Key Points
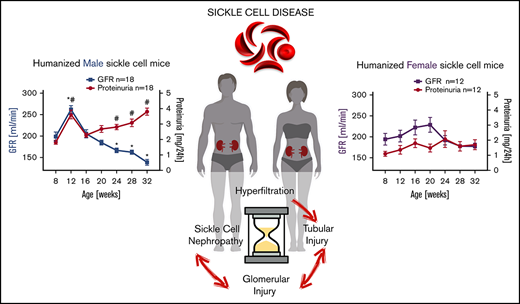
In HbSS mice, hyperfiltration predicts long-term kidney damage and tubular injury occurs prior to glomerular damage.
Sex contributes to the progression of kidney injury in HbSS mice.

Abstract
We previously reported that humanized sickle cell (HbSS) mice develop spontaneous nephropathy, a major cause of morbidity and mortality in sickle cell disease (SCD). Because sex-dependent protective mechanisms in SCD have been reported, we examined the course of nephropathy in male and female HbSS mice to determine contributors and/or predictors of disease severity. In male HbSS mice, glomerular filtration rate was characterized by a rapid onset of hyperfiltration and subsequent progressive decline of renal function over 20 weeks. Early tubular injury presented with increased excretion of kidney injury marker 1 (KIM-1), progressive loss of tubular brush border, and interstitial fibrosis that preceded the onset of glomerular damage, suggesting a tubuloglomerular mechanism of kidney injury in these mice. Additionally, we observed a strong association between the magnitude of hyperfiltration and the degree of long-term kidney injury in male HbSS mice. Unlike males, female HbSS mice did not demonstrate a significant loss of renal function or severe kidney damage during the time course of the study. These results suggest that magnitude of hyperfiltration predicts the onset of chronic kidney damage in male HbSS mice, whereas protective mechanisms in female HbSS mice delay the onset of SCD nephropathy.
Introduction
Sickle cell disease (SCD) is a hematologic disorder that leads to extensive pathology in diverse organ systems. The kidney is among the most frequently affected organs, as evidenced by albuminuria that occurs in approximately two-thirds of SCD patients.1 Renal involvement begins in early childhood and can progress to end-stage renal disease (ESRD) at a median age of 23 years.2 Furthermore, SCD patients that progress to ESRD demonstrate heightened mortality, with a median survival rate of 2 years.2,3 Despite significant improvement in the management of SCD and associated prolongation of life expectancy, SCD patients suffer from progressive organ damage, including chronic kidney disease.4,5 Therefore, there is an urgent need to better understand the sequence of events that initiate renal injury in SCD in order to identify at-risk patients and intervene before irreversible damage has occurred.
The early renal phenotype of SCD is marked by a period of elevated glomerular filtration rate (GFR), termed the hyperfiltration phase, as well as a urinary concentrating defect and abnormalities in tubular function.6,7 Specifically, the importance of the hyperfiltration phase as a potential mediator of early renal damage in SCD has generated considerable discussion.8-10 Hyperfiltration has been demonstrated to be highly prevalent in children with SCD.11 During adolescence, SCD patients begin to encounter a decline in estimated GFR (eGFR), including some young adults who progress to chronic kidney disease (CKD).7,12 Despite this, the importance of hyperfiltration as a risk factor for future progression to ESRD is unknown. Moreover, additional cross-sectional studies have demonstrated the presence of elevated renal injury biomarkers in young SCD patients.7,13-15 Elevations in tubular injury markers are associated with glomerular injury,16 highlighting potential tubular alterations happening prior to glomerulopathy, and suggesting a tubuloglomerular mechanism of kidney injury in SCD. However, critically absent is evidence to demonstrate the characteristics of primary pathophysiological processes involved in kidney injury and the natural course of the renal disease in SCD.
Sex differences in the frequency and severity of many renal diseases, including ischemic acute kidney injury and CKD, have been well documented in animal models and humans.17-22 In 2015, 62% of CKD patients in the United States who progressed to ESRD were men, whereas only 38% were women23 : women with CKD presented with a slower decline in renal function with time when compared with men with CKD.24,25 Also, women are more resistant to ischemia/reperfusion induced kidney injury,21 and present with greater renal protection against chronic nitric oxide inhibition induced CKD than men.19 Sex differences were also reported in diabetes,22 another kidney disease with multiple similarities to SCD nephropathy. Moreover, increased hemolysis is a risk factor for hyperfiltration, proteinuria, and CKD in SCD.9,26,27 Recently, Raslan et al first observed reduced hemolysis and hemolytic markers in female SCD patients, suggesting potential sex-dependent protection and survival advantage from chronic SCD complications.28 Despite this, sex differences in sickle nephropathy have been largely ignored and understudied in SCD. Therefore, we performed a longitudinal study to determine the course of the renal phenotype in male and female humanized sickle cell (HbSS) mice and examined potential contributors and predictors of long-term renal outcomes.
Materials and methods
Animal model
Studies used male and female genetic control (HbAA) and HbSS mice originally generated and characterized by Townes and colleagues.29 Homozygous experimental sickle cell disease knockout, knock-in mice, on a mixed genetic background (C57BL/6 and 129S1/SvlmJ), with notation B6; 129-Hbatm1(HBA)TowHbbtm2(HBG1,HBB*)Tow/Hbbtm3(HBG1,HBB)Tow/J, harbor a mutant β-globin gene construct expressing human hemoglobin S. Homozygous control mice were derived from the same colony and harbor wild-type β-globin construct expressing human hemoglobin A. To obtain homozygous HbSS mice, the breeding strategy involves crossing heterozygous (HbAS) female and homozygous (HbSS) male with a reticulocyte count of ∼70%. Genetic control mice are obtained by crossing HbAS male and female. Mice were housed under conditions of constant temperature, humidity, 12-hour light/dark cycle, and they were provided with water and food (Harlan Teklad) ad libitum. Longitudinal GFR and kidney damage analyses were performed in HbSS and age-matched HbAA mice, starting at 8 weeks of age. Separate groups of mice (8, 12, 20, and 32 weeks old) were used for blood and histological analysis. All mice were maintained and studied in accordance with the National Institutes of Health Guide for the Care and use of Laboratory Animals following a protocol reviewed and approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committees.
Metabolic cage studies
Metabolic cage studies were performed in 4-week intervals for 24 weeks starting at 8 weeks of age. Mice were allowed to adapt to metabolic cages for 1 day prior to collection of 24-hour urine samples.
GFR measurement
GFR was measured in 4-week intervals for 24 weeks starting at 8 weeks of age using transcutaneous measurement of a fluorescein isothiocyanate (FITC)–labeled sinistrin technique (NIC-kidney device; MediBeacon GmbH) (FITC-sinistrin: 0.15 mg per gram of body weight; Fresenius Kabi Austria GmbH) as previously described.30
Glomeruli isolation
Glomeruli were isolated on ice by a consecutive sieving technique as previously described.31
Plasma and urine analysis
All urine analyses were performed on 24-hour urine samples collected in metabolic cages. Urinary protein concentration was measured using the Bradford assay (Bio-Rad Laboratories). Urinary albumin concentration was determined using an immunoperoxidase assay (GenWay Biotech Inc). Urinary nephrin concentration was measured using a mouse NPHN (Nephrin) ELISA kit (Elabscience). Urinary kidney injury molecule-1 (KIM-1) concentration was measured using a mouse KIM-1 ELISA kit (TIM-1; Abcam). Hematological parameters of EDTA–whole blood were measured on a HemaVet 1700 hematology analyzer (CDC Technology). The University of Alabama at Birmingham–University of California at San Diego (UAB-UCSD) O’Brien Center for Acute Kidney Injury Research bioanalytical core facility used liquid chromatography–tandem mass spectrometry to measure plasma creatinine levels.32
RNA extraction and quantitative real-time polymerase chain reaction
Total RNA extraction and real-time polymerase chain reaction were performed as previously described.31 The TaqMan primer gene-expression assay (Applied Biosystems) of Wilms tumor antigen 1 (WT-1; Mn01337048), nephrin (Mn00497828_m1), synaptopodin (Mn03413333_m1), megalin (Mn01328171_m1), neutrophil gelatinase–associated lipocalin (NGAL; Mn01324470_m1), hypoxia-inducible factor 1-α (HIF-1α; Mm01236112_m1) was used according to the manufacturer’s instructions. Caspase-3 primers were synthesized by Integrated DNA Technologies.
