

Key Points
Genetically matched MDS-RS and normal patient-specific iPSC-HSPCs are used to derive a mutant SF3B1 splicing signature.
Integrated transcriptomics and chromatin accessibility nominate TEAD as a putative novel transcriptional regulator of SF3B1K700E cells.

Abstract
SF3B1K700E is the most frequent mutation in myelodysplastic syndrome (MDS), but the mechanisms by which it drives MDS pathogenesis remain unclear. We derived a panel of 18 genetically matched SF3B1K700E- and SF3B1WT-induced pluripotent stem cell (iPSC) lines from patients with MDS with ring sideroblasts (MDS-RS) harboring isolated SF3B1K700E mutations and performed RNA and ATAC sequencing in purified CD34+/CD45+ hematopoietic stem/progenitor cells (HSPCs) derived from them. We developed a novel computational framework integrating splicing with transcript usage and gene expression analyses and derived a SF3B1K700E splicing signature consisting of 59 splicing events linked to 34 genes, which associates with the SF3B1 mutational status of primary MDS patient cells. The chromatin landscape of SF3B1K700E HSPCs showed increased priming toward the megakaryocyte- erythroid lineage. Transcription factor motifs enriched in chromatin regions more accessible in SF3B1K700E cells included, unexpectedly, motifs of the TEA domain (TEAD) transcription factor family. TEAD expression and transcriptional activity were upregulated in SF3B1-mutant iPSC-HSPCs, in support of a Hippo pathway-independent role of TEAD as a potential novel transcriptional regulator of SF3B1K700E cells. This study provides a comprehensive characterization of the transcriptional and chromatin landscape of SF3B1K700E HSPCs and nominates novel mis-spliced genes and transcriptional programs with putative roles in MDS-RS disease biology.
Introduction
Myelodysplastic syndromes (MDS) are myeloid malignancies characterized by ineffective hematopoiesis, blood cytopenias, and an increased risk of progression to secondary acute myeloid leukemia.1 Recurrent somatic mutations in genes encoding splicing factors (SFs) were discovered a decade ago as a novel class of driver mutations in MDS, collectively occurring in more than 50% of patients with MDS.2-5 Mutations in splicing factor 3B, subunit 1 (SF3B1), are present in approximately 30% of patients with MDS and define a distinct MDS clinical subgroup, termed MDS with ring sideroblasts (MDS-RS), characterized by erythroblasts with abnormal iron accumulation in mitochondria that form a ring around the cell nucleus (ring sideroblasts), ineffective erythropoiesis, macrocytic anemia, and favorable prognosis.3-7
SF3B1 is a core spliceosomal protein (a key component of the U2 small nuclear ribonucleoprotein complex [snRNP]) that binds upstream of the branch point and is required to facilitate 3′ splice site recognition of most introns.8 Nearly all mutations in SF3B1 are heterozygous, most commonly target the K700 hotspot, and result in altered RNA-binding specificity of mutant SF3B1. SF3B1 mutations are associated with preferential use of cryptic 3′ splice sites, leading to nonsense-mediated decay (NMD) or generation of different isoforms of multiple transcripts.9-11
Some recent studies implicated specific mis-splicing events associated with SF3B1 mutations in the pathogenesis of MDS or other malignancies. An alternative erythroferrone (ERFE) transcript in SF3B1-mutant erythroid lineage cells was linked to disruption of iron homeostasis.12
Decrease in expression of BRD9, a component of the noncanonical BRG1-associated factors (BAF) chromatin-remodeling complex, through inclusion of a “poison exon,” was also shown to confer oncogenic properties in uveal melanoma models.13 Despite these insights, the mechanisms by which mutant SF3B1 drives MDS, and malignancy in general, remain incompletely understood, and the critical mis-splicing events that mediate these effects are not well characterized. Importantly, mis-splicing events have thus far been cataloged either in primary patient cells or murine or cellular models, each with distinct limitations. Patient samples are heterogeneous in terms of clonality, presence of co-occurring mutations, and cell type composition. Conversely, murine models have the important limitation that alternative splicing events are largely nonconserved between mouse and human.14 Finally, previous cellular models of SF3B1 mutations consisted of engineered immortalized leukemia cell lines (such as K562), which harbor mutations not related to MDS pathogenesis, and result in abnormal levels and stoichiometry of mutant and wild-type (WT) SF3B1 because of aneuploidy and/or use of overexpression systems.
Here, we leveraged MDS patient cell reprogramming to generate a panel of karyotypically normal diploid-induced pluripotent stem cell (iPSC) lines with an isolated SF3B1K700E mutation, as well as genetically matched WT iPSCs, from patients with MDS-RS. By integrating splicing, gene expression, and transcript usage analyses, we derived a splicing signature of mutant SF3B1 that we validated in datasets of patients with MDS. Furthermore, we characterized the chromatin landscape of SF3B1 K700E iPSC-derived hematopoietic stem/progenitor cells (iPSC-HSPCs) and identify increased transcriptional activity of the TEAD family of transcription factors (TFs) in mutant cells. This study provides a refined view of the altered mis-spliced transcriptome of human SF3B1K700E HSPCs and characterizes for the first time their chromatin landscape, pinpointing TEAD as a potential regulator of SF3B1K700E HSPCs.
Methods
Human samples
Human bone marrow (BM) mononuclear cell samples from 3 MDS-RS patients (supplemental Table 1) were obtained with informed consent under protocols approved by a local institutional review board at Karolinska Institute.
