

Key Points
PTT generated a stronger abscopal response compared with RT when combined with a toll-like receptor 9 agonist.
PTT increased antilymphoma T-cell response both locally and systemically, suggesting a strong in situ vaccination effect.

Abstract
In preclinical studies, we investigated a novel mechanism of in situ vaccination in lymphoma. Radiation therapy (RT) can induce abscopal responses in lymphoma models, but this has not translated into clinical efficacy. We hypothesized that immune stimulation with cytosine guanine dinucleotide (CpG) deoxynucleotides could enhance abscopal effects induced by RT or photothermal therapy (PTT), which has been shown to have an immune stimulatory effect in solid tumors but has not been studied in lymphoma. We designed a branched gold nanoparticle (NP) platform to carry CpG deoxynucleotides while maintaining PTT function and compared the immunologic profile of the tumor microenvironment after PTT or RT in a dual-flank lymphoma model. One flank was treated with CpG deoxynucleotides with RT or PTT, and the other tumor was left untreated. We found that the CpG deoxynucleotide/PTT group had significant reduction in growth in both treated (primary) and untreated (secondary) tumors, suggesting an improved abscopal response, with a concomitant increase in CD8/CD4 and cytotoxic T-cell/regulatory T-cell ratios in both primary and secondary tumors compared with CpG deoxynucleotides/RT. Dendritic cells in primary and secondary draining lymph nodes had increased maturation markers in the CpG deoxynucleotide/PTT group, and the effector memory T cells (both CD4 and CD8) in the secondary tumor and spleen were increased, suggesting a systemic vaccination effect. These data suggest that in a lymphoma model, PTT using a CpG deoxynucleotide NP platform resulted in enhanced in situ vaccination and abscopal response compared with RT.
Introduction
There are ∼80 000 new cases of non-Hodgkin lymphoma (NHL) in the United States each year.1 Ninety percent are B-cell lymphomas, which cause an estimated 20 000 annual deaths. Diffuse large B-cell lymphoma (DLBCL) is the most common type of NHL. Two-thirds of these cases are cured with standard chemoimmunotherapy. However, translocations in MYC or MYC with BCL2 or BCL6 (high-grade DLBCL) worsen 5-year overall survival, estimated at 33% vs 72% with nonrearranged DLBCLs.2,3 Despite the success of transplantation or chimeric antigen receptor (CAR) T-cell therapy, only 30% to 40% of patients have long term remission.4-8 There are currently no standard treatment options for progression after stem cell transplantation or CAR T-cell therapy. The design and implementation of innovative immune based approaches to improve these outcomes represent important unmet needs.
Although radiation therapy (RT) has a limited clinical role in advanced-stage lymphoma, it has been used as a method to generate an abscopal effect by stimulation of local and systemic immune responses, resulting in in situ vaccination.9,10 However, the success of this approach has so far been limited. MacManus et al11 described 4 of 29 patients with follicular lymphoma receiving palliative RT who had some degree of abscopal effect (at least 1 untreated site with response). They also described patients with mantle cell lymphoma, Hodgkin lymphoma, and Richter’s transformation who experienced varying degrees of abscopal effect after palliative RT. However, when RT was combined with immune stimulants, such as toll-like receptor 9 agonist CpG deoxynucleotides, for the treatment of advanced-stage indolent lymphoma, the response rates increased but compete response rates remained low.12
Unlike external-beam RT, photothermal therapy (PTT) using gold nanoparticles (GNPs) can generate a strong in situ vaccination effect by heat ablation of melanoma tumors.13 The local high-heat ablation stimulates a cascade of proinflammatory cytokines, including interleukin-6 (IL-6), IL-1β, tumor necrosis factor-α, G-CSF, granulocyte-macrophage colony-stimulating factor, and CCL2, that are able to not only promote the expansion of both proinflammatory (CD4+ and CD8+) cells but also increase suppressive immune cells. In a melanoma model, PTT also increased CD8+ T-cell infiltration in nontreated tumors and dendritic cell (DC) maturation in the draining lymph nodes, as well as increasing systemic immune suppression via increased myeloid-derived suppressor cells (MDSCs). In addition, CpG deoxynucleotides can directly inhibit the immunosuppressive functions of MDSCs and cause them to differentiate into macrophages with antitumor activity.14 The increase in MDSCs from PTT can be mitigated with the incorporation of CpG deoxynucleotides.15 CpG deoxynucleotides are especially attractive in lymphoma because they stimulate immune responses and have direct cytotoxic effects against malignant B cells.16-19 Furthermore, PTT has also been evaluated in combination with checkpoint inhibitors with success in solid tumor murine models,20-22 but little is known about the immune effects of PTT in lymphoma.
Therefore, we hypothesized that CpG deoxynucleotides could enhance immune stimulation and lead to systemic tumor response induced by PTT. To address this hypothesis, we designed a branched GNP (BNP) platform to carry CpG deoxynucleotides while maintaining PTT capabilities (Figure 1A). BNPs have grooves to carry and protect CpG deoxynucleotides from degradation.23 We also compared the immunologic profile of the local tumor microenvironment of treated and untreated tumors as well as systemic responses after PTT or RT in a dual-flank lymphoma model.
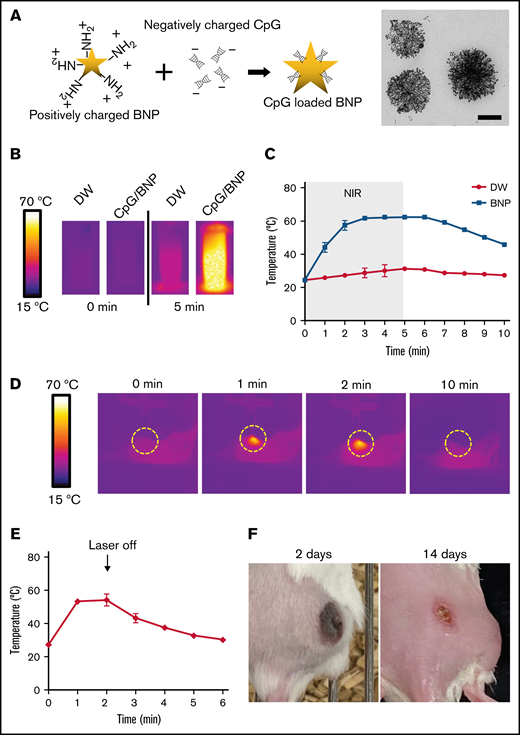
Synthesis and characterization of BNPs with CpG deoxynucleotide complex (CpG/BNP). (A) Schematic of coating positively charged BNPs with negatively charged CpG deoxynucleotides. On the right is transmission electron microscopic image of BNP. Scale bar, 100 nm. (B) Photothermal moiety of CpG/BNP. CpG/BNP (4 mg/50 μL) was irradiated for 5 minutes with near-infrared (NIR) laser (808 nm) with 2-W output (361.2 mW/cm2). Temperature was monitored by NIR camera. (C) Temperature tracing of deionized water (DW) and BNP during and after 5 minutes of laser exposure. (D) In vivo thermal image of PTT of single flank A20 lymphoma tumor on BALB/c mouse. After injection of 1 mg of CpG/BNP to right flank, mice were irradiated for 2 minutes with NIR laser (808 nm) with 2-W output (361.2 mW/cm2). (E) Temperature curve of lymphoma tumor during and after PTT. (F) Photothermal ablation–mediated necrosis of mice skin. Scar notable 2 days after PTT that resolved after 14 days with no recurrence of the tumor.
Synthesis and characterization of BNPs with CpG deoxynucleotide complex (CpG/BNP). (A) Schematic of coating positively charged BNPs with negatively charged CpG deoxynucleotides. On the right is transmission electron microscopic image of BNP. Scale bar, 100 nm. (B) Photothermal moiety of CpG/BNP. CpG/BNP (4 mg/50 μL) was irradiated for 5 minutes with near-infrared (NIR) laser (808 nm) with 2-W output (361.2 mW/cm2). Temperature was monitored by NIR camera. (C) Temperature tracing of deionized water (DW) and BNP during and after 5 minutes of laser exposure. (D) In vivo thermal image of PTT of single flank A20 lymphoma tumor on BALB/c mouse. After injection of 1 mg of CpG/BNP to right flank, mice were irradiated for 2 minutes with NIR laser (808 nm) with 2-W output (361.2 mW/cm2). (E) Temperature curve of lymphoma tumor during and after PTT. (F) Photothermal ablation–mediated necrosis of mice skin. Scar notable 2 days after PTT that resolved after 14 days with no recurrence of the tumor.
Methods
Materials
Hydrochloroauric acid (Sigma, St Louis, MO), silver nitrate (Sigma), L-ascorbic acid (Sigma), and Milli-Q water were used to synthesize the BNPs) during the study. Sodium cholic acid was purchased from Pierce (Thermo Fisher Scientific, Waltham, MA). Amine-PEG-thiol (Creative PEGWorks, Chapel Hill, NC) was used to stabilize the BNP structure and create a positive charge. CpG 2395 oligodeoxynucleotides (InvivoGen, San Diego) were used to formulate CpG deoxynucleotides/BNPs. All the chemicals were analytic grade reagents and used without further purification.
Synthesis of BNPs with CpG deoxynucleotides
Highly branched GNPs (BNPs) were synthesized with a previously optimized bile acid–mediated high-yield 1-pot synthesis.24 During the magnetic stirring of 10 mL of sodium cholic acid in Milli-Q water (2 mM) for 350 rpm, 1 mL of hydrochloroauric solution (10 mM) and 150 μL of silver nitrate solution (10 mM) were subsequently added. Then, 150 μL of a 200-mM L-ascorbic acid solution was added to synthesize BNPs. The reaction mixture was stirred for an extra 20 seconds and left undisturbed for 2 hours. BNPs were purified by centrifugation 3 times in ethanol and 3 times in water (12 000 rpm; 15 minutes). BNPs were redispersed in fresh water with thiol-PEG–carboxylic acid and then dialyzed in a dialysis membrane (molecular weight cutoff, 3500 Da) against fresh water for 1 day.
Loading efficiency of CpG deoxynucleotides on BNPs
Various weight/weight ratios of CpG deoxynucleotides/BNPs (0.1, 0.4, and 0.8 mg of CpG oligodeoxynucleotides to 0.8 mg of BNPs) were mixed and incubated overnight at 4°C, shielded from light. Then, CpG deoxynucleotides/BNPs were removed by ultrafiltration (molecular weight cutoff, 10 kDa; Amicon-ultra; Sigma-Aldrich, Burlington, MA), and the residual CpG deoxynucleotides were collected (from the bottom of the tube). Residual CpG deoxynucleotides were quantified by measuring the optical density at 260 nm, corresponding to a standard curve.
NIR laser–induced photothermal properties of BNPs
A medical laser system (BWF5; B&W Tek, Inc., Newark, DE) was used to investigate photothermal characteristics in vitro and in vivo. Each sample was illuminated with a fiber-coupled NIR (808 nm) diode-laser (2 W/cm2) for 2 minutes. An infrared thermal camera (ICI7320P; Infrared Cameras Inc., Beaumont, TX) was used to measure the temperature of the samples.
Tumor response and immune characterization after treatment with CpG deoxynucleotides/PTT and CpG deoxynucleotides/RT in a dual-flank lymphoma model
For the dual-flank lymphoma model, A20 lymphoma cells in Matrigel were injected into the right flank of BALB/c mice or athymic nude mice (Charles River Laboratory) to establish a primary tumor. Three days later, a secondary tumor was established in the left flank. When the tumor reached 5 mm in diameter, PTT with CpG deoxynucleotides (sequence 2395) or RT (10 Gy) with CpG deoxynucleotides of the primary tumor was administered. Control groups included PTT without CpG deoxynucleotides, CpG deoxynucleotides only, BNPs with CpG deoxynucleotides without PTT, and phosphate-buffered saline (PBS). PTT was administered with an NIR laser (808 nm). Tumor sizes were recorded for 2 weeks every 2 days. Sixteen days after PTT or RT, BALB/c mice were euthanized, and flow cytometry was performed on the primary tumor, secondary tumor, spleen, and draining lymph nodes, analyzing for T cells (CD3, CD4, CD8, CD44, CD62L, CD25, FoxP3, and interferon-γ (IFN-γ); BD Biosciences) and DCs (CD11c, CD80, and CD86; BD Biosciences). Parts of the primary tumor, secondary tumor, and spleen were also collected and fixed for histologic sections and stained with hematoxylin and eosin by the Cancer Center pathology core facility. The slides were reviewed by an expert hematopathologist (A.B.).
Winn assay
T cells from spleens from untreated or CpG deoxynucleotide/PTT–treated A20 lymphoma tumor–bearing mice were isolated using EasySep Mouse T-Cell Isolation Kit (STEMCELL Technologies, Cambridge, MA). The T cells were mixed with A20 cells at a 25:1 ratio and implanted in naïve BALB/c mice. Tumor growth was monitored and measured every 2 to 3 days.
Statistics
Results are described as mean ± standard deviation, representing at least 3 independent experiments. A 2-tailed Student t test was applied to evaluate the significance of experiments by using GraphPad Prism software (La Jolla, CA).
Results
Characterization of CpG deoxynucleotides/BNPs for PTT
The positive surface charge of amine-terminated BNPs was +9.6 mV, and CpG deoxynucleotide loading changed the surface charge to approximately −20 mV in various CpG deoxynucleotide/BNP weight ratios (supplemental Figure 1). However, the hydrodynamic size, which is important for cell uptake and distribution, was not different at various ratios. Average hydrodynamic size of the CpG deoxynucleotide/BNP formulation remained ∼120 nm. CpG deoxynucleotide loading can be marginally increased by increasing the weight ratio of CpG deoxynucleotides/BNPs, but the loading efficiency of CpG deoxynucleotides is maximized at ∼50% (supplemental Figure 1).
Importantly, CpG deoxynucleotides/BNPs had photothermal transduction properties. When irradiated with an NIR laser (at 808 nm), the aqueous CpG deoxynucleotide/BNP solution had an increased temperature of 41.3°C (Figure 1B). CpG deoxynucleotide/BNP–mediated photothermal heating was remarkable compared with laser-only controls (Figure 1C). Furthermore, the CpG deoxynucleotide/BNP complex was able to create enough heat to result in PTT in lymphoma-bearing BALB/c mice (Figure 1D). CpG/PTT was highly localized only to the region where the laser was irradiated and the NPs were located. PTT increased temperatures in the tumors up to 54.2°C after 2 minutes of laser exposure (Figure 1E). The initial heating rate of CpG deoxynucleotides/BNPs was 0.73°C/s, which was similar to that of bare BNPs when irradiated by the NIR laser (808 nm; 0.47 W/cm2). The calculated photothermal transduction efficiency for the CpG deoxynucleotides/BNPs was 61%, which is ∼2 times higher than that of alternative photothermal agents. The sufficient CpG deoxynucleotide/BNP–mediated PTT left a burn mark on the tumor after 2 weeks (Figure 1F).
Evaluation of tumor response and immunologic profile in the microenvironment after PTT using CpG/BNPs and CpGs with RT
Prepared CpG deoxynucleotides/BNPs were used to investigate the potential abscopal effect of the combination of CpG deoxynucleotides and PTT treatment. Subsequently, immune and tumor responses were compared with those seen with the combination of CpG deoxynucleotides and RT in the A20 lymphoma dual-flank mouse model described above (Figure 2A). A primary tumor was implanted in the left flank on day −14, and a secondary tumor was implanted in the right flank on day −7. Then, on day 0, the primary tumor was treated with CpG deoxynucleotides/PTT, CpG deoxynucleotides/RT, or a control, such as PBS (control), PTT only, or CpG deoxynucleotides only (Figure 2A). For the primary tumor, CpG deoxynucleotides/PTT resulted in a significantly lower tumor volume at 0.17 ± 0.18 cm3 on day 16 compared with CpG deoxynucleotides/RT (0.50 cm3; P < .05), PTT only (0.54 cm3; P < .01), CpG deoxynucleotides only (1.00 cm3; P < .05), and control (1.94 cm3; P < .001; Figure 2B). Interestingly, for the secondary tumor, which was untreated, the CpG deoxynucleotide/PTT group again had the lowest tumor volume (0.29 cm3) compared with the CpG deoxynucleotide/RT (0.83 cm3; P < .001), PTT-alone (0.687 cm3; P < .1), CpG deoxynucleotide–alone (0.85 cm3; P < .05), and control groups (1.65 cm3; P < .001; Figure 2C). The tumors treated with CpG deoxynucleotides/PTT were also visually smaller compared with the other treatment groups (Figure 2D).
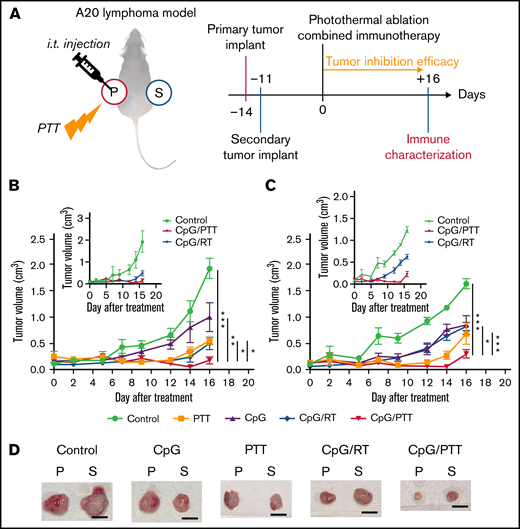
PTT in dual-flank A20 lymphoma model. (A) Schematic illustration and schedule of the experimental model. Tumor-inhibition efficacy was monitored for 16 days after photothermal ablation combined immunotherapy. Local and systemic immunotherapies were evaluated from primary and secondary tumors. (B) Tumor growth curves of primary tumor (treated). Inset graph focuses on difference between PTT and RT. (C) Tumor growth of secondary tumor (untreated). Inset graph focuses on difference between PTT and RT. (D) Representative resected primary (p) and secondary (s) tumors for each experimental group. Scale bar, 1 cm. Data were obtained from at least 6 independent samples. Data are shown as means ± standard errors of the mean. *P < .05, **P < .01, ***P < .001 (2-tailed paired t test). i.t., intratumoral; Treg, regulatory T cell.
PTT in dual-flank A20 lymphoma model. (A) Schematic illustration and schedule of the experimental model. Tumor-inhibition efficacy was monitored for 16 days after photothermal ablation combined immunotherapy. Local and systemic immunotherapies were evaluated from primary and secondary tumors. (B) Tumor growth curves of primary tumor (treated). Inset graph focuses on difference between PTT and RT. (C) Tumor growth of secondary tumor (untreated). Inset graph focuses on difference between PTT and RT. (D) Representative resected primary (p) and secondary (s) tumors for each experimental group. Scale bar, 1 cm. Data were obtained from at least 6 independent samples. Data are shown as means ± standard errors of the mean. *P < .05, **P < .01, ***P < .001 (2-tailed paired t test). i.t., intratumoral; Treg, regulatory T cell.
Sections of the primary and secondary tumors were harvested for pathologic evaluation. Upon review by a hematopathologist (A.B.), the histology of these murine A20 tumors was morphologically similar to those of aggressive large-cell lymphomas in humans (supplemental Figure 2). Compared with the control group, the primary tumors of the CpG deoxynucleotide/PTT group showed mainly fibrosis and immune infiltration.
Taken together, these data demonstrate that PTT has stronger antilymphoma effects compared with RT when combined with immune stimulants, not only at the treated site but also at untreated sites, suggesting an enhanced in situ vaccination response.
Adaptive immune response in primary tumor: CpG deoxynucleotides/PTT to the primary tumor induced an increase in CTL tumor infiltration and reduction in CD4+ T cells and Tregs
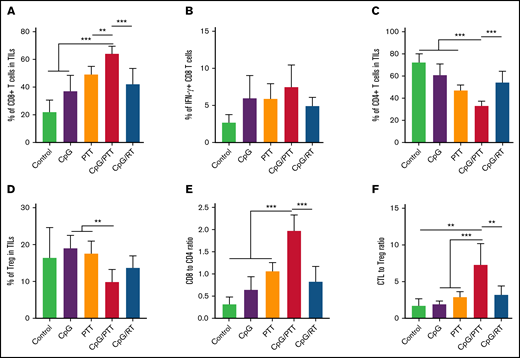
To further investigate the mechanism for the improved antilymphoma effect in the primary tumor after CpG deoxynucleotides/PTT, we compared the differences in immune response between the CpG deoxynucleotide/PTT and other treatment groups at day 16 (Figure 3). After CpG deoxynucleotide/PTT treatment, there was an increase in the CD8+ T-cell count (70.78%) in the primary tumor compared with CpG deoxynucleotides/RT treatment (48.97%; P < .001) and various controls (PBS, 23.24%; CpG deoxynucleotides, 28.82%; and PTT, 47.86%; Figure 3A). Within the CD8+ T-cell population, there was an increase in the percentage of IFN-γ+ CD8+ T cells in the CpG deoxynucleotide/PTT–treated sites (25.53%) compared with the CpG deoxynucleotide/RT–treated sites (16.88%; P < .01), suggesting an increase in CTL tumor infiltration (Figure 3B). Furthermore, the CpG deoxynucleotide/PTT–treated sites had a lower CD4+ T-cell count (29.87%) compared with those treated with CpG deoxynucleotides/RT (53.65%; P < .001) and controls (PBS, 71.64%; CpG deoxynucleotides, 65.74%; and PTT, 46.90%; Figure 3C).

Immune profile of primary tumor at day 16. (A) Population of CD8+ T cells in tumor-infiltrating lymphocytes (TILs). (B) Population of IFN-γ+ cytotoxic T lymphocytes (CTLs) in CD8+ population. (C) Population of CD4+ T cells in TILs. (D) Population of CD3+CD4+CD25+FoxP3+ Tregs in TILs. (E) CD8+/CD4+ T-cell ratio in TILs. (F) CTL/Treg ratio in TILs to evaluate immunologic balance of tumor after therapy. Data were obtained from at least 5 independent samples. Data are shown as means ± standard deviations. *P < .05, **P < .01, ***P < .001 (2-tailed paired t test).
Immune profile of primary tumor at day 16. (A) Population of CD8+ T cells in tumor-infiltrating lymphocytes (TILs). (B) Population of IFN-γ+ cytotoxic T lymphocytes (CTLs) in CD8+ population. (C) Population of CD4+ T cells in TILs. (D) Population of CD3+CD4+CD25+FoxP3+ Tregs in TILs. (E) CD8+/CD4+ T-cell ratio in TILs. (F) CTL/Treg ratio in TILs to evaluate immunologic balance of tumor after therapy. Data were obtained from at least 5 independent samples. Data are shown as means ± standard deviations. *P < .05, **P < .01, ***P < .001 (2-tailed paired t test).
Within the CD4+ T cells, there were no differences in Th1 (IFN-γ+), Th2 (IL-4+), and Th17 (IL-17+) populations between untreated (4% ± 2.1%, 5.2% ± 3.5%, and 0.55% ± 0.3%, respectively) and CpG deoxynucleotide/PTT–treated (3.7% ± 2.4%, 4.6% ± 1%, and 0.83% ± 0.26%, respectively) mice (supplemental Figure 3A-C). The results represented here are shown as percentages of immune cells in the parent populations. Future work will include absolute numbers to better understand the changes in the tumor immune microenvironment from the CpG deoxynucleotide/PTT therapy.
The CpG deoxynucleotide/PTT–treated sites also had the lowest percentage of Tregs (7.09%) compared with those treated with CpG deoxynucleotides/RT (12.04%; P < .05; Figure 3D). Therefore, CpG deoxynucleotide/PTT treatment resulted in significantly higher CD8/CD4 ratio (2.44; Figure 3E) and CTL/Treg ratio (10.47), especially when compared with CpG deoxynucleotide/RT treatment (0.94 and 4.61, respectively; Figure 3F). In addition, CpG deoxynucleotide/PTT treatment resulted in the highest percentage of mature DCs (measured as CD80+CD86+ in CD11c+ cells) compared with all other groups (Figure 4A).

DC maturation increases with PTT. (A) Percentage of DC maturation in primary (treated) tumor. (B) Percentage of DC maturation in secondary (untreated) tumor. Data were obtained from at least 5 independent samples. Data are shown as means ± standard deviations. *P < .05, ***P < .001 (2-tailed paired t test).
DC maturation increases with PTT. (A) Percentage of DC maturation in primary (treated) tumor. (B) Percentage of DC maturation in secondary (untreated) tumor. Data were obtained from at least 5 independent samples. Data are shown as means ± standard deviations. *P < .05, ***P < .001 (2-tailed paired t test).
To investigate T-cell dependence in the CpG deoxynucleotide/PTT treatment, we used an athymic nude mouse model implanted with A20 lymphoma cells. On the secondary tumor side, we found no difference between the CpG deoxynucleotide/PTT treatment group and the control group (supplemental Figure 4). Furthermore, the primary tumor growth after CpG deoxynucleotide/PTT therapy was not as suppressed compared with that in the control mice in this immunodeficient model. In addition, we combined T cells isolated from splenocytes after CpG deoxynucleotide/PTT therapy in A20 lymphoma–bearing BALB/c mice with A20 cells and implanted the mixture subcutaneously (Winn assay). None of the mice with CpG deoxynucleotide/PTT–treated T cells grew tumors. All 4 mice with control mouse T cells had tumor growth (P < .01; supplemental Figure 5). These 2 experiments suggest that the abscopal effect from PTT deoxynucleotide/CpG therapy is T-cell dependent.
Taken together, these data suggest that the burn from PTT with CpG deoxynucleotide–loaded BNPs was T-cell dependent and generated a stronger antilymphoma response and more favorable immune microenvironment compared with that from RT with CpG deoxynucleotide–loaded BNPs.
Adaptive immune response in the secondary untreated tumor
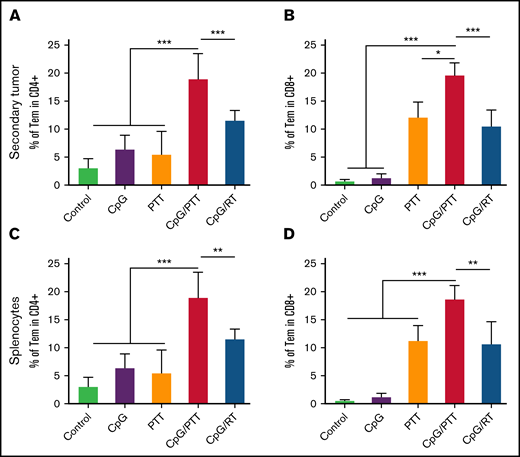
As with the primary tumor, we profiled the immune population of the secondary untreated tumor to evaluate the improved abscopal effect with CpG deoxynucleotides/PTT (Figure 5). We found that CpG deoxynucleotides/PTT led to an increase in CD8+ T-cell tumor infiltration (64.45%) compared with CpG deoxynucleotides/RT (42.27%; P < .001) and other controls (PBS, 22.12%; CpG deoxynucleotides, 37.36%; and PTT, 49.54%; Figure 5A). Concomitantly, there was a reduction in CD4+ T cells (33.28%) in the CpG deoxynucleotide/PTT group compared with that in the CpG deoxynucleotide/RT (54.43%; P < .001) and control groups (PBS, 72.68%; CpG deoxynucleotides, 61.14%; and PTT, 47.32%; Figure 5C). As in the primary tumor, within the CD4+ T cells, there was no difference in Th1, Th2, and Th 17 populations between control (2.6% ± 1.4%, 3.4% ± 1.6%, and 0.48% ± 0.16%, respectively) and CpG deoxynucleotide/PTT–treated (4.5% ± 1.5%, 5.4% ± 2%, and 0.54% ± 0.25%, respectively) mice (supplemental Figure 3D-F). In contrast, to the primary tumor, the percentages of IFN-γ CD8+ T cells and Tregs were not significantly different between the CpG deoxynucleotide/PTT and CpG deoxynucleotide/RT group (Figure 5B). However, the CpG deoxynucleotide/PTT group showed the highest CD8/CD4 ratio (1.89; Figure 5E). When looking at the Treg and CTL/Treg ratio, the CpG deoxynucleotide/PTT group had a significantly higher CTL/Treg ratio (7.29) compared with CpG deoxynucleotide/RT (3.22), PBS, (1.71), CpG deoxynucleotide (1.94), and PTT (2.91) groups (P < .01; Figure 5D-F). The CpG deoxynucleotide/PTT group also had the highest percentage of mature DCs in the untreated secondary tumor compared with all other groups (Figure 4B). Overall, these data indicate that PTT resulted in an increased systemic immune response and improved antilymphoma effect in the secondary untreated tumor that was related to an improved T-cell response, especially compared with RT.
Immune profile of secondary tumor at day 16. Systemic memory immune response of primary A20 lymphoma affects immunologic status of secondary A20 lymphoma. (A) Population of CD8+ T cells in tumor-infiltrating lymphocytes (TILs). (B) Population of IFN-γ+ CTLs in CD8+ population. (C) Population of CD4+ T cells in TILs. (D) Population of CD3+CD4+CD25+FoxP3+ Tregs in TILs. (E) CD8+/CD4+ T-cell ratio in TILs. (F) CTL/Treg ratio in TILs to evaluate immunologic balance of tumor after therapy. Data were obtained from at least 5 independent samples. Data are shown as means ± standard deviations. **P < .01, ***P < .001 (2-tailed paired t test).
Immune profile of secondary tumor at day 16. Systemic memory immune response of primary A20 lymphoma affects immunologic status of secondary A20 lymphoma. (A) Population of CD8+ T cells in tumor-infiltrating lymphocytes (TILs). (B) Population of IFN-γ+ CTLs in CD8+ population. (C) Population of CD4+ T cells in TILs. (D) Population of CD3+CD4+CD25+FoxP3+ Tregs in TILs. (E) CD8+/CD4+ T-cell ratio in TILs. (F) CTL/Treg ratio in TILs to evaluate immunologic balance of tumor after therapy. Data were obtained from at least 5 independent samples. Data are shown as means ± standard deviations. **P < .01, ***P < .001 (2-tailed paired t test).
Measurement of systemic CD4+ and CD8+ effector memory T cells
To further analyze the systemic response of CpG deoxynucleotides/PTT, we measured effector memory T cells in secondary untreated tumor and spleen and found increased secondary memory T cells with CD3+, CD4+, CD44+, and CD62L− lymphocytes (Figure 6), which is important for systemic antitumor responses. In the secondary tumor, the CD4+ effector memory T cells (18.95%) were significantly higher compared with those in CpG deoxynucleotide/RT (11.56%; P < .01) and control groups (PBS, 3.04%; CpG deoxynucleotides, 6.39%; and PTT, 5.48%; Figure 6A). The CD8+ effector memory T cells were also higher (18.69%) compared with those in CpG deoxynucleotide/RT (10.68%; P < .01) and control groups (PBS, 0.54%; CpG deoxynucleotides, 1.17%; and PTT, 11.26%; Figure 6B). We found the same results in the spleen, with an increase in CD4+ effector memory T cells (15.08%) compared with CpG deoxynucleotide/RT (9.95%; P < .05) and control groups (control, 3.18%; CpG deoxynucleotides, 5.85%; and PTT, 10.29%; Figure 6C), as well as in CD8+ effector memory T cells (30.18%) compared with CpG deoxynucleotide/RT (20.83%; P < .001) and control groups (PBS, 0.82%; CpG deoxynucleotides, 2.17%; and PTT, 23.37%; Figure 6D).
Systemic immunologic memory response from photothermal ablation combined immunotherapy. (A) Population of CD3+CD4+CD44highCD62Llow effector memory CD4 T cells (CD4 Tems) in tumor-infiltrating lymphocytes (TILs) of secondary tumor. (B) Population of CD3+CD8+CD44highCD62Llow CD8 Tems in TILs secondary tumor. (C-D) Population of CD4 Tems (C) and CD8 Tems (D) in the spleen. Data were obtained from at least 5 independent samples. Data are shown as means ± standard deviations. *P < .05, **P < .01, ***P < .001 (2-tailed paired t test).
Systemic immunologic memory response from photothermal ablation combined immunotherapy. (A) Population of CD3+CD4+CD44highCD62Llow effector memory CD4 T cells (CD4 Tems) in tumor-infiltrating lymphocytes (TILs) of secondary tumor. (B) Population of CD3+CD8+CD44highCD62Llow CD8 Tems in TILs secondary tumor. (C-D) Population of CD4 Tems (C) and CD8 Tems (D) in the spleen. Data were obtained from at least 5 independent samples. Data are shown as means ± standard deviations. *P < .05, **P < .01, ***P < .001 (2-tailed paired t test).
Within the spleen, there was an increase in CD8+ T cells (32.4% ± 1%) and a decrease in CD4+ T cells (68% ± 1.8%) after CpG deoxynucleotide/PTT therapy compared with untreated controls (28.7% ± 2.8% and 63% ± 1%, respectively; Figure 7A,F). Among the CD4+ T cells, there was a notable reduction in the Th2 population (3% vs 1.7% controls) and Tregs (5.3% vs 2.8% controls), whereas no difference was noted in the Th1 (1% vs 1.2% control) and Th17 populations (0.32% vs 0.34% control; Figure 7B-E). The reductions in Th2 and Tregs suggest a more favorable proinflammatory systemic environment. In addition, within the CD8+ T cells, there was an increase in granzyme-expressing (1.84% ± 0.3% vs 1.3% ± 0.3% control) and dual granzyme and IFN-γ–expressing CTLs (0.86% ± 0.08% vs 0.67% ± 0.13%; Figure 7G-I). Granzyme secretion is important for direct killing of tumors and anticancer effects,25,26 whereas the increase in polyfunctional CD8+ T cells suggests a stronger antilymphoma effect.27,28 Overall, these data show an exaggerated T-cell education, which consequently results in suppression of lymphoma tumor growth.
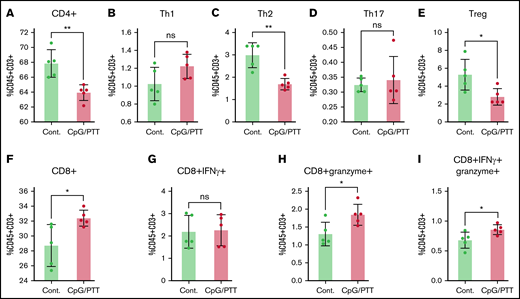
T-cell profiling of splenocytes after CpG deoxynucleotide/PTT therapy. (A-I) Populations of CD4+ T cells (A), Th1 T cells (CD4+ IFN-γ) (B), Th2 T cells (CD4+ IL-4) (C), Th17 T cells (CD4+ IL-17) (D), Tregs (CD4+CD25+FOXP3+) (E), CD8+ T cells in total T cells (F), IFN-γ–expressing CD8+ T cells (G), granzyme-expressing CD8+ T cells (H), and dual-expressing CD8+ T cells in total T cells (CD45+CD3+) (I). *P < .05, **P < .01 (2-tailed paired t test). ns, not significant.
T-cell profiling of splenocytes after CpG deoxynucleotide/PTT therapy. (A-I) Populations of CD4+ T cells (A), Th1 T cells (CD4+ IFN-γ) (B), Th2 T cells (CD4+ IL-4) (C), Th17 T cells (CD4+ IL-17) (D), Tregs (CD4+CD25+FOXP3+) (E), CD8+ T cells in total T cells (F), IFN-γ–expressing CD8+ T cells (G), granzyme-expressing CD8+ T cells (H), and dual-expressing CD8+ T cells in total T cells (CD45+CD3+) (I). *P < .05, **P < .01 (2-tailed paired t test). ns, not significant.
Discussion
In the studies described herein, we found that CpG deoxynucleotides/PTT enhanced lymphoma response in an A20 syngeneic model compared with CpG deoxynucleotides/RT in both the primary treated tumor in one flank and secondary untreated tumor in the other flank. This was accompanied by an improved abscopal response, with a concomitant increase in CD8/CD4 and CTL/Treg ratios in both primary and secondary tumors compared with CpG deoxynucleotides/RT. DCs in the primary and secondary draining lymph nodes had increased maturation markers in the CpG deoxynucleotide/PTT group, and the effector memory T cells (both CD4 and CD8) in the secondary tumor and spleen were increased, suggesting a systemic vaccination effect.
The lack of systemic immune response to local RT (ie, abscopal response) is secondary to the immune-suppressive tumor microenvironment.29 Frank et al12 combined low-dose RT with CpG deoxynucleotides for the treatment of advanced-stage indolent lymphoma and saw encouraging responses. In that trial, the overall response rate was high (89.6%), but 7 of 29 patients (24%) had a partial response, and only 1 had a complete response (3%). CpG deoxynucleotides are synthetic oligodeoxynucleotides containing the unmethylated CpG motif that potently stimulates the innate immune system by activating toll-like receptor 9. CpG deoxynucleotides have been studied in depth for lymphoma therapy, pioneered by Sagiv-Barfi et al30 and several other research groups.31,32 In addition, CpG deoxynucleotides have been tested in other clinical trials in combination with anti-CD20 targeted therapies,33,34 with limited success because of the difficulty in delivering the CpG deoxynucleotides systemically. NP–CpG deoxynucleotide conjugates are currently being tested to improve the delivery of CpG deoxynucleotides and have been found to have improved cytotoxicity toward lymphomas,35 and we found that they resulted in greater immune stimulation.36
Our focus was to determine differences in the immune profile of the microenvironment in a lymphoma model with RT compared with PTT, both on a CpG deoxynucleotide–loaded BNP platform. We found that CpG deoxynucleotides/PTT had significant antilymphoma effects compared with CpG deoxynucleotides/RT, not only on the treated tumors but also on distant untreated tumors.
PTT is no longer a laboratory-only concept. A clinical pilot device has been used in prostate cancer with silica gold nanoshells producing localized photothermal ablation.37 CpG deoxynucleotides/PTT in our study generated a more favorable antilymphoma T-cell response with increased CD8/CD4 and CTL/Treg ratios in both primary and secondary tumors. This is in line with previous work showing an increased CD8/CD4 T-cell ratio after PTT in melanoma models.13 Furthermore, we found increased effector memory T cells (CD4+ and CD8+) in both the secondary untreated tumor and spleen, suggesting a successful systemic immune effect. Clinically, higher levels of memory T cells were found to correlate with better overall and disease-free survival in patients with colon cancer.38
Tumor-infiltrating effector memory T-cell levels correlated with response to checkpoint inhibitors in a murine model.39 Also, an increase in circulating memory T cells was found to result in improved clinical responses to checkpoint inhibitors in non–small-cell lung cancer.40 The combination of checkpoint inhibition with PTT has shown great promise in several solid tumor mouse models.41-44 Therefore, further investigation of CpG deoxynucleotides/PTT in combination with checkpoint inhibitor treatment seems warranted.
Prior studies using PTT have focused on solid tumor animal models, and there is very little information about the role of PTT in hematologic malignancies. Here, we describe immune profiles after PTT using a BNP platform in a lymphoma model, but there are many NP types that can enhance PTT, such as silica gold nanoshells, hollow gold nanoshells, nanorods, and NIR absorbing dyes. We were able to characterize and fabricate an NP platform that could sufficiently carry CpG deoxynucleotides while maintaining the ability to generate heat with PTT.
The BNP synthesis process used here is biocompatible, because there are no synthetic surfactants, there is high photothermal conversion efficiency, and there is a high payload for therapeutic delivery, all of which translate to feasible clinical trials. The synthesis of BNPs was scaled up to allow the production of a large amount of colloidal BNPs in a short reaction period of <1 minute, which is important for large-batch manufacturing for clinical use. Multibranches of BNPs provide a space for the DNA to be carried and hide from DNases to prevent degradation. This extraordinarily high photothermal transduction should be particularly advantageous for targeted treatment of tumors.
The immune status of A20 tumor–bearing mice can be an important consideration for the validity of the animal model. A20 lymphoma models with or without Matrigel have been established in order to evaluate various immune-based therapeutics, and the tumor immune environment is well described.30,45 The reported immune status (measured by CD4/CD8 ratio) was comparable to our results (2:1) in the tumor (Figures 3 and 4).46 In addition, stroma cells in the tumor microenvironment are important for lymphoma growth.47 Bascuas et al46 noted that A20 lymphoma is a reasonable model to mimic human NHL, and the histologic appearance of the A20 lymphoma tumors resembled that of aggressive large-cell lymphomas in humans. However, even though the A20 lymphoma model is widely used, there is a reasonable argument that the A20 lymphoma model is not fully representative of the complex NHL ecosystem. Because PTT has the capability to destroy the extracellular matrix, this observation suggests that the combination of PTT with drugs that target the stromal cells would be a fruitful path for future studies.
Although our data suggest that the abscopal response is a T cell–mediated process, further characterization, in addition to the athymic mouse model and Winn assays that we describe, is warranted. Studies using cytokine neutralization or cell-depletion strategies will help elucidate the specific immune mechanism for the antilymphoma response and are part of future experimental designs. We also plan to use other more clinically relevant syngeneic murine lymphoma models, including transgene-driven models (Emu-Myc or EZH2 mutated), to delineate the specific molecular or cellular pathways involved with the PTT platform.
Finally, we note that the improved antitumor effects were transient, and the tumors eventually grew out without additional treatment. We noticed a slight increase in tumor growth at day 16 (when we had to euthanize the animals based on our Institutional Animal Care and Use Committee protocol). This was an aggressive lymphoma model, and treatment with CpG deoxynucleotides requires at least 3 doses, whereas the CpGdeoxynucleotide/PTT treatment occurred only once.30 The response to the CpG deoxynucleotide/PTT therapy would likely have been more durable for indolent lymphomas, which are more sensitive to in situ vaccination strategies.12 Similar to other vaccines in cancer therapy, PTT technology will need to be combined other immune therapeutics, such as checkpoint inhibitors, CAR T cells, or bispecific antibodies, because CpG deoxynucleotides/PTT improve antilymphoma T-cell responses. Combination therapy is the major focus of our future work.
In conclusion, we demonstrated that CpG deoxynucleotides/PTT generated a stronger immune response in the microenvironment of a lymphoma model when compared with CpG deoxynucleotides/RT. In addition, we showed increased DC maturation and T cell–mediated immune responses. Additional studies, including dosing schedule, combination therapy with checkpoint inhibitors or other immune-based treatments, and experiments to better delineate the specific molecular and cellular pathways involved, are warranted.
Acknowledgments
This work was supported by Robert H. Lurie Comprehensive Cancer Center (RHLCCC) Support Grant CA060553 from the National Cancer Institute, National Institutes of Health, the Translational Bridge Program, and National Institute of Biomedical Imaging and Bioengineering (NIBIB) grant R01EB026207. A.Y.L. was supported by the American Society of Hematology Research Training Award for Fellows and the Lymphoma Research Foundation Postdoctoral Fellowship Grant. L.I.G. was supported by the RHLCCC Support Grant and the Shannahan, Brookstone, and Mander Foundations.
Authorship
Contribution: A.Y.L. was the lead scientist for the project and initiated the project idea, designed and oversaw the experiments, and wrote most of this manuscript; B.C. performed the animal and flow cytometric experiments and analyzed the data; E.Y. performed additional animal and flow cytometric experiments and analyzed the data; T.S. performed the nanoparticle characterization; H.C. assisted with the animal experiments; A.B. reviewed and commented on the histologic slides; and D.-H.K. and L.I.G. funded the project and assisted in experiment design, interpretation of the data, and editing of this manuscript.
Conflict-of-interest disclosure: A.Y.L. has consulted for PresiCa. A.B. has received honoraria for speakers’ bureau from Eli Lilly, Foundation Medicine/Roche China, Thermo Fisher Scientific, and Bayer. L.I.G. has served on advisory boards for Kite/Gilead, Juno/Celgene/Bristol Myers Squibb, and Bayer, has received research funding from Juno/Celgene/Bristol Myers Squibb, and is a cofounder of Zylem, Inc. The remaining authors declare no competing financial interests.
Correspondence: Dong-Hyun Kim, Department of Radiology, Northwestern University Feinberg School of Medicine, 737 N Michigan, Suite 1600, Chicago, IL 60611; e-mail: dhkim@northwestern.edu; and Leo I. Gordon, Division of Hematology Oncology, Department of Medicine, Northwestern University Feinberg School of Medicine, Arkes Family Pavilion Suite 850, 676 N Saint Clair, Chicago, IL 60611; e-mail: l-gordon@northwestern.edu.