Histological analysis
Kidneys isolated from control and HbSS mice were immersed in 10% formalin and embedded in paraffin. Four-micrometer-thick kidney sections were stained with Masson trichrome, hematoxylin and eosin, periodic-acid Schiff–hematoxylin, Picro Sirius red, and Prussian blue. Tissues were evaluated blindly according to the criteria used for quantification of the changes in renal structures, as previously described.31 Tubulointerstitial fibrosis, presented as a percentage of red positive area, was quantified using Picro Sirius red–stained sections and ImageJ Fiji software. A representative field of the kidney section was evaluated under ×400 magnification. Results were averaged for each group and time point.
Statistical analysis
Statistical analysis was performed using Prism 7.0 software (GraphPad). Data were analyzed using 1-way analysis of variance (ANOVA) with repeated measurements with the Tukey post hoc test, 2-way ANOVA with the Tukey post hoc test, linear regression, or the unpaired Student t test. Results are expressed as means ± standard error of the mean (SEM), with P < .05 being considered statistically significant.
Results
Longitudinal study of kidney function in control and HbSS mice
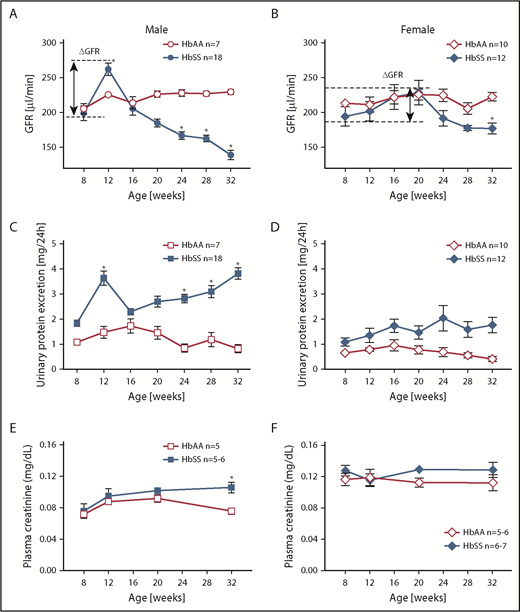
To determine the time course of renal function in HbAA and HbSS mice, we analyzed GFR by measuring clearance of FITC-sinistrin. At 8 weeks of age, male HbSS mice exhibited no differences in GFR or urinary protein excretion compared with HbAA mice (Figure 1A,C). Glomerular hyperfiltration and proteinuria were observed at 12 weeks of age, and at subsequent ages, we observed a progressive loss of renal function associated with a twofold increase in proteinuria in male HbSS mice (Figure 1A,C).
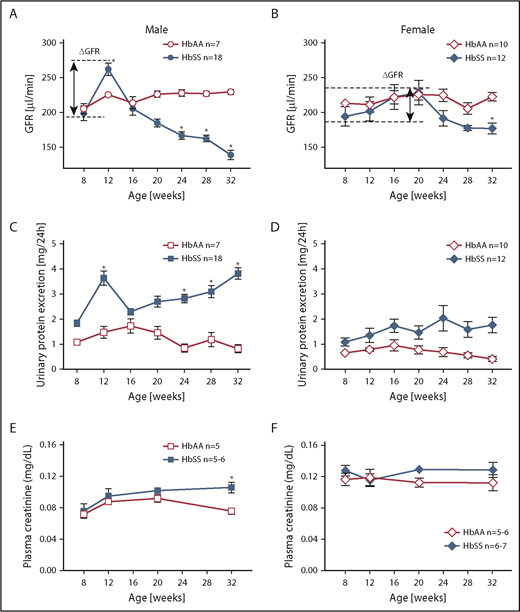
Longitudinal assessment of kidney function in male and female HbSS and HbAA mice. Trajectory of glomerular filtration rate (GFR) in male (A) and female (B) genetic control (HbAA) and HbSS mice. Trajectory of urinary protein excretion in male (C) and female (D) HbAA and HbSS mice. Plasma creatinine in male (E) and female (F) HbAA and HbSS mice. Data are mean ± SEM; *P < .05 vs 8-week HbSS mice; Analysis by 1-way ANOVA with repeated measurements with Tukey post hoc analysis. (A) Genotype and age, P < .0001. (B) Genotype, P = .0030; age, P = .0040. (C) Genotype, P = .0155; age, P = .0196. (D) Genotype, P < .0001; age, P = .5008, or 2-way ANOVA with the Tukey post hoc test. (E) Interaction, P = .1899; genotype, P = .0078; age, P = .0069. (F) Interaction, P = .5871; genotype, P = .9415; age, P = .0899.
Longitudinal assessment of kidney function in male and female HbSS and HbAA mice. Trajectory of glomerular filtration rate (GFR) in male (A) and female (B) genetic control (HbAA) and HbSS mice. Trajectory of urinary protein excretion in male (C) and female (D) HbAA and HbSS mice. Plasma creatinine in male (E) and female (F) HbAA and HbSS mice. Data are mean ± SEM; *P < .05 vs 8-week HbSS mice; Analysis by 1-way ANOVA with repeated measurements with Tukey post hoc analysis. (A) Genotype and age, P < .0001. (B) Genotype, P = .0030; age, P = .0040. (C) Genotype, P = .0155; age, P = .0196. (D) Genotype, P < .0001; age, P = .5008, or 2-way ANOVA with the Tukey post hoc test. (E) Interaction, P = .1899; genotype, P = .0078; age, P = .0069. (F) Interaction, P = .5871; genotype, P = .9415; age, P = .0899.
Unlike males, female HbSS mice demonstrated considerable variation in the time course of GFR between individual mice ranging from initial values of 194 ± 14 μL/min at 8 weeks of age then peaking at 20 weeks of age at 229 ± 18 μL/min and decreasing to 177 ± 7 μL/min at the end of the study (Figure 1B). These changes were not significantly different. If we consider the first time point of the study as a reference GFR value for these mice (representing normal kidney function), then we observe a long-lasting hyperfiltration phase in female HbSS mice with a peak at 20 weeks of age, and subsequent slow decline in kidney function (Figure 1B). Over the course of our experiments, female HbSS mice manifested mild but not statistically significant proteinuria (Figure 1D). The routine renal function marker, plasma creatinine, increased significantly only in male HbSS mice in the late stage of renal injury (Figure 1E-F). These results demonstrate a sex-dependent progression and severity of kidney disease in HbSS mice and highlight the limitations of using plasma creatinine as a marker of renal function decline in mice. None of the measured variables changed significantly throughout the course of study in male and female control HbAA mice (Figure 1). In general, no significant differences in hematological parameters of both male and female HbSS mice within the time course of the study were observed (Table 1).
Longitudinal study of glomerular injury in control and HbSS mice
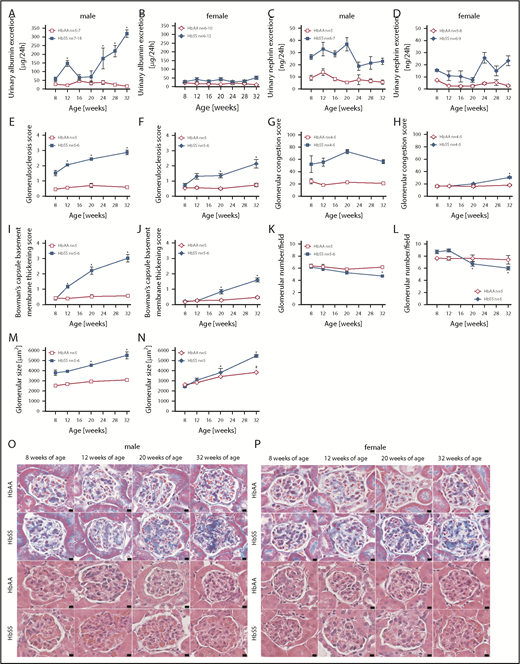
A commonly accepted concept is that hyperfiltration leads to direct glomerular damage, thus we performed analysis of glomerular structure and function at different time points of the study. Following hyperfiltration, individual male HbSS mice developed varying degrees of progressive glomerular damage as evidenced by albuminuria and glomerulopathy, with glomerular sclerosis, congestion, hypertrophy, basement membrane of Bowman capsule thickening, and glomerular loss (Figure 2A,C,E,G,I,K,M,O). Female HbSS mice demonstrated mild but not statistically significant albuminuria, nephrinuria, and a minor degree of glomerular injury (Figure 2B,D,F,H,J,L,N,P). Control mice did not display these pathologic features (Figure 2).
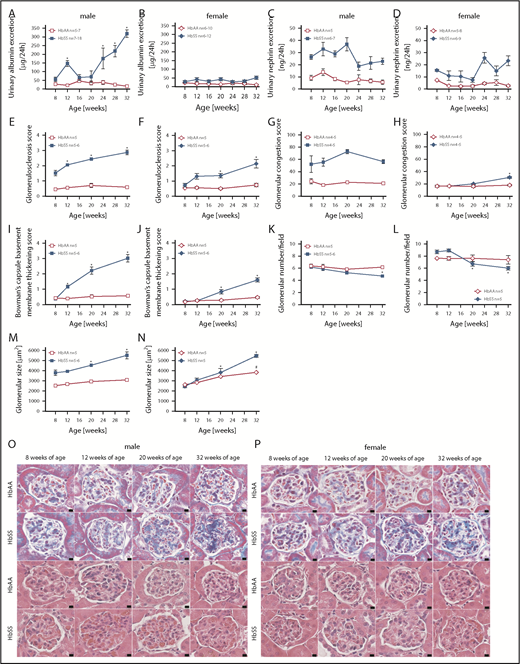
Measures of glomerular injury in male and female HbSS and HbAA mice. Trajectory of urinary albumin excretion in male (A) and female (B) HbAA and HbSS mice. Trajectory of urinary nephrin excretion in male (C) and female (D) HbAA and HbSS mice. Quantification of glomerulosclerosis represented as sclerosis index score in male (E) and female (F) HbAA and HbSS mice. Quantification of glomerular vascular congestion represented as the sum of the glomerular congestion index score in male (G) and female (H) HbAA and HbSS mice. Quantification of the Bowman's capsule basement membrane thickening score in male (I) and female (J) HbAA and HbSS mice. Number of glomeruli per field in male (K) and female (L) HbAA and HbSS mice. Glomerular size represented as mean area of glomeruli from male (M) and female (N) HbAA and HbSS mice. (O) Representative Masson trichrome– and hematoxylin and eosin–stained sections of glomeruli from male HbAA and HbSS mice. Original magnification ×40; scale bar = 50 μm. (P) Representative Masson trichrome– and hematoxylin and eosin—stained sections of glomeruli from female HbAA and HbSS mice. Original magnification ×40; scale bars = 50 μm. Data are mean ± SEM; *P < .05 vs 8-week HbSS mice. Analysis by 1-way ANOVA with repeated measurements with Tukey post hoc analysis. (A) Genotype and age, P < .0001. (B) Genotype, P < .0001; age, P = .9348. (C) Genotype, P < .0001; age, P = .0202. (D) Genotype, P < .0001; age, P = .0279, or 2-way ANOVA with Tukey post hoc test. (E) Interaction, genotype, and age, P < .0001. (F) Interaction, P = .0039; genotype and age, P < .0001. (G) Interaction, P = .3446; genotype, P < .0001; age, P = .2662. (H) Interaction, P = .0003; genotype, P = .0002; age, P < .0001. (I-) Interaction, genotype, and age, P < .0001. (K) Interaction, P = .0571; genotype, P = .0050; age, P = .0038. (L) Interaction, P = .0032; genotype, P = .9450; age, P = .0009. (M) Interaction, P = .0118; genotype and age, P < .0001. (N) Interaction, P = .0005; genotype, P = .0006; age, P = .0001.
Measures of glomerular injury in male and female HbSS and HbAA mice. Trajectory of urinary albumin excretion in male (A) and female (B) HbAA and HbSS mice. Trajectory of urinary nephrin excretion in male (C) and female (D) HbAA and HbSS mice. Quantification of glomerulosclerosis represented as sclerosis index score in male (E) and female (F) HbAA and HbSS mice. Quantification of glomerular vascular congestion represented as the sum of the glomerular congestion index score in male (G) and female (H) HbAA and HbSS mice. Quantification of the Bowman's capsule basement membrane thickening score in male (I) and female (J) HbAA and HbSS mice. Number of glomeruli per field in male (K) and female (L) HbAA and HbSS mice. Glomerular size represented as mean area of glomeruli from male (M) and female (N) HbAA and HbSS mice. (O) Representative Masson trichrome– and hematoxylin and eosin–stained sections of glomeruli from male HbAA and HbSS mice. Original magnification ×40; scale bar = 50 μm. (P) Representative Masson trichrome– and hematoxylin and eosin—stained sections of glomeruli from female HbAA and HbSS mice. Original magnification ×40; scale bars = 50 μm. Data are mean ± SEM; *P < .05 vs 8-week HbSS mice. Analysis by 1-way ANOVA with repeated measurements with Tukey post hoc analysis. (A) Genotype and age, P < .0001. (B) Genotype, P < .0001; age, P = .9348. (C) Genotype, P < .0001; age, P = .0202. (D) Genotype, P < .0001; age, P = .0279, or 2-way ANOVA with Tukey post hoc test. (E) Interaction, genotype, and age, P < .0001. (F) Interaction, P = .0039; genotype and age, P < .0001. (G) Interaction, P = .3446; genotype, P < .0001; age, P = .2662. (H) Interaction, P = .0003; genotype, P = .0002; age, P < .0001. (I-) Interaction, genotype, and age, P < .0001. (K) Interaction, P = .0571; genotype, P = .0050; age, P = .0038. (L) Interaction, P = .0032; genotype, P = .9450; age, P = .0009. (M) Interaction, P = .0118; genotype and age, P < .0001. (N) Interaction, P = .0005; genotype, P = .0006; age, P = .0001.
Longitudinal study of podocyte injury in control and HbSS mice
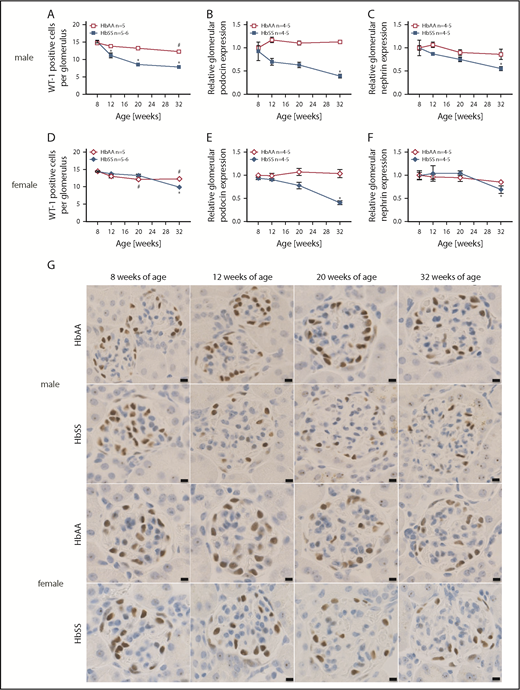
We sought to investigate podocyte number and structure as these glomerular epithelial cells are critical components of the glomerular filtration barrier’s structure and function. WT-1 immunostaining progressively decreased (Figure 3A,D,G) along with loss of podocin and nephrin expression (Figure 3B-C,E-F) in both male and female HbSS mice. Podocyte injury occurred subsequent to hyperfiltration in both sexes. However, male HbSS mice present with greater severity of podocyte and glomerular damage when compared with age-matched female HbSS mice. No structural defects of podocytes were present in control male and female HbAA mice throughout the study (Figure 3).
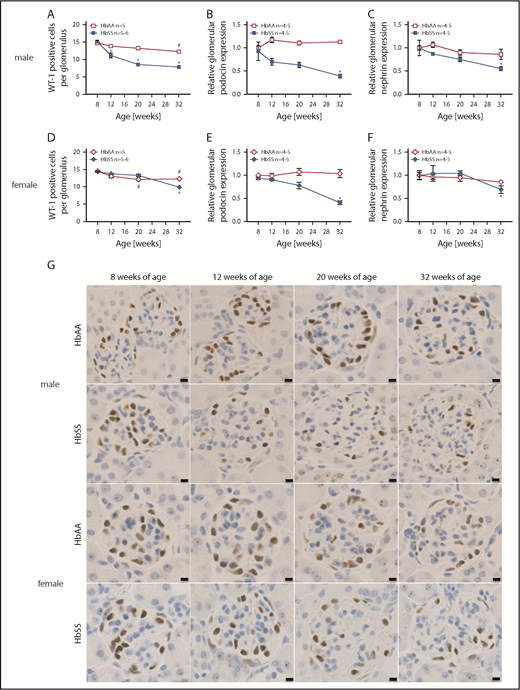
Data showing the time course of glomerular injury in male and female HbSS and HbAA mice. (A) Quantification of WT-1+–stained glomerular sections from male HbAA and HbSS mice represented as number of WT-1+–stained cells per glomerulus. (B) Relative glomerular podocin expression in glomeruli isolated from male HbAA and HbSS mice. (C) Relative glomerular nephrin expression in glomeruli isolated from male HbAA And HbSS mice. (D) Quantification of WT-1+–stained glomerular sections from female HbAA and HbSS mice represented as number of WT-1+–stained cells per glomerulus. (E) Relative glomerular podocin expression in glomeruli isolated from female HbAA and HbSS mice. (F) Relative glomerular nephrin expression in glomeruli isolated from female HbAA and HbSS mice. (G) Representative WT-1+–stained sections of glomeruli from male and female HbAA and HbSS mice. Original magnification ×40; scale bars = 50 μm. Data are mean ± SEM; *P < .05 vs 8-week HbSS; #P < .05 vs 8-week HbAA. Analysis with 2-way ANOVA with Tukey post hoc analysis. (A) Interaction, genotype, and age, P < .0001. (B) Interaction and age, P < .0001; genotype, P = .4236. (C) Interaction, P = .0028; genotype, P < .0001; age, P = .0739. (D) Interaction and genotype, P < .0001; age, P = .0004. (E) Interaction, P = .2687; genotype, P = .0039; age, P = .0017. (F) Interaction, P = .5020; genotype, P = 9953; age, P = .0411.
Data showing the time course of glomerular injury in male and female HbSS and HbAA mice. (A) Quantification of WT-1+–stained glomerular sections from male HbAA and HbSS mice represented as number of WT-1+–stained cells per glomerulus. (B) Relative glomerular podocin expression in glomeruli isolated from male HbAA and HbSS mice. (C) Relative glomerular nephrin expression in glomeruli isolated from male HbAA And HbSS mice. (D) Quantification of WT-1+–stained glomerular sections from female HbAA and HbSS mice represented as number of WT-1+–stained cells per glomerulus. (E) Relative glomerular podocin expression in glomeruli isolated from female HbAA and HbSS mice. (F) Relative glomerular nephrin expression in glomeruli isolated from female HbAA and HbSS mice. (G) Representative WT-1+–stained sections of glomeruli from male and female HbAA and HbSS mice. Original magnification ×40; scale bars = 50 μm. Data are mean ± SEM; *P < .05 vs 8-week HbSS; #P < .05 vs 8-week HbAA. Analysis with 2-way ANOVA with Tukey post hoc analysis. (A) Interaction, genotype, and age, P < .0001. (B) Interaction and age, P < .0001; genotype, P = .4236. (C) Interaction, P = .0028; genotype, P < .0001; age, P = .0739. (D) Interaction and genotype, P < .0001; age, P = .0004. (E) Interaction, P = .2687; genotype, P = .0039; age, P = .0017. (F) Interaction, P = .5020; genotype, P = 9953; age, P = .0411.
Longitudinal study of tubular injury in control and HbSS mice
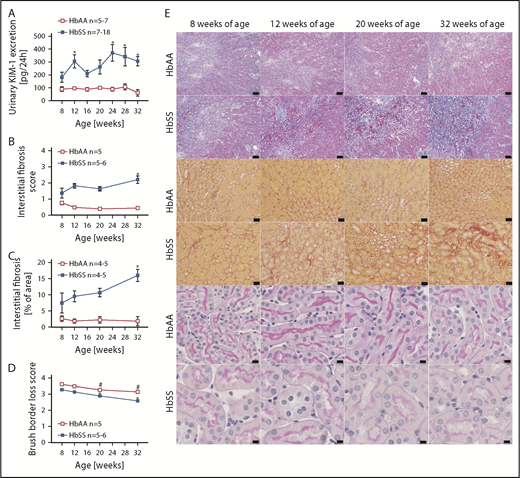
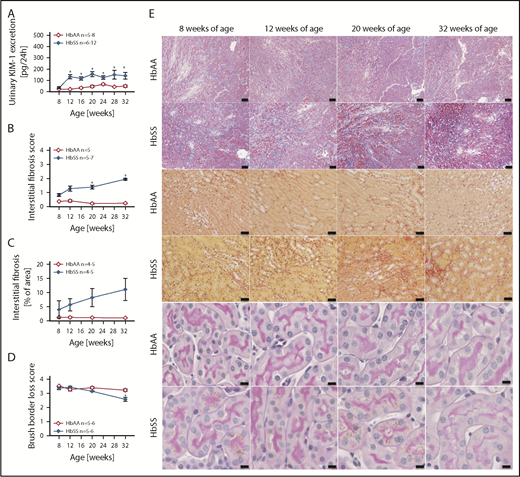
Increased levels of urinary KIM-1, an established marker of proximal tubule damage, were observed in both male and female HbSS mice at 8 weeks of age, and remained elevated throughout the study (Figures 4A and 5A). Histological analysis demonstrated a gradual loss of brush border and progressive extension of interstitial fibrosis in both sexes of HbSS mice (Figures 4B-E and 5B-E), implicating tubular injury as a potential contributor to the progression of SCD nephropathy.
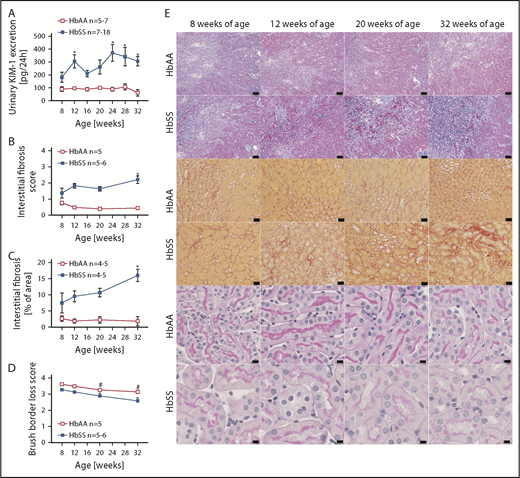
Measures of renal tubular injury progression in male HbSS mice compared with genetic controls (HbAA). (A) Trajectory of kidney injury marker 1 (KIM-1) in male HbAA and HbSS mice. (B) Quantification of interstitial fibrosis index score in male HbAA and HbSS mice. (C) Quantification of interstitial fibrosis index score in male HbAA and HbSS mice. (D) Quantification of brush border loss index score in male HbAA and HbSS mice. (E) Representative Masson trichrome–, Picro Sirius Red–, and periodic-acid Schiff–hematoxylin-stained sections of renal cortex and medulla from male HbAA and HbSS mice. Original magnification ×10 (scale bars = 100 μm) for Masson trichrome staining and ×40 (scale bars = 50 μm) for Picro Sirius Red and periodic-acid Schiff–hematoxylin stainings. Data are mean ± SEM; *P < .05 vs 8-week HbSS mice. #P < .05 vs 8-week HbAA mice. Analysis by 1-way ANOVA with repeated measurements with Tukey post hoc analysis. (A) Genotype, P < .0001; age, P = .0437, or 2-way ANOVA with Tukey post hoc test. (B) Interaction, P = .0140; genotype, P < .0001; age, P = .2854. (C) Interaction, P = .1065; genotype, P < .0001; age, P = .2016. (D) Interaction, P = .3515; genotype and age, P < .0001.
Measures of renal tubular injury progression in male HbSS mice compared with genetic controls (HbAA). (A) Trajectory of kidney injury marker 1 (KIM-1) in male HbAA and HbSS mice. (B) Quantification of interstitial fibrosis index score in male HbAA and HbSS mice. (C) Quantification of interstitial fibrosis index score in male HbAA and HbSS mice. (D) Quantification of brush border loss index score in male HbAA and HbSS mice. (E) Representative Masson trichrome–, Picro Sirius Red–, and periodic-acid Schiff–hematoxylin-stained sections of renal cortex and medulla from male HbAA and HbSS mice. Original magnification ×10 (scale bars = 100 μm) for Masson trichrome staining and ×40 (scale bars = 50 μm) for Picro Sirius Red and periodic-acid Schiff–hematoxylin stainings. Data are mean ± SEM; *P < .05 vs 8-week HbSS mice. #P < .05 vs 8-week HbAA mice. Analysis by 1-way ANOVA with repeated measurements with Tukey post hoc analysis. (A) Genotype, P < .0001; age, P = .0437, or 2-way ANOVA with Tukey post hoc test. (B) Interaction, P = .0140; genotype, P < .0001; age, P = .2854. (C) Interaction, P = .1065; genotype, P < .0001; age, P = .2016. (D) Interaction, P = .3515; genotype and age, P < .0001.
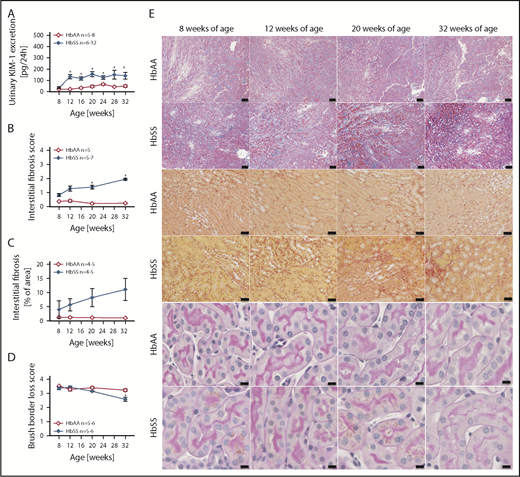
Measures of renal tubular injury progression in female HbSS mice compared with genetic controls (HbAA). (A) Trajectory of KIM-1 in female HbAA and HbSS mice. (B) Quantification of interstitial fibrosis index score in female HbAA and HbSS mice. (C) Quantification of interstitial fibrosis in female HbAA and HbSS mice. (D) Quantification of brush border loss index score in male HbAA and HbSS mice. (E) Representative Masson trichrome–, Picro Sirius Red–, and periodic-acid Schiff–hematoxylin-stained sections of renal cortex and medulla from female HbAA and HbSS mice. Original magnification ×10 (scale bars = 100 μm) for Masson trichrome staining and ×40 (scale bars = 50 μm) for Picro Sirius Red and periodic-acid Schiff–hematoxylin stainings. Data are mean ± SEM; *P < .05 vs 8-week HbSS mice. Analysis by 1-way ANOVA with repeated measurements with Tukey post hoc analysis (A) Genotype, P < .0001; age, P = .0049, or 2-way ANOVA with the Tukey post hoc test. (B) Interaction, P = .0140; genotype, P < .0001; age, P = .2854. (C) Interaction, P = .3947; genotype, P = .0004; age, P = .4610. (D) Interaction, P = .0008; genotype, P < .0001; age, P = .0011.
Measures of renal tubular injury progression in female HbSS mice compared with genetic controls (HbAA). (A) Trajectory of KIM-1 in female HbAA and HbSS mice. (B) Quantification of interstitial fibrosis index score in female HbAA and HbSS mice. (C) Quantification of interstitial fibrosis in female HbAA and HbSS mice. (D) Quantification of brush border loss index score in male HbAA and HbSS mice. (E) Representative Masson trichrome–, Picro Sirius Red–, and periodic-acid Schiff–hematoxylin-stained sections of renal cortex and medulla from female HbAA and HbSS mice. Original magnification ×10 (scale bars = 100 μm) for Masson trichrome staining and ×40 (scale bars = 50 μm) for Picro Sirius Red and periodic-acid Schiff–hematoxylin stainings. Data are mean ± SEM; *P < .05 vs 8-week HbSS mice. Analysis by 1-way ANOVA with repeated measurements with Tukey post hoc analysis (A) Genotype, P < .0001; age, P = .0049, or 2-way ANOVA with the Tukey post hoc test. (B) Interaction, P = .0140; genotype, P < .0001; age, P = .2854. (C) Interaction, P = .3947; genotype, P = .0004; age, P = .4610. (D) Interaction, P = .0008; genotype, P < .0001; age, P = .0011.
Potential mechanisms of glomerulotubular injury in HbSS mice
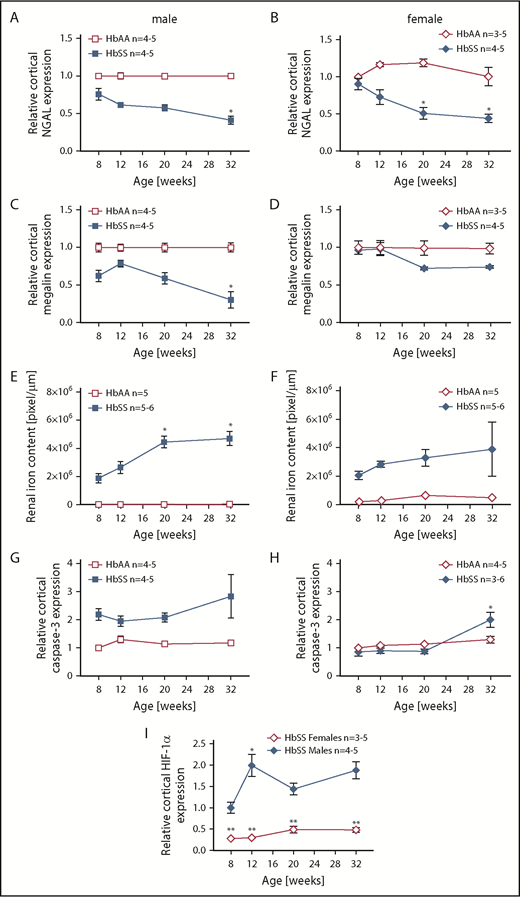
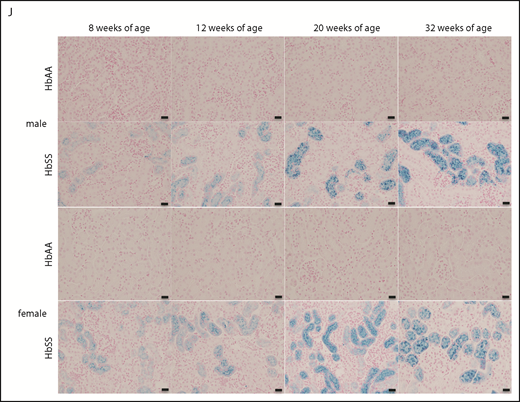
The mechanisms that initiate the cascade of renal involvement in SCD are poorly understood. Histological analysis showed progressive tubular iron deposition in HbSS mice (Figure 6E,F,J). In male HbSS mice, expression of NGAL, a constitutive tubular molecule, was decreased as early as at 8 weeks of age (Figure 6A). Interestingly, progressive tubular iron accumulation occurred with increased cortical expression of caspase-3, a marker of cell death, in 8-week-old HbSS mice (Figure 6G). In contrast, slow gradual reduction in NGAL and caspase-3 expression were observed in female HbSS mice (Figure 6B,H). Moreover, the pattern of megalin expression, a proximal tubule-specific albumin handling receptor, was consistent with the degree of albuminuria in both HbSS sexes, with an increase during the hyperfiltration phase at 12 weeks of age in males followed by a slow decline (Figure 6C) whereas females displayed a trend toward a decrease at 20 and 32 weeks of age (Figure 6D).
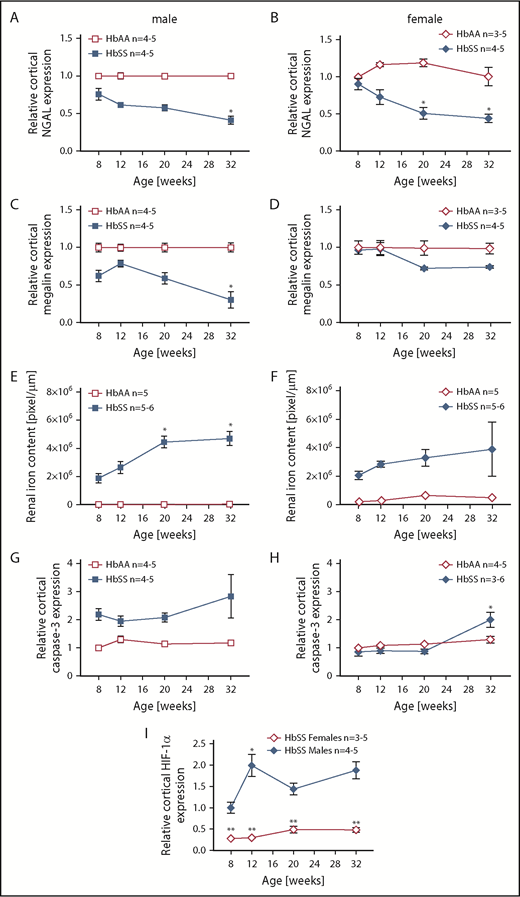
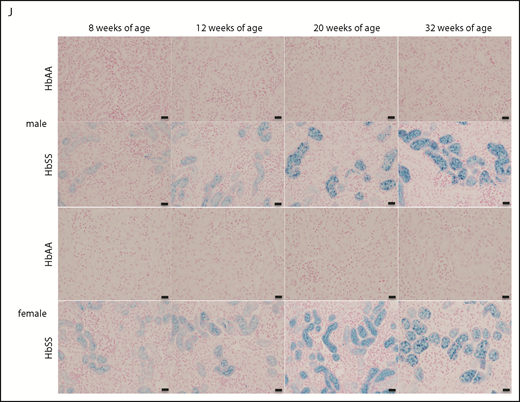
Time course in additional measures of renal tubular injury in male and female HbSS mice compared with genetic controls (HbAA). (A) Relative NGAL expression in renal cortex of male HbAA and HbSS mice. (B) Relative NGAL expression in renal cortex of female HbAA and HbSS mice. (C) Relative megalin expression in renal cortex of male HbAA and HbSS mice. (D) Relative megalin expression in renal cortex of female HbAA and HbSS mice. (E) Quantification of iron deposition in the whole kidney scans (represented as megapixels per micrometer) in male HbAA and HbSS mice. Original magnification ×40; scale bar = 50 μm. (F) Quantification of iron deposition in the whole kidney scans (represented as megapixels per micrometer) in male HbAA and HbSS mice. (G) Relative caspase-3 expression in renal cortex of male HbAA and HbSS mice. (H) Relative caspase-3 expression in renal cortex of female HbAA and HbSS mice. (I) Relative HIF-1α expression in renal cortex of male and females HbSS mice. (J) Representative Prussian blue–stained sections of renal cortex from male and female HbAA and HbSS mice. Original magnification ×10; scale bar = 100 μm. Data are mean plus or minus SEM; *P < .05 vs 8-week HbSS mice; **P < .05 vs age-matched males HbSS. Analysis by 2-way ANOVA with the Tukey post hoc test. (A) Interaction, P = .0099; genotype, P < .0001; age, P = .0097. (B) Interaction, P = .0031; genotype, P < .0001; age, P = .0234. (C) Interaction, P = .0176; genotype, P < .0001; age, P = .0184. (D) Interaction, P = .2074; genotype, P = .0119; age, P = .1529. (E) Interaction, P = .0002; genotype, P < .0001; age, P = .0002. (F) Interaction, P = .4335; genotype, P < .0001; age, P = .1189. (G) Interaction, P = .3509; genotype, P < .0001; age, P = .3981. (H) Interaction, P = .0007; genotype, P < .0001; age, P = .7121, or unpaired Student t test (I).
Time course in additional measures of renal tubular injury in male and female HbSS mice compared with genetic controls (HbAA). (A) Relative NGAL expression in renal cortex of male HbAA and HbSS mice. (B) Relative NGAL expression in renal cortex of female HbAA and HbSS mice. (C) Relative megalin expression in renal cortex of male HbAA and HbSS mice. (D) Relative megalin expression in renal cortex of female HbAA and HbSS mice. (E) Quantification of iron deposition in the whole kidney scans (represented as megapixels per micrometer) in male HbAA and HbSS mice. Original magnification ×40; scale bar = 50 μm. (F) Quantification of iron deposition in the whole kidney scans (represented as megapixels per micrometer) in male HbAA and HbSS mice. (G) Relative caspase-3 expression in renal cortex of male HbAA and HbSS mice. (H) Relative caspase-3 expression in renal cortex of female HbAA and HbSS mice. (I) Relative HIF-1α expression in renal cortex of male and females HbSS mice. (J) Representative Prussian blue–stained sections of renal cortex from male and female HbAA and HbSS mice. Original magnification ×10; scale bar = 100 μm. Data are mean plus or minus SEM; *P < .05 vs 8-week HbSS mice; **P < .05 vs age-matched males HbSS. Analysis by 2-way ANOVA with the Tukey post hoc test. (A) Interaction, P = .0099; genotype, P < .0001; age, P = .0097. (B) Interaction, P = .0031; genotype, P < .0001; age, P = .0234. (C) Interaction, P = .0176; genotype, P < .0001; age, P = .0184. (D) Interaction, P = .2074; genotype, P = .0119; age, P = .1529. (E) Interaction, P = .0002; genotype, P < .0001; age, P = .0002. (F) Interaction, P = .4335; genotype, P < .0001; age, P = .1189. (G) Interaction, P = .3509; genotype, P < .0001; age, P = .3981. (H) Interaction, P = .0007; genotype, P < .0001; age, P = .7121, or unpaired Student t test (I).
To investigate the potential determinant of delayed renal injury in female HbSS mice, we measured cortical expression of HIF-1α. Female HbSS mice had significantly lower HIF-1α messenger RNA expression when compared with males (Figure 6I), suggesting a decreased level of renal ischemia in females.
Association of glomerular hyperfiltration with long-term kidney damage in HbSS mice
The degree to which hyperfiltration predicts, or contributes to, chronic renal injury in SCD is poorly understood. Thus, we sought to determine whether the magnitude of glomerular hyperfiltration predicts the degree of long-term kidney damage in HbSS mice. The degree of rise in GFR during the hyperfiltration phase (from 8 to 12 weeks in males, and 8 to 20 weeks in females) was significantly different from control GFR level only in male HbSS mice (Figure 7A-B).
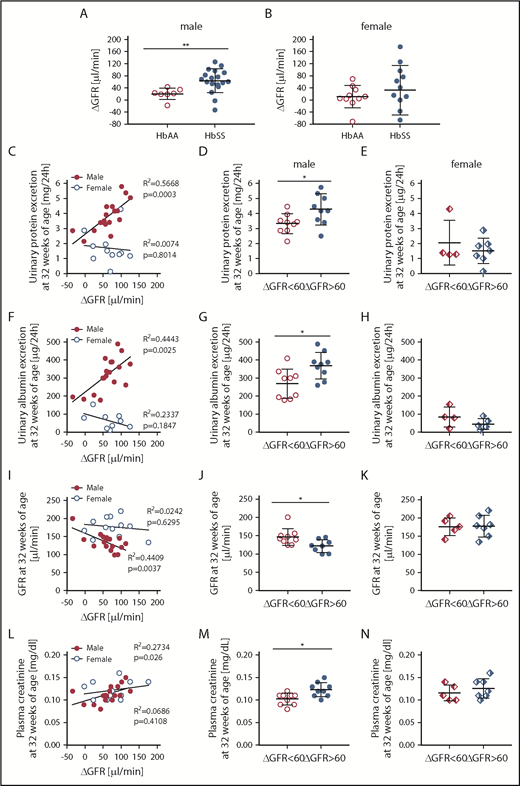
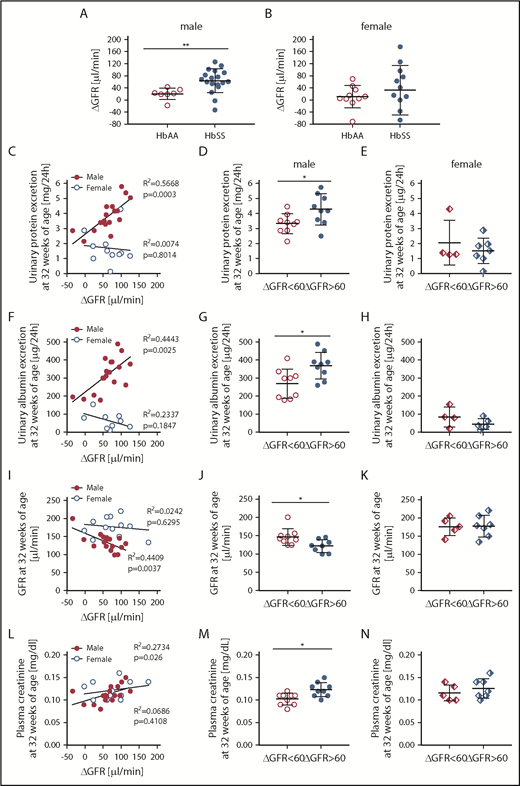
Evidence that the magnitude of hyperfiltration phase correlates with kidney injury only in male, but not female, HbSS mice. (A) The degree of rise in GFR from 8 to 12 weeks (ΔGFR) in male HbAA and HbSS mice. (B) The degree of rise in GFR from 8 to 20 weeks in (ΔGFR) female HbAA and HbSS mice. (C) Correlation of glomerular hyperfiltration (ΔGFR) with urinary protein excretion at 32 weeks of age in male and female HbSS mice. (D) Urinary protein excretion at 32 weeks of age in male HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (E) Urinary protein excretion at 32 weeks of age in female HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (F) Correlation of glomerular hyperfiltration (ΔGFR) with urinary albumin excretion at 32 weeks of age in male and female HbSS mice. (G) Urinary albumin excretion at 32 weeks of age in male HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (H) Urinary albumin excretion at 32 weeks of age in female HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (I) Correlation of glomerular hyperfiltration (ΔGFR) with GFR at 32 weeks of age in male and female HbSS mice. (J) GFR at 32 weeks of age in male HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (K) GFR at 32 weeks of age in female HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (L) Correlation of glomerular hyperfiltration (ΔGFR) with plasma creatinine at 32 weeks of age in male and female HbSS mice. (M) Plasma creatinine at 32 weeks of age in male HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (N) Plasma creatinine at 32 weeks of age in female HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. Data are mean ± SEM; *P < .05 vs ΔGFR <60; **P < .05 vs HbAA mice. Analysis by linear regression (C,F,I,L) or unpaired Student t test (A-B,D-E,G-H,J-K,M-N).
Evidence that the magnitude of hyperfiltration phase correlates with kidney injury only in male, but not female, HbSS mice. (A) The degree of rise in GFR from 8 to 12 weeks (ΔGFR) in male HbAA and HbSS mice. (B) The degree of rise in GFR from 8 to 20 weeks in (ΔGFR) female HbAA and HbSS mice. (C) Correlation of glomerular hyperfiltration (ΔGFR) with urinary protein excretion at 32 weeks of age in male and female HbSS mice. (D) Urinary protein excretion at 32 weeks of age in male HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (E) Urinary protein excretion at 32 weeks of age in female HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (F) Correlation of glomerular hyperfiltration (ΔGFR) with urinary albumin excretion at 32 weeks of age in male and female HbSS mice. (G) Urinary albumin excretion at 32 weeks of age in male HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (H) Urinary albumin excretion at 32 weeks of age in female HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (I) Correlation of glomerular hyperfiltration (ΔGFR) with GFR at 32 weeks of age in male and female HbSS mice. (J) GFR at 32 weeks of age in male HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (K) GFR at 32 weeks of age in female HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (L) Correlation of glomerular hyperfiltration (ΔGFR) with plasma creatinine at 32 weeks of age in male and female HbSS mice. (M) Plasma creatinine at 32 weeks of age in male HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. (N) Plasma creatinine at 32 weeks of age in female HbSS mice with the magnitude of glomerular hyperfiltration <60 μL/min and >60 μL/min. Data are mean ± SEM; *P < .05 vs ΔGFR <60; **P < .05 vs HbAA mice. Analysis by linear regression (C,F,I,L) or unpaired Student t test (A-B,D-E,G-H,J-K,M-N).
Hyperfiltration at 12 weeks of age in male HbSS mice was significantly correlated with kidney injury at 32 weeks of age, as evidenced by proteinuria, albuminuria, GFR decline, and elevated plasma creatinine (Figure 7C,F,I,L). Additionally, the magnitude of hyperfiltration in male HbSS mice was associated with the degree of kidney damage at the end of the study. Male mice with a rise in GFR >60 μL/min, at 12 weeks of age, had significantly greater proteinuria, albuminuria, plasma creatinine, and significantly lower GFR (Figure 7D,G,J,M) when compared with the group with a rise in GFR <60 μL/min. Surprisingly, in female HbSS mice, hyperfiltration failed to correlate with any of the functional markers of kidney injury at the end of the study (Figure 7C,F,I,L), nor was the magnitude of hyperfiltration associated with subsequent renal injury (Figure 7E,H,K,N) in female HbSS mice. These data demonstrate that the extent of hyperfiltration is associated with the degree of subsequent renal injury, specifically in male HbSS mice, and suggest that the magnitude and duration of hyperfiltration may be a predictor of CKD risk in SCD.
Discussion
The current study demonstrated that the pattern of the renal phenotype in humanized sickle mouse (HbSS knockout–knock-in) largely matches that of SCD patients,33 with a hyperfiltration phase that is followed by a progressive decline in renal function, specifically in male mice. Importantly, 2 major findings of this study are that (1) the degree of hyperfiltration was a predictor of the degree of subsequent renal injury in male mice only and (2) renal tubular injury precedes the increase in indices of glomerular injury in both sexes. Evidence that hyperfiltration predicts subsequent GFR decline is further supported by data in female HbSS mice where the pattern of hyperfiltration and progression of renal dysfunction and injury was attenuated and delayed. We conclude that this model provides an excellent tool for identifying processes involved in long-term renal outcomes, and furthermore, suggest that future studies need to further explore sex differences in renal involvement in SCD.
The prevalence of hyperfiltration in SCD patients varies between 30% and 76%, and greater prevalence of hyperfiltration is associated with younger age.7,9,34 Differences in the reported values for hyperfiltration status are compounded by the use of various methods for measuring and calculating GFR. Haymann et al examined the prevalence of glomerular hyperfiltration, defined as GFR >130 for women, and >140 mL/min per 1.73 m2 for men, in SCD.9 Hyperfiltration, as measured by urinary 51Cr EDTA clearance and assessed using the Modification of Diet in Renal Disease eGFR, was present in 66% of selected SCD patients. Moreover, 49% of the patients with hyperfiltration presented with no albuminuria. In these patients, the risk factors for hyperfiltration were young age and markers of chronic hemolysis, such as low hemoglobin or fetal hemoglobin levels.9 Although this study importantly highlights the prevalence of hyperfiltration in SCD, these data do not characterize the time course of renal disease progression. Although it is possible that glomerular hyperfiltration leads to focal segmental glomerulosclerosis and CKD, evidence that glomerular hyperfiltration is indeed a reliable determinant and predictor of subsequent CKD in SCD is unknown. To address this question, longitudinal basic and clinical studies examining a direct contribution of hyperfiltration to renal involvement in SCD are needed. This need provided the rationale for our sex-specific longitudinal studies to evaluate the pathophysiology of renal complications using a humanized mouse model of SCD, as well as to address the gap in knowledge among studies analyzing only a snapshot of the prevalence but not the course of progression. Using the sensitive and specific plasma FITC-sinistrin clearance approach, we eliminate a potential overestimation of GFR related to tubular secretion that is very high in mice.35 Our study demonstrates a significant correlation between the presence and the magnitude of hyperfiltration with subsequent severe renal damage in male HbSS mice. This finding provides evidence that early glomerular hyperfiltration is a determinant and predictor of subsequent progression of SCD nephropathy.
Interestingly, Haymann et al also reported that only 15.6% of male and 23.3% of female patients were free of SCD-associated nephropathy, and hyperfiltration occurred at a significantly different frequency between sexes (60% for men vs 42% for women).9 In accordance with these results, we observed a less severe renal phenotype in female SCD mice. A potential explanation of our observations may involve the pattern of the hyperfiltration phase. Because we did not observe a dramatic loss in glomerular number, thereby in nephron number, our results suggest that renal hemodynamic alterations are most likely underlying mechanisms of glomerular hyperfiltration in HbSS mice. However, considering the difference in magnitude of hyperfiltration observed between sexes, it is probably that the trajectory of hyperfiltration contributes to resultant renal injury. Male HbSS mice display a rapid increase in GFR, peaking at ∼12 weeks of age and an equally rapid reversal of the trajectory of GFR with a subsequent progressive decline in kidney function. Female mice present with a slower onset of hyperfiltration, reaching a peak at roughly 20 weeks of age that is followed by an equally slow subsequent normalization of GFR. Given that HbSS females present with a moderate but extended hyperfiltration phase, it is likely that these mice will have an eventual loss of GFR and associated renal injury. Importantly, our study exposes important and neglected sex-specific differences in CKD in SCD.
Urinary KIM-1 is a biomarker of renal ischemia-reperfusion injury and proximal tubule damage.36-39 Here, we demonstrate that an increase in urinary KIM-1 occurs as early as 8 weeks of age, regardless of sex, in HbSS mice, implicating early tubular injury as a possible initial contributor to the progression of SCD nephropathy. Urinary KIM-1 excretion is elevated and strongly associated with albuminuria, a risk factor for CKD in SCD.16 KIM-1 is considered a sensitive biomarker of early diabetic renal injury that may precede albuminuria, suggesting that tubular dysfunction may develop prior to glomerular injury in sickle nephropathy.28,40,41 Other investigators provided similar evidence in diabetic nephropathy, a kidney disease with important similarities to SCD-associated nephropathy, such as early hyperfiltration and glomerular injury characterized by focal segmental glomerulosclerosis.
One of the potential mechanisms of a primary tubular pathology involved in kidney injury is iron deposition. We observed that progressive tubular iron overload with accompanying increased cell death suggests cytotoxic and proinflammatory effects of heme/iron that may promote tubular injury in SCD. Heme exposure has been shown to cause severe tubulopathy and a significant increase in urinary KIM-1 in HbSS mice,42 as well as in SCD patients.43 Decreased expression of other markers of tubular injury, such as NGAL and megalin, were observed prior to most of the glomerular injury manifestations. Considering that chronic hemolysis leads to increased kidney iron accumulation, mostly limited to proximal tubules, it is unlikely that iron toxicity directly injures the glomerulus, as suggested by Hirschberg.44 It is interesting that only some of the patients in Haymann’s cohort, presented with hyperfiltration, developed albuminuria; however, it is unknown whether this is associated with iron accumulation. Tubular damage, possibly mediated by iron accumulation that occurs prior to glomerular damage, may exacerbate hyperfiltration-associated mechanisms of progressive kidney disease. The question of whether tubular damage contributes to the onset of hyperfiltration or vice versa still remains to be answered, nevertheless our findings foster further studies to investigate the role of renal tubular dysfunction in poor long-term renal outcomes in SCD.
Although multiple studies provide evidence for a slower rate of progression to ESRD in women compared with men, regardless of etiology,24 we are unaware of any studies that report sex-associated differences in the prevalence of ESRD in SCD. However, the median age at death among all SCD patients is significantly greater in women compared with men, but still 20 to 30 years shorter than the general population.45 One potential explanation could be that men, due to a more rapid disease progression, die of other SCD-related complications before they reach ESRD. Contrary to previous studies that reported no sex-related differences in SCD nephropathy,1,10,16 our data show that female mice with SCD most likely have delayed onset of CKD. Our findings demonstrate that sex contributes to the progression of kidney injury in SCD mice and highlight the potential importance of considering sex when diagnosing and managing SCD nephropathy.
One of the potential mechanisms of sex differences in renal progression in HbSS mice could be that males have a greater degree of hypoxia and therefore may have accelerated injury and progression of renal involvement in SCD. Our results show that HbSS males have higher cortical expression of HIF-1α when compared with females. As vascular stasis results in ischemia-reperfusion, the possibility remains that male HbSS mice present with higher HIF-1α expression due to a greater degree of glomerular vascular stasis, resulting in higher propensity for cyclical episodes of renal ischemia-reperfusion and recurrent acute kidney injury. Testosterone has been shown to aggravate ischemia-reperfusion–driven kidney injury.46 Orchiectomized male mice present with decreased ischemia-reperfusion–induced renal damage, whereas females administrated with testosterone show increase in kidney susceptibility to ischemia and exacerbated rate of injury.46
There are a number of additional pathways that could account for sex differences in SCD nephropathy. These include possible differences in the degrees of hemolysis, intrarenal ischemic-driven injury resistance, fetal hemoglobin levels, or the timeline of the study. Distinct sex differences in hemolytic markers in SCD women vs men has been recently reported.28 Female SCD patients have significantly lower absolute reticulocyte count, bilirubin, and lactate dehydrogenase compared with male patients. Hemolysis status may be associated with renal function in SCD, particularly albuminuria and eGFR,47 and it may provide a prospective explanation for differences in the renal phenotype. Also, prominently occurring in SCD chronic hemolysis results in intrinsic vasculopathy. Red blood cell–derived lysis products induce oxidative stress48 and inflammation,49 and inhibit nitric oxide signaling.50,51 Recent studies by Kanias et al identified sex as a risk factor for hemolysis in hemolytic disease.52 In particular, testosterone increased susceptibility to hemolysis in males across 22 mouse strains and SCD. In support of in vivo study showing increased receptivity of male red blood cells to oxidative and osmotic stress, the authors have also demonstrated testosterone-driven increased predisposition to osmotic and oxidative hemolysis in testosterone repletion study in orchiectomized mice.52 Thus, delayed onset of SCD nephropathy may be due to potentially lower levels of hemolysis in SCD female mice. Another potential contributor to the observed renal female phenotype may be fetal hemoglobin. Higher fetal hemoglobin levels have been reported in healthy and SCD women vs men. Also, studies elucidating genetic regulators of fetal hemoglobin production revealed that the F-cell production locus is partially controlled by loci on X chromosome.53 Further studies are needed to explore these potential mechanisms.
In summary, these longitudinal studies identified hyperfiltration as a determinant as well as a predictor of the onset of long-term kidney damage in SCD. Additionally, our results provide evidence that both tubular and glomerular mechanisms could account for potential sex differences in the progression of kidney injury in SCD. These findings provide important rationale for long-term longitudinal studies in SCD patients to address the issue of the potential benefit of nephroprotective therapeutic approaches that could influence the natural course of SCD nephropathy.
Acknowledgments
The authors thank Xiaofen Liu for assistance with histology and Binli Tao for support with breeding and genotyping.
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (U01 HL117684 [D.M.P. and J.S.P.]; R00 HL127178 [J.S.S.]; and K23 HL127100 [J.D.L.]); the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (K01 DK105038 [K.A.H.]; F30 DK107194 [B.M.F.]; and P30 DK079337 [the core resource of The University of Alabama at Birmingham–University of California at San Diego O'Brien Center]); the American Society of Nephrology Joseph A. Carlucci Research Fellowship (M.K.); and the American Society of Hematology (ASH Scholar) (J.D.L.).
Authorship
Contribution: M.K., J.D.L., and D.M.P. designed the study; M.K., J.D.L., K.A.H., and J.S.S. carried out experiments; M.K., J.D.L., J.S.S., K.A.H., and B.M.F. analyzed the data; M.K., K.A.H., and B.M.F. made the figures; M.K., J.D.L., B.M.F., J.S.S., J.S.P., and D.M.P. drafted and revised the paper; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David M. Pollock, Cardio-Renal Physiology and Medicine, Division of Nephrology, Department of Medicine, University of Alabama at Birmingham, KAUL 802, 720 20th St S, Birmingham, AL 35233; e-mail: davidpollock@uabmc.edu.