Human iPSC generation and culture
Cryopreserved BM mononuclear cells from 3 MDS-RS patients were thawed and cultured in X-VIVO 15 media with 1% nonessential amino acids, 1 mM l-glutamine, and 0.1 mM β-mercaptoethanol, supplemented with 100 ng/mL stem cell factor (SCF), 100 ng/mL Flt3 ligand, 100 ng/mL thrombopoietin (TPO), and 20 ng/mL interleukin 3 (IL-3) for at least 1 and for up to 7 days to induce cell proliferation. For induction of reprogramming, 200 000 cells were plated on a retronectin-coated well of a 24-well plate and transduced with the viral cocktail CytoTune-iPS 2.0 Sendai reprogramming kit (Invitrogen), containing KLF4, OCT4, and SOX2 virus, the c-MYC virus, and the KLF4 virus. One or 2 days later, the cells were harvested and plated on mitotically inactivated mouse embryonic fibroblasts (MEFs) in 6-well plates and centrifuged at 500 rpm for 30 minutes at room temperature. The next day and every day thereof, half of the medium was gently replaced with human embryonic stem cell (hESC) medium with 0.5 mM valproic acid. After 3 to 4 weeks, colonies with human pluripotent stem cell (hPSC) morphology were manually picked and expanded. Culture of human iPSCs on mitotically inactivated MEFs was performed as previously described.15
Hematopoietic differentiation
Hematopoietic differentiation was performed using a spin-embryoid body (EB) protocol previously described.16 Briefly, cells were dissociated into single cells with accutase and plated at 3500 cells per well in round-bottom low-attachment 96-well plates in Albumin Polyvinylalcohol Essential Lipids 2 (APEL2) medium containing 5% protein-free hybridoma medium (PFHM-II), 30 ng/mL bone morphogenetic protein 4, and 10 μM Y-27632. The plates were centrifuged at 800 rpm for 5 minutes to induce EB aggregation. After 24 hours, the medium was replaced by APEL2 medium containing 5% PFHM-II, 30 ng/mL bone morphogenetic protein 4, and 50 ng/mL fibroblast growth factor 2 (FGF2). After 2 days, the cytokine cocktail was changed to 5% PFHM-II, 20 ng/mL vascular endothelial growth factor, 10 ng/mL FGF2, 100 ng/mL SCF, 20 ng/mL Flt3 ligand, 20 ng/mL TPO, and 40 ng/mL IL-3. On day 8, EBs were collected and resuspended in StemPro34 SFM medium with 1% nonessential amino acids, 1 mM l-glutamine, and 0.1 mM β-mercaptoethanol, supplemented with 100 ng/mL SCF, 20 ng/mL Flt3 ligand, 20 ng/mL TPO, and 40 ng/mL IL-3. The medium was thereafter replaced every 2 days. At the end of the differentiation culture, the cells were collected and dissociated with accutase into single cells and used for flow cytometry or clonogenic assays, as previously described.15 Further erythroid differentiation in liquid culture was performed as previously described.17
RNA-sequencing analysis
RNA was extracted with the Direct-zol RNA purification kit (Zymo R2061). Sequencing libraries were prepared using the TruSeq Stranded mRNA library prep kit (Illumina 20020594) from 500 ng input RNA. Samples were barcoded and run on a Hi-seq 4000 in a 100-bp/100-bp paired-end run, using the Hi-seq 3000/4000 SBS kit (Illumina).
HSPC samples from 16 iPSC lines were included in the RNA-seq analyses after quality control of the raw data (supplemental Table 2). RNA-seq reads from the fastq files were mapped to the GRCh37 assembly of the human genome using the STAR aligner.18 The Ensembl GRCh37 gene and transcript annotations were used. Salmon19 was used to perform transcript quantification, and gene counts were generated from the transcript level abundances using the tximport function of the tximport R package.20 Differential gene expression analysis was performed using DESeq2.21 Genes with a false discover rate (FDR) < 0.05 and absolute expression log2fc > 1 in SF3B1K700E vs SF3B1WT cells were considered as differentially expressed.
Differential transcript usage between the SF3B1K700E and SF3B1WT iPSC-HSPCs was performed using the DEXSeq22 and stageR23 R-packages. Transcripts with a relative abundance proportion <5% in all samples were filtered. Transcripts were considered to have differential usage if absolute usage log2fc was >1 and overall FDR was <0.05 (supplemental Methods).
Differential alternative splicing was performed using the rMATS tool24 using the aligned BAM files. The relative expression (inclusion level) of alternatively spliced isoforms was estimated by the fraction of reads mapping to an alternative splicing event over the total reads.24 Events with FDR < 0.05 and absolute inclusion level difference > 10% were considered as differentially spliced between the SF3B1K700E and SF3B1WT iPSC-HSPCs.
Integration framework of differential gene expression, transcript usage, and splicing
To generate a SF3B1K700E signature, we combined differential gene expression, differential transcript usage, and differential splicing analyses. First, we identified the set of transcripts that contain the exons present in each differential splicing event using the maser R-package. We then filtered out nondifferentially used transcripts and paired each differential splicing event with the remaining set of differentially used transcripts. The pairs that belonged to genes with a statistically significant expression log2fc and contained a differential splicing event with an FDR value within the 20 lowermost FDR values were considered as the “tier 1” set, from which the mutant SF3B1 signature events and genes were derived.
ATAC-sequencing analysis
Nuclear pellets (supplemental Methods) were subjected to transposase reaction using the Illumina Nextera DNA Sample Preparation Kit. The libraries were quantified using the Agilent BioAnalyzer. Sequencing of 75 nucleotide-long paired-end reads was performed in a NextSeq-500 (Illumina).
HSPC samples from 15 iPSC lines were included in the ATAC-seq analyses after quality control of the raw data (supplemental Table 2). ATAC-seq reads from the fastq files were trimmed with the TrimGalore tool to remove adaptor sequences and then aligned to the GRCh37 reference genome using the Bowtie225 aligner. Reads with a mapping quality (MAPQ) score < 10 were removed using samtools. Duplicate reads were removed using Picard. All aligned reads were shifted to remove Tn5 transposase artifacts, as previously described26 using deeptools.27 Peaks were called using MACS228 (supplemental Methods) and then filtered using the irreproducible discovery rate28,29 framework with a cutoff of 0.05. Then, we merged all reproducible peaks to create an ATAC-seq atlas. Differential accessibility analysis was performed using DESeq2. Peaks with an FDR cutoff of 0.05 and absolute log2fc > 1 were considered differentially accessible.
Results
Generation of MDS patient-derived SF3B1K700E and genetically matched WT iPSC lines
From a previous population genome profiling study,3 we identified 3 BM mononuclear cell samples from 3 patients with MDS-RS (P21, P22, P23) harboring isolated SF3B1K700E mutations with high variant allele frequencies (VAFs; range, 37%-42%; Figure 1A; supplemental Table 1). Upon reprogramming, we obtained both SF3B1K700E and SF3B1WT iPSC lines from all patients. Despite the high VAF of SF3B1K700E in the starting samples, most iPSC colonies (12 of 18, 66.7%; 47 of 51, 92%; and 34 of 41, 83% from patients P21, P22, and P23, respectively) were WT and thus originated from normal cells (Figure 1B). This reprogramming advantage of normal over mutant cells has previously been observed by us and others in reprogramming of MDS and acute myeloid leukemia samples with other mutations.15,30 We established 3 independent SF3B1K700E and 3 SF3B1WT iPSC lines from each patient (total lines = 18), to serve as biological replicates (supplemental Table 2). We excluded the presence of any karyotypic abnormalities in any of these lines (supplemental Figure 1). We also excluded the presence of any other MDS/acute myeloid leukemia driver mutations in all lines or in the starting cells with next-generation sequencing of a panel of 126 genes implicated in myeloid malignancy.31
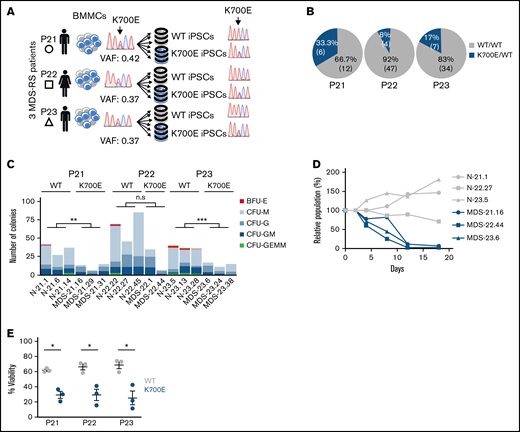
Derivation and phenotypic characterization of MDS-RS patient-derived SF3B1K700E and SF3B1WT iPSCs. (A) Schematic overview of the derivation of iPSC lines with isolated SF3B1K700E mutation and genetically matched normal WT lines from 3 patients with MDS-RS (BMMCs, bone marrow mononuclear cells). (B) Percentage and absolute number of iPSC colonies of each genotype derived from each patient sample. (C) Methylcellulose assays on day 14 of hematopoietic differentiation. The number of colonies from 5000 seeded cells is shown per iPSC line (CFU-GEMM, colony-forming unit-granulocyte, erythrocyte, monocyte, megakaryocyte; CFU-GM, CFU-granulocyte, monocyte; CFU-G, colony-forming unit-granulocyte; CFU-M, CFU-monocyte; BFU-E, burst-forming unit-erythrocyte); n.s., not significant; **P ≤ .01; ***P ≤ .001. (D) Competitive growth assay. The cells were mixed 1:1.5 at the onset of hematopoietic differentiation with a genetically matched WT iPSC line stably expressing green fluorescent protein (GFP). The relative population size was estimated as the percentage of GFP− cells (measured by flow cytometry) at each time point (days 4-18 of differentiation), relative to the percentage of GFP− cells on day 2. (E) Percent viable (4′,6-diamidino-2-phenylindole−) cells on day 14 of hematopoietic differentiation of the indicated SF3B1K700E and SF3B1WT iPSC-HSPCs. Mean and standard error of the mean (SEM) of 1 to 3 independent differentiation experiments with 2 to 3 iPSC lines from each group are shown. *P ≤ .05.
Derivation and phenotypic characterization of MDS-RS patient-derived SF3B1K700E and SF3B1WT iPSCs. (A) Schematic overview of the derivation of iPSC lines with isolated SF3B1K700E mutation and genetically matched normal WT lines from 3 patients with MDS-RS (BMMCs, bone marrow mononuclear cells). (B) Percentage and absolute number of iPSC colonies of each genotype derived from each patient sample. (C) Methylcellulose assays on day 14 of hematopoietic differentiation. The number of colonies from 5000 seeded cells is shown per iPSC line (CFU-GEMM, colony-forming unit-granulocyte, erythrocyte, monocyte, megakaryocyte; CFU-GM, CFU-granulocyte, monocyte; CFU-G, colony-forming unit-granulocyte; CFU-M, CFU-monocyte; BFU-E, burst-forming unit-erythrocyte); n.s., not significant; **P ≤ .01; ***P ≤ .001. (D) Competitive growth assay. The cells were mixed 1:1.5 at the onset of hematopoietic differentiation with a genetically matched WT iPSC line stably expressing green fluorescent protein (GFP). The relative population size was estimated as the percentage of GFP− cells (measured by flow cytometry) at each time point (days 4-18 of differentiation), relative to the percentage of GFP− cells on day 2. (E) Percent viable (4′,6-diamidino-2-phenylindole−) cells on day 14 of hematopoietic differentiation of the indicated SF3B1K700E and SF3B1WT iPSC-HSPCs. Mean and standard error of the mean (SEM) of 1 to 3 independent differentiation experiments with 2 to 3 iPSC lines from each group are shown. *P ≤ .05.
To assess the effects of the SF3B1K700E mutation on hematopoiesis, we used an in vitro directed differentiation protocol that produces definitive-type HSPCs. Differentiation of 2 to 3 independent SF3B1K700E and SF3B1WT lines from each patient revealed no defects in hematopoietic specification, as indicated by the emergence of CD34+ and CD45+ HSPCs (supplemental Figure 2A-C). However, the number of hematopoietic colonies generated from SF3B1K700E iPSC-HSPCs in methylcellulose assays was significantly lower than that generated from genetically matched normal lines (Figure 1C). Moreover, SF3B1K700E iPSC-HSPCs exhibited reduced proliferation and lower viability, as well as impaired erythroid maturation, compared with matched normal iPSC-HSPCs (Figure 1D-E; supplemental Figure 2D-F). Reduced growth has previously been reported in various cellular and murine models of splicing factor mutations.16,32 Decreased erythroid maturation and formation of RSs from patient-derived iPSCs harboring a SF3B1 mutation has also been previously shown.33 Taken together, these results demonstrate that the SF3B1K700E mutation causes a differentiation and proliferation defect, recapitulating hallmark phenotypes in MDS patient-derived iPSCs and ex vivo primary MDS cells.15-17,33,34
Global gene expression, mis-splicing, and differential transcript usage in SF3B1K700E HSPCs
To examine the effects of SF3B1K700E in the transcriptome, we performed RNA sequencing in sorted CD34+/CD45+ iPSC-HSPCs from 3 SF3B1K700E and 3 SF3B1WT iPSC lines from each patient (total 18 lines; supplemental Figure 3; supplemental Table 2). Samples MDS-22.1 and MDS-22.43 did not pass quality control at the library preparation stage and were not included in the analyses. Principal component analysis (PCA) and hierarchical clustering based on gene expression grouped the iPSC lines by genotype (eg, SF3B1K700E vs SF3B1WT; Figure 2A-B).

Integrative gene expression, alternative splicing, and transcript usage analyses categorize gene targets of mutant SF3B1. (A) PCA plots based on gene expression of the 3000 most highly variable genes color-coded by SF3B1 mutation status and sign-coded by patient ID. (B) Heatmap showing distance of the indicated iPSC-HSPCs based on pairwise Pearson correlation of their gene expression profiles, color-coded by SF3B1 mutation status and patient ID. (C) PCA plot based on inclusion levels of the differentially spliced events between SF3B1K700E and SF3B1WT iPSC-HSPCs. (D) Scatterplots comparing the mean inclusion levels of the differentially spliced events in SF3B1K700E vs SF3B1WT iPSC-HSPCs with different event types broken down by color, as indicated. (E) Schematic summarizing the integrative analysis used to derive a mutant SF3B1 signature of splicing events and genes and scatterplot showing inclusion level difference of all 59 signature splicing events, corresponding to 34 genes. A positive y axis value indicates that the event is more frequently found in SF3B1K700E vs SF3B1WT cells.
Integrative gene expression, alternative splicing, and transcript usage analyses categorize gene targets of mutant SF3B1. (A) PCA plots based on gene expression of the 3000 most highly variable genes color-coded by SF3B1 mutation status and sign-coded by patient ID. (B) Heatmap showing distance of the indicated iPSC-HSPCs based on pairwise Pearson correlation of their gene expression profiles, color-coded by SF3B1 mutation status and patient ID. (C) PCA plot based on inclusion levels of the differentially spliced events between SF3B1K700E and SF3B1WT iPSC-HSPCs. (D) Scatterplots comparing the mean inclusion levels of the differentially spliced events in SF3B1K700E vs SF3B1WT iPSC-HSPCs with different event types broken down by color, as indicated. (E) Schematic summarizing the integrative analysis used to derive a mutant SF3B1 signature of splicing events and genes and scatterplot showing inclusion level difference of all 59 signature splicing events, corresponding to 34 genes. A positive y axis value indicates that the event is more frequently found in SF3B1K700E vs SF3B1WT cells.
Differential gene expression analysis revealed 2737 differentially expressed genes in the SF3B1K700E mutant vs WT lines, 1821 of which were upregulated in the SF3B1K700E cells (supplemental Figure 4A-B). Gene set enrichment analysis showed enrichment of gene sets related to metabolism and cell morphology in genes upregulated in SF3B1K700E cells and enrichment of genes related to myeloid lineage differentiation in the downregulated genes (supplemental Figure 4C-E).
To examine the effects of the SF3B1K700E mutation on splicing, we characterized alternative splicing (AS) events in the SF3B1K700E and SF3B1WT cells, classified as alternative 3′ splice site use (A3SS), alternative 5′ splice site use (A5SS), mutually exclusive exons (MXE), retention of introns (RI), and skipping (inclusion or exclusion) of cassette exons (SE). A total of 1829 differential splicing events were detected between SF3B1K700E and SF3B1WT cells, which included 983 SE, 338 mutually MXE, 265 A3SS, 173 RI, and 70 A5SS events (supplemental Figure 4F; supplemental Table 3). Hierarchical clustering, as well as PCA, based on the inclusion levels of the differential splicing events, also separated the cells based on genotype, as expected (Figure 2C; supplemental Figure 4G). Consistent with previous studies, we found increased exclusion of cassette exons, increased use of alternative 3′ splice sites, and decreased retention of introns in SF3B1K700E cells (Figure 2D; supplemental Figure 4H).9,11
To evaluate the effects of the SF3B1K700E mutation at the transcript level, we performed differential transcript usage analysis, which identified 1086 differentially used transcripts between SF3B1K700E and SF3B1WT cells (547 more used and 539 less used in SF3B1K700E compared with SF3B1WT cells). These differentially used transcripts belong to 865 genes, 198 of which were also found to be differentially expressed (supplemental Figure 4I).
In summary these analyses demonstrate that SF3B1K700E mutations are associated with distinct gene expression, splicing, and transcript usage signatures.
Integration framework categorizes mutant SF3B1 gene targets by linking differential splicing to differential transcript usage and differential gene expression
Most previous studies have prioritized candidate target genes of mis-splicing by mutant SF3B1 in cancer cells by selecting splicing events based on the size of differences in inclusion level of the isoforms between mutant and control cells.11 To categorize splicing effects of the SF3B1K700E mutation in MDS, we developed a computational approach combining analyses at 3 different transcriptomic levels: gene expression, splicing, and transcript usage. This framework was used to classify the splicing events into 5 tier-based classes (supplemental Figure 5A; supplemental Table 4).
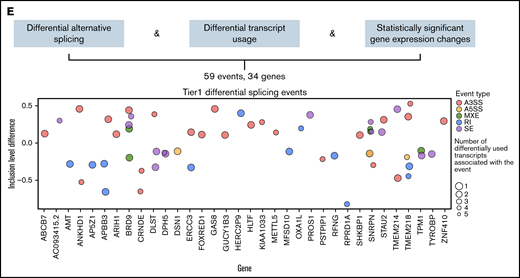
Of 1829 total differential splicing events between SF3B1K700E and SF3B1WT HSPCs, 215 were associated with at least 1 differentially used transcript. Of these 215 events, 95 belong to genes with a statistically significant (FDR < 0.05) expression log2 fold change (log2fc) between SF3B1K700E and SF3B1WT HSPCs. Of these 95 events, we selected the top 59 differentially spliced events (with the lowest 20 FDR values). These tier 1 59 events belong to 34 genes: 19 downregulated and 15 upregulated in SF3B1K700E vs SF3B1WT cells (Figures 2E and 3; supplemental Figure 5B; supplemental Table 5). Fifty-one (86%) of these 59 tier 1 events are A3SS, RI, or SE events (supplemental Figure 5C). This set of 59 events contained more A3SS events with increased use in SF3B1K700E vs SF3B1WT cells and more RI events that were less retained in SF3B1K700E vs SF3B1WT cells, reflecting the event distribution among all differential splicing events (Figure 3). We observed that several of the transcripts used preferentially in SF3B1K700E vs SF3B1WT HSPCs were annotated as NMD (Figure 3). Notably, this increased use of NMD transcripts was also associated with decreased expression of the corresponding genes (DLST, BRD9, KIAA1033, SHKBP1, GAS8). This is consistent with previous findings showing that SF3B1 mutations induce widespread use of abnormal cryptic 3′ splice sites, leading to NMD of multiple transcripts.13,35
![Events of the mutant SF3B1 splicing signature. Heatmap showing the row normalized inclusion levels of the 59 signature events across HSPCs from all iPSC lines. For each row, color-coded side panels present metadata relevant to each event, including the log2fc of expression of the respective genes, the biotypes of the up- and downregulated transcripts that are associated with the splicing events, and the presence of the events in the MDS patient dataset of Pellagatti et al,9 encoded as not present (signature events not present in any comparison); present but not significant (signature events that were not statistically significant or/and had an absolute inclusion level difference < 0.1 in both comparisons [SF3B1mut vs SF-WT and SF3B1mut vs WT, ie, healthy individuals]); present only in SF3B1mut vs SF-WT (signature events statistically significant [FDR < 0.05] and with an absolute inclusion level difference > 0.1 only in the SF3B1mut vs SF-WT MDS patient comparison); and present in SF3B1mut vs SF-WT and SF3B1mut vs WT (signature events statistically significant [FDR < 0.05] and with an absolute inclusion level difference > 0.1 in both comparisons). The annotations of the transcript biotypes are derived from the Ensembl GRCh37 gtf annotation file. Each row represents one event labeled with the respective gene name followed by a number indicating distinct events.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/10/10.1182_bloodadvances.2021006325/4/m_advancesadv2021006325f3.png?Expires=1769105012&Signature=i~VuanxxeSQu0JrMpEiH2XRvL6rH81Cuzi98pE5IwHDewQC9-n5wG2dgOKxHICjavWpVWCkoxtDOfpJNsz2xCOMUvywBejH77JsQvVBSJDALZjDoKYwMgzqB1fem0~~p2Cf4IsCev-yrdhZR~Atu0fAr9FbsOB5-pPiR0aVK6uovPtCeH326kHbo59dKZNGPwjLI3RFprAWFHgqA9OSakj0fzvfFnfHa-~-yKRbC32AbCC1aDoyKmaLjJd8auccfZTA9sfKsh0jYK2-3q96pHSFOPGaEnVm3avq20BFmd5uIqBxrg5nM~kN-PpGDYqTDiRlv1lThqMNm8p-vVBHysQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Events of the mutant SF3B1 splicing signature. Heatmap showing the row normalized inclusion levels of the 59 signature events across HSPCs from all iPSC lines. For each row, color-coded side panels present metadata relevant to each event, including the log2fc of expression of the respective genes, the biotypes of the up- and downregulated transcripts that are associated with the splicing events, and the presence of the events in the MDS patient dataset of Pellagatti et al,9 encoded as not present (signature events not present in any comparison); present but not significant (signature events that were not statistically significant or/and had an absolute inclusion level difference < 0.1 in both comparisons [SF3B1mut vs SF-WT and SF3B1mut vs WT, ie, healthy individuals]); present only in SF3B1mut vs SF-WT (signature events statistically significant [FDR < 0.05] and with an absolute inclusion level difference > 0.1 only in the SF3B1mut vs SF-WT MDS patient comparison); and present in SF3B1mut vs SF-WT and SF3B1mut vs WT (signature events statistically significant [FDR < 0.05] and with an absolute inclusion level difference > 0.1 in both comparisons). The annotations of the transcript biotypes are derived from the Ensembl GRCh37 gtf annotation file. Each row represents one event labeled with the respective gene name followed by a number indicating distinct events.
Events of the mutant SF3B1 splicing signature. Heatmap showing the row normalized inclusion levels of the 59 signature events across HSPCs from all iPSC lines. For each row, color-coded side panels present metadata relevant to each event, including the log2fc of expression of the respective genes, the biotypes of the up- and downregulated transcripts that are associated with the splicing events, and the presence of the events in the MDS patient dataset of Pellagatti et al,9 encoded as not present (signature events not present in any comparison); present but not significant (signature events that were not statistically significant or/and had an absolute inclusion level difference < 0.1 in both comparisons [SF3B1mut vs SF-WT and SF3B1mut vs WT, ie, healthy individuals]); present only in SF3B1mut vs SF-WT (signature events statistically significant [FDR < 0.05] and with an absolute inclusion level difference > 0.1 only in the SF3B1mut vs SF-WT MDS patient comparison); and present in SF3B1mut vs SF-WT and SF3B1mut vs WT (signature events statistically significant [FDR < 0.05] and with an absolute inclusion level difference > 0.1 in both comparisons). The annotations of the transcript biotypes are derived from the Ensembl GRCh37 gtf annotation file. Each row represents one event labeled with the respective gene name followed by a number indicating distinct events.
The 59-splicing event signature is associated with SF3B1 mutational status
To test whether the SF3B1 signature derived in iPSC-HSPCs is also found in primary patient samples, we interrogated transcriptome data from CD34+ BM cells from 68 patients with MDS and 8 healthy individuals from a published dataset.9 Thirty-one of the 59 tier 1 events (53%) were found differentially spliced (FDR < 0.05, |inclusion level difference| > 0.1) between SF3B1-mutated patients (SF3B1mut, n = 28) and patients with MDS without any SF mutations (SF-WT, n = 40). Twenty-eight of those were also found differentially spliced between SF3B1-mutated patients and healthy individuals (WT; n = 8; Figure 3; supplemental Figure 5D). This splicing signature was not found in events differentially spliced between MDS primary cells harboring other splicing factor mutations (SRSF2, U2AF1) and SF-WT MDS or healthy individuals and is thus specific to SF3B1 mutations (supplemental Figure 5E). PCA based on the inclusion level of the mutant SF3B1 signature splicing events separated all samples (SF3B1mut; SF-WT; WT) based on SF3B1 genotype, with the exception of 1 sample, annotated as SF-WT, which clustered together with the SF3B1-mutated samples (Figure 4). Examination of the RNA-seq data for sequence alterations in the SF3B1 locus in this specific patient revealed a previously overlooked 6-bp in-frame deletion spanning the K700E hotspot (SF3B1p.K700_V701delKV; Figure 4). This demonstrates that the splicing signature derived in iPSC-HSPCs is also present in HSPCs of patients with MDS. Furthermore, patients with SF3B1 mutations other than K700E clustered together with the SF3B1K700E-mutated patients (Figure 4), which indicates that our signature is representative of a broader spectrum of SF3B1 mutations.

Splicing event signature separates SF3B1-mutated MDS cases. PCA plot based on the inclusion levels of the signature splicing events in the patient samples of Pellagatti et al,9 separating MDS SF3B1K700E-mutated patients (K700E SF3B1mut MDS) and patients with SF3B1 mutations other than K700E (non-K700E SF3B1mut MDS) from patients without SF mutations (SF-WT MDS) and healthy individuals (WT). The asterisk marks 1 patient annotated as SF-WT. Clustering of this sample together with the SF3B1-mutated cases prompted us to more closely interrogate the sequence of the SF3B1 locus for any previously unidentified mutations. We thus discovered an in-frame 6-bp deletion (SF3B1p.K700_V701delKV) removing 2 amino acids, including the K700 hotspot.
Splicing event signature separates SF3B1-mutated MDS cases. PCA plot based on the inclusion levels of the signature splicing events in the patient samples of Pellagatti et al,9 separating MDS SF3B1K700E-mutated patients (K700E SF3B1mut MDS) and patients with SF3B1 mutations other than K700E (non-K700E SF3B1mut MDS) from patients without SF mutations (SF-WT MDS) and healthy individuals (WT). The asterisk marks 1 patient annotated as SF-WT. Clustering of this sample together with the SF3B1-mutated cases prompted us to more closely interrogate the sequence of the SF3B1 locus for any previously unidentified mutations. We thus discovered an in-frame 6-bp deletion (SF3B1p.K700_V701delKV) removing 2 amino acids, including the K700 hotspot.
Chromatin accessibility landscape of SF3B1K700E HSPCs
To investigate the chromatin landscape of SF3B1K700E cells, we performed ATAC sequencing (supplemental Methods) in sorted CD34+/CD45+ iPSC-HSPC samples paired to those used for RNA sequencing (3 SF3B1K700E and 3 SF3B1WT iPSC lines from each patient; supplemental Figure 3; supplemental Table 2) resulting in an ATAC-seq atlas of 56 420 peaks. (Samples MDS-22.1, MDS-22.43, and N-21.1 did not pass quality control at the library preparation stage and were not included in the analyses.) PCA and hierarchical clustering based on chromatin accessibility grouped the iPSC lines by genotype (Figure 5A-B). Differential accessibility analysis revealed 3737 differentially accessible peaks between the SF3B1K700E and SF3B1WT HSPCs, 1527 of which were more accessible in the mutants (Figure 5C; supplemental Figure 6A). Differentially accessible peaks were predominantly localized in intronic and intergenic regions (supplemental Figure 6B). Chromatin accessibility changes correlated with gene expression changes in both directions (more accessible and upregulated; less accessible and downregulated; Figure 5D-E; supplemental Figure 6C). Next, we compared the chromatin accessibility profiles of the SF3B1K700E and SF3B1WT iPSC-HSPCs to those defined in primary human hematopoietic cell types along the hematopoietic hierarchy36 (supplemental Methods). Of the 56 420 total ATAC-seq peaks called in the iPSC-HSPC dataset, 40 568 overlapped with the peaks from Corces et al36 (total, 98 525). Differential accessibility analysis on these 40 568 peaks resulted in 2757 differentially accessible peaks between SF3B1K700E and SF3B1WT iPSC-HSPCs. The pairwise Pearson correlation between read counts of these 2757 peaks in iPSC-HSPCs and the hematopoietic populations of Corces et al36 showed that the chromatin landscapes of SF3B1K700E cells resembled more those of megakaryocyte–erythroid progenitor cells and erythroid cells, whereas the chromatin landscape of SF3B1WT cells resembled more that of granulocyte-monocyte progenitors and monocytes (Figure 5F). These results suggest a potential chromatin priming of SF3B1K700E CD34+ HSPCs toward the erythroid rather than the myeloid lineage and may reflect the more prominent involvement of the erythroid lineage in the pathology and clinical presentation of MDS-RS.
![SF3B1K700E HSPCs have altered chromatin landscapes. (A) PCA based on accessibility of all peaks in the ATAC-seq atlas color-coded by SF3B1 mutation status and sign-coded by patient ID. (B) Heatmap showing the distance of the HSPCs from the indicated iPSC lines, based on pairwise Pearson correlation of their chromatin accessibility landscapes, color-coded by SF3B1 mutation status and patient ID. (C) Scatterplot showing the accessibility log2fc and the adjusted P value of the differentially accessible peaks between SF3B1K700E and SF3B1WT iPSC-HSPCs per chromosome, color-coded by the adjusted P value. Each point represents a peak. (D) Cumulative distribution function (CDF) of the expression log2fc of genes more accessible in SF3B1K700E HSPCs, genes less accessible in SF3B1K700E HSPCs, and all genes, showing that genes more accessible in SF3B1K700E HSPCs are upregulated (Kolmogorov–Smirnov [KS] test, P = 1.17e-07) and genes less accessible in SF3B1K700E HSPCs are downregulated (KS test, P = 3.13e-16) compared with background. (E) Scatterplot showing the log2fc accessibility value of the differentially accessible peaks and the log2fc expression value of the linked gene (genes for which P value could not be calculated were excluded). (F) Heatmap showing Pearson correlation values of normalized read counts for ATAC-seq peaks that overlap between the indicated iPSC-HSPCs and primary normal hematopoietic cell subpopulations (hematopoietic stem cells [HSC], multipotent progenitors [MPP], common myeloid progenitors [CMP], lymphoid-primed multipotent progenitors [LMPP], granulocyte- monocyte progenitors [GMP], megakaryocyte-erythrocyte progenitors [MEP], common lymphoid progenitors [CLP], monocytes [Mono], erythroid cells [Ery], natural killer cells [NK], and B cells) from Corces et al.36](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/10/10.1182_bloodadvances.2021006325/4/m_advancesadv2021006325f5.png?Expires=1769105012&Signature=TrSrOZRa4TeUG3yPN7Z6BwOQdvtGzRq5U-Xs42tZCIafjXNjyr1p2TZDBUPdRjCndoehxdL1eKpkkMYz4bSdWk1o6t9cBjqAcaJJUiBp~sW1rFMaoCPrCunhklaPJ6Q2u3AYO4Yqe3NH~-wq4BADAcBU7nTISDRa1weGnBBSHoCxFSANIM1hr8M8nFiFvfg3BW8~FmIR31SnmhmzjwbADqlqdHmRU0--AMbbqKYz4omZaZks0UKRYFVOnj8Yh2mK04dUTw~1R-JDIZBD1sqgmyrcBmwYQg6PmkPtrjq4KNfLHWsfbk7M~tCP0gokG~pLzLrPa7U2oUqPLV4X0yvVhA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
SF3B1K700E HSPCs have altered chromatin landscapes. (A) PCA based on accessibility of all peaks in the ATAC-seq atlas color-coded by SF3B1 mutation status and sign-coded by patient ID. (B) Heatmap showing the distance of the HSPCs from the indicated iPSC lines, based on pairwise Pearson correlation of their chromatin accessibility landscapes, color-coded by SF3B1 mutation status and patient ID. (C) Scatterplot showing the accessibility log2fc and the adjusted P value of the differentially accessible peaks between SF3B1K700E and SF3B1WT iPSC-HSPCs per chromosome, color-coded by the adjusted P value. Each point represents a peak. (D) Cumulative distribution function (CDF) of the expression log2fc of genes more accessible in SF3B1K700E HSPCs, genes less accessible in SF3B1K700E HSPCs, and all genes, showing that genes more accessible in SF3B1K700E HSPCs are upregulated (Kolmogorov–Smirnov [KS] test, P = 1.17e-07) and genes less accessible in SF3B1K700E HSPCs are downregulated (KS test, P = 3.13e-16) compared with background. (E) Scatterplot showing the log2fc accessibility value of the differentially accessible peaks and the log2fc expression value of the linked gene (genes for which P value could not be calculated were excluded). (F) Heatmap showing Pearson correlation values of normalized read counts for ATAC-seq peaks that overlap between the indicated iPSC-HSPCs and primary normal hematopoietic cell subpopulations (hematopoietic stem cells [HSC], multipotent progenitors [MPP], common myeloid progenitors [CMP], lymphoid-primed multipotent progenitors [LMPP], granulocyte- monocyte progenitors [GMP], megakaryocyte-erythrocyte progenitors [MEP], common lymphoid progenitors [CLP], monocytes [Mono], erythroid cells [Ery], natural killer cells [NK], and B cells) from Corces et al.36
SF3B1K700E HSPCs have altered chromatin landscapes. (A) PCA based on accessibility of all peaks in the ATAC-seq atlas color-coded by SF3B1 mutation status and sign-coded by patient ID. (B) Heatmap showing the distance of the HSPCs from the indicated iPSC lines, based on pairwise Pearson correlation of their chromatin accessibility landscapes, color-coded by SF3B1 mutation status and patient ID. (C) Scatterplot showing the accessibility log2fc and the adjusted P value of the differentially accessible peaks between SF3B1K700E and SF3B1WT iPSC-HSPCs per chromosome, color-coded by the adjusted P value. Each point represents a peak. (D) Cumulative distribution function (CDF) of the expression log2fc of genes more accessible in SF3B1K700E HSPCs, genes less accessible in SF3B1K700E HSPCs, and all genes, showing that genes more accessible in SF3B1K700E HSPCs are upregulated (Kolmogorov–Smirnov [KS] test, P = 1.17e-07) and genes less accessible in SF3B1K700E HSPCs are downregulated (KS test, P = 3.13e-16) compared with background. (E) Scatterplot showing the log2fc accessibility value of the differentially accessible peaks and the log2fc expression value of the linked gene (genes for which P value could not be calculated were excluded). (F) Heatmap showing Pearson correlation values of normalized read counts for ATAC-seq peaks that overlap between the indicated iPSC-HSPCs and primary normal hematopoietic cell subpopulations (hematopoietic stem cells [HSC], multipotent progenitors [MPP], common myeloid progenitors [CMP], lymphoid-primed multipotent progenitors [LMPP], granulocyte- monocyte progenitors [GMP], megakaryocyte-erythrocyte progenitors [MEP], common lymphoid progenitors [CLP], monocytes [Mono], erythroid cells [Ery], natural killer cells [NK], and B cells) from Corces et al.36
Increased transcriptional activity of the TEAD family of transcription factors in SF3B1K700E HSPCs
To identify transcriptional programs of potential importance to SF3B1K700E HSPCs, we performed TF motif enrichment analysis in ATAC-seq peaks more accessible in SF3B1K700E cells that were linked to genes upregulated in SF3B1K700E cells (Figure 5E). This analysis revealed enrichment of motifs of several prototypical hematopoietic lineage TFs, such as those of the GATA, ETS, and AP-1 families. Unexpectedly, motifs of the TEAD family were also enriched (Figure 6A-C). Furthermore, regions more accessible and linked to upregulated genes in SF3B1K700E cells that contained TEAD motifs overlapped with annotated TEAD binding sites (supplemental Figure 7A).
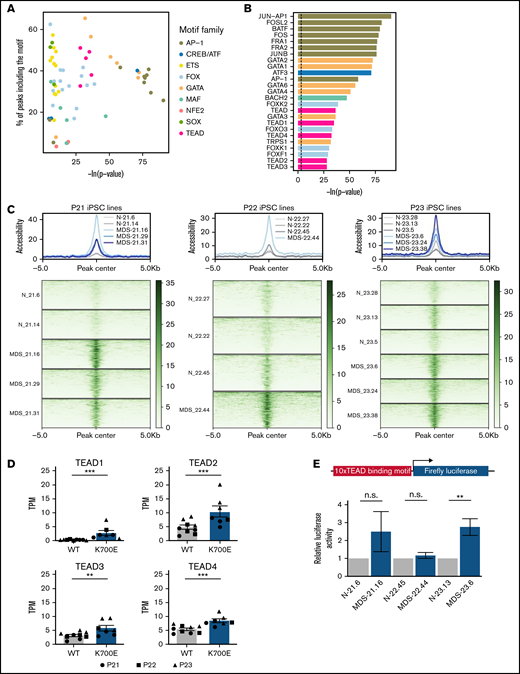
Increased transcriptional activity of TEAD TFs in SF3B1K700E HSPCs. (A) TF motifs enriched in peaks more accessible in SF3B1K700E compared with SF3B1WT HSPCs and linked to upregulated genes. (B) Most statistically significant TF motifs enriched in peaks more accessible in SF3B1K700E compared with SF3B1WT HSPCs and linked to upregulated genes. (C) Tornado plots showing the normalized accessibility signal in peaks more accessible in SF3B1K700E compared with SF3B1WT HSPCs and linked to upregulated genes that contain TEAD motifs. (D) Expression levels of TEAD family genes in iPSC-HSPCs. Mean and SEM of transcripts per million (TPM) values from RNA-seq are shown. **Padj ≤ .01; ***Padj ≤ .001. (E) TEAD reporter activity in HSPCs from the indicated iPSC lines. Mean and SEM of 2 to 5 independent differentiation and transduction experiments per line are shown. n.s., not significant; **P ≤ .01.
Increased transcriptional activity of TEAD TFs in SF3B1K700E HSPCs. (A) TF motifs enriched in peaks more accessible in SF3B1K700E compared with SF3B1WT HSPCs and linked to upregulated genes. (B) Most statistically significant TF motifs enriched in peaks more accessible in SF3B1K700E compared with SF3B1WT HSPCs and linked to upregulated genes. (C) Tornado plots showing the normalized accessibility signal in peaks more accessible in SF3B1K700E compared with SF3B1WT HSPCs and linked to upregulated genes that contain TEAD motifs. (D) Expression levels of TEAD family genes in iPSC-HSPCs. Mean and SEM of transcripts per million (TPM) values from RNA-seq are shown. **Padj ≤ .01; ***Padj ≤ .001. (E) TEAD reporter activity in HSPCs from the indicated iPSC lines. Mean and SEM of 2 to 5 independent differentiation and transduction experiments per line are shown. n.s., not significant; **P ≤ .01.
The TEAD family of TFs are best known as effectors of the Hippo signaling pathway, with important roles in various biological processes and malignancies, albeit no previous links to hematologic disease.37,38 To further investigate a potential role for TEAD TFs in SF3B1K700E HSPCs, we examined expression of the 4 members of the TEAD family TEAD1-4 in SF3B1K700E and SF3B1WT cells. TEAD2 and TEAD4 were the TEAD family members expressed at the highest levels in both SF3B1K700E and SF3B1WT cells, including iPSC-HSPCs, as well as patient cells (Figure 6D; supplemental Figure 7B). All 4 TEAD genes were upregulated in the SF3B1K700E compared with SF3B1WT iPSC-HSPCs (Figure 6D).
To experimentally test whether TEAD transcriptional activity is higher in SF3B1K700E cells, we transduced SF3B1K700E and SF3B1WT iPSC-HSPCs with a luciferase construct (supplemental Methods) reporting TEAD activity. Reporter activity was higher or trended higher in SF3B1K700E compared with SF3B1WT iPSC-HSPCs from 2 of the 3 patients (Figure 6E). TEAD is best known as an effector of the Hippo signaling pathway and is bound to DNA as a complex with YAP or TAZ transcriptional coactivators.39 To test the activity of the Hippo pathway in our cells, we performed immunoblots in SF3B1K700E and SF3B1WT iPSC-HSPCs from 2 of the patients. Although we confirmed TEAD expression at the protein level, we did not detect YAP activation (phosphorylated form pYAPS127) or expression of YAP or TAZ (supplemental Figure 7C). These results, collectively, support a Hippo pathway-independent increase of TEAD expression and transcriptional activity in SF3B1K700E HSPCs.
Discussion
In summary, here we harnessed somatic cell reprogramming to derive genetically faithful models of SF3B1K700E mutation. The genetically matched and strictly clonal conditions, the ability to derive relatively homogeneous cell populations through in vitro differentiation, and the availability of biological replicates to control for any effects of the reprogramming process (nongenetic line-to-line variability) and of the patient’s genetic background on the transcriptome were all critical components of this study. Although our differentiation protocol generates definitive-type hematopoiesis, iPSC-derived blood cells may resemble fetal more than adult cells.40
Nonetheless, we and others have shown that iPSC models of myeloid malignancies capture phenotypic and molecular characteristics of disease and can be used to discover new disease mechanisms and therapeutic vulnerabilities, further corroborated by the present study.16,17,41,42
Our study was also powered by a data integration framework with which we were able to assess the combination of the effects of the SF3B1K700E mutation across parallel levels of deregulation of the transcriptome (gene, transcript, splicing) toward deriving a SF3B1K700E splicing signature. These integrated analyses validated several known gene candidates, such as ANKHD19 ,METTL5 (A3SS event),9,13 ABCB7 (A3SS event),11,43,44 and BRD9 (SE event).13 Additionally, the genes DPH5, COASY, ZDHHC16, TMEM214, and EI24, previously cataloged as mis-spliced in SF3B1 mutant cells, are also included in our tier 2 and tier 1 set.13 Furthermore, we nominate several new splicing events in genes not previously reported mis-spliced by mutant SF3B1 that warrant further investigation for their relevance to the pathogenesis of MDS. The diversity of mis-splicing events, many of which are found across different models of SF3B1K700E mutation, may suggest a multifactorial disease pathogenesis. In addition, the specificity of the mutant SF3B1 signature, derived from the iPSC lines and validated in primary patient samples, identifies atypical mutations involving the K700 hotspot, such as the SF3B1p.K700_V701delKV that we report here, as functionally equivalent to the K700E mutation, and can thus be further used to evaluate the role of putative pathogenic variants in SF3B1.45
Our study is the first to characterize the chromatin landscape of SF3B1K700E HSPCs. Interestingly, we report potential “priming” at the chromatin level of SF3B1K700E HSPCs toward the erythroid over the myeloid lineage, a finding that may be related to the preferential involvement of the erythroid lineage in MDS and, in particular, MDS-RS. It is unclear whether any of the global chromatin accessibility changes that we report here are a direct consequence of mis-splicing (for example, of a chromatin regulator gene, such as BRD9,13 or a pioneer transcription factor). Likely, at least some of them reflect differences in differentiation state and lineage priming as an indirect consequence of the SF3B1K700E mutation. Because reprogramming to pluripotency effectively erases the epigenome of the somatic cell, differences found between mutant and WT cells across replicates can be solely attributed to genotype.
Several master hematopoietic lineage TF motifs were present in chromatin regions that were differentially accessible between SF3B1K700E and SF3B1WT iPSC-HSPCs, which may underlie the differentiation and colony formation impairment of these cells. Interestingly, our chromatin accessibility analyses, followed by functional studies, lend support to a putative role for the TEAD TFs in the context of SF3B1K700E mutation. The relevance of elevated TEAD activity to the pathogenesis of MDS-RS and its link to SF3B1 mutations will need to be validated in further studies involving assessment of TEAD binding to DNA and functional experiments, such as genetic perturbation of TEAD factors, in our iPSC models, as well as potential validation of the findings in primary patient cells. Pending further investigation, this novel finding may point to a new disease mechanism and possible therapeutic vulnerabilities specific to SF3B1K700E cells.
Acknowledgments
This work was supported by an Edward P. Evans Foundation discovery research grant and a Geoffrey Beene Cancer Research Center grant. Work in the E.P.P. laboratory was also supported by National Institutes of Health grants R01HL137219 and R01CA225231, the New York State Stem Cell Board, the Pershing Square Sohn Cancer Research Alliance, and a Leukemia and Lymphoma Society scholar award. Work in the E.P. laboratory was also supported by the Josie Robertson Foundation. E.B. was supported by the Edward P. Evans Foundation. L.M. was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC; investigator grant 20125, AIRC 5x1000 project 21267) and Cancer Research United Kingdom, Fundación Científica de la Asociación Española Contra el Cáncer (FC AECC), and AIRC under the Accelerator Award Program (projects C355/A26819 and 22796).
Authorship
Contribution: A.G.D., A.G.K., D.E., M.O., and N.S. performed experiments; G.A., E.B., J.A.O., and R.K. analyzed data; T.M.-B. and E.H.-L. provided patient samples; Y.N., L.M., S.O., and M.C. provided MDS patient datasets; S.A.A. designed experiments; E.P. and E.P.P. conceived, designed, and supervised the study; and G.A., E.P., and E.P.P. wrote the manuscript.
Conflict-of-interest disclosure: E.P. is founder and equity holder of Isabl Inc., a cancer whole genome diagnostics company. E.P.P. has received honoraria from Celgene and Merck and research support from Incyte for research not related to this study. The remaining authors declare no competing financial interests.
Correspondence: Eirini P. Papapetrou, Department of Oncological Sciences, Icahn School of Medicine at Mount Sinai, 1470 Madison Ave, New York, NY 10029; e-mail: eirini.papapetrou@mssm.edu; and Elli Papaemmanuil, Center for Computational Oncology, Department of Epidemiology and Biostatistics, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; e-mail: papaemme@mskcc.org.

