: zuzana_tothova@dfci.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal