Key Points
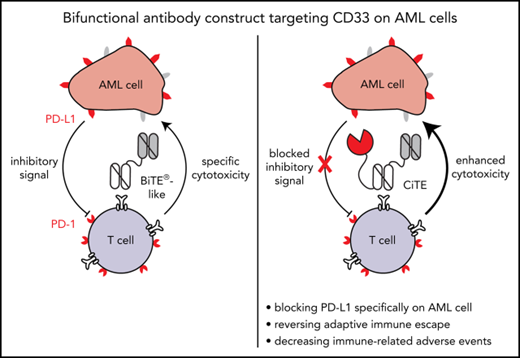
Characterization of an αPD-L1 × αCD3 × αCD33 antibody construct with bifunctional activity against AML cells.
Strong cytotoxicity against primary AML cells in vitro and high selectivity in a xenograft mouse model.

Abstract
The CD33-targeting bispecific T-cell engager (BiTE) AMG 330 proved to be highly efficient in mediating cytolysis of acute myeloid leukemia (AML) cells in vitro and in mouse models. Yet, T-cell activation is correlated with upregulation of programmed cell death-ligand 1 (PD-L1) and other inhibitory checkpoints on AML cells that confer adaptive immune resistance. PD-1 and PD-L1 blocking agents may counteract T-cell dysfunction, however, at the expense of broadly distributed immune-related adverse events (irAEs). We developed a bifunctional checkpoint inhibitory T cell–engaging (CiTE) antibody that combines T-cell redirection to CD33 on AML cells with locally restricted immune checkpoint blockade. This is accomplished by fusing the extracellular domain of PD-1 (PD-1ex), which naturally holds a low affinity to PD-L1, to an αCD3.αCD33 BiTE-like scaffold. By a synergistic effect of checkpoint blockade and avidity-dependent binding, the PD-1ex attachment increases T-cell activation (3.3-fold elevation of interferon-γ) and leads to efficient and highly selective cytotoxicity against CD33+PD-L1+ cell lines (50% effective concentration = 2.3-26.9 pM) as well as patient-derived AML cells (n = 8). In a murine xenograft model, the CiTE induces complete AML eradication without initial signs of irAEs as measured by body weight loss. We conclude that our molecule preferentially targets AML cells, whereas high-affinity blockers, such as clinically approved anticancer agents, also address PD-L1+ non-AML cells. By combining the high efficacy of T-cell engagers with immune checkpoint blockade in a single molecule, we expect to minimize irAEs associated with the systemic application of immune checkpoint inhibitors and suggest high therapeutic potential, particularly for patients with relapsed/ refractory AML.
Introduction
The treatment of acute myeloid leukemia (AML) remains challenging in 2018. Only one-half of the patients are eligible for curative intensive induction chemotherapy, and the majority will relapse because of the persistence of chemoresistant leukemic stem cells. Allogeneic hematopoietic stem cell transplantation as postremission therapy is able to lower this risk, yet it is correlated with a significant incidence of transplant-related morbidity and mortality.1-4 Particularly patients with relapsed or refractory (r/r) disease as well as patients that are medically not fit for intensive treatment regiments urgently require new therapeutic approaches.
In acute lymphoblastic leukemia (ALL), several targeted immunotherapies have already reached clinical implementation as standard treatment. With the approval of the bispecific T-cell engager (BiTE) blinatumomab in 2014, the utilization of T cells as immune effectors also entered clinical mainstream.5 This bispecific molecule addresses CD19 on B cells and thus redirects antigen-experienced T cells to leukemic cells.6,7 In AML, the myeloid lineage antigen CD33 has been the focus of immunotherapeutic strategies for decades. Targeting CD33 by the antibody-drug conjugate gemtuzumab ozogamicin (Mylotarg) has proven to be safe and led to reapproval for the treatment of adults at primary diagnosis as well as adults and children with r/r disease.8 Also, the preclinical evaluation of the BiTE antibody AMG 330 indicated efficient cytotoxic lysis of primary AML patient samples in allogeneic and autologous settings and entered clinical trials in August 2015 (NCT02520427).9-12
However, T-cell–recruiting immunotherapy is accompanied by the induction of adaptive immune escape mechanisms such as programmed cell death-ligand 1 (PD-L1) upregulation in response to proinflammatory cytokines.13,14 Recent studies were able to directly correlate PD-L1 expression on cell lines with a decrease in AMG 330–mediated cytotoxicity and demonstrated that this effect could be abrogated by PD-L1 blockade.15 We could underpin these findings ex vivo on primary AML patient cells, in which the efficacy of AMG 330 was enhanced by complementation with PD-1/PD-L1 blocking monoclonal antibodies (mABs).13
PD-1 and PD-L1 inhibitors are approved for the treatment of solid cancers, and clinical trials are currently exploring these agents in hematologic malignancies.16-18 So far, monotherapy has shown limited clinical benefit and current strategies explore combinatorial approaches with hypomethylating agents. First data of a clinical phase 1B/2 study with the αPD-1 mAB nivolumab in combination with azacytidine in patients with r/r AML demonstrated encouraging median overall survival rates of 5.7 months (NCT02397720).19
Yet, the clinical application of PD-1 and PD-L1 blocking mABs is hampered by the frequent occurrence of immune-related adverse events (irAEs). These include skin disorders, colitis, hepatitis, endocrinopathies, pneumonitis, and myocarditis and range from weak to severe or fatal toxicity.20-25 Medical intervention can require treatment interruption or discontinuation and immune suppression with corticosteroids.26
To combine the benefits of bispecific T cell–engaging molecules with PD-1/PD-L1 checkpoint blockade and prevent on-target off-leukemia events, we have developed a novel immunotherapeutic format. Bifunctional checkpoint inhibitory T cell–engaging (CiTE) antibodies consist of a high-affinity αCD33 single-chain variable fragment (scFv) fused to an αCD3ε scFv in 1 polypeptide chain. Additionally, we attached the extracellular domain (amino acid 33-149) of PD-1 (PD-1ex), which intrinsically holds a low-affinity to PD-L1. We hypothesized that the PD-1ex domain is not sufficient to directly target PD-L1–expressing cells and does not block PD-1/PD-L1 interactions unspecifically. Instead, we aimed that PD-L1 blockade is thus dependent on αCD33 scFv-mediated targeting, which would consequently restrict checkpoint blockade to the surface of leukemic cells. A single-chain triplebody (sctb),27 in which the PD-1ex module is replaced by an αPD-L1 scFv, served as high-affinity control.
Our data reveal that the CiTE antibody binds to AML and T cells, increases T-cell effector functions compared with a BiTE-like molecule, and induces efficient cancer cell eradication. Notably, in vitro the CiTE demonstrates a high selectivity for CD33+PD-L1+ cells, whereas PD-L1+ cells are not affected. This is further supported in a murine model system, where no indication for the development of irAEs because of on-target off-leukemia binding of the cross-reactive PD-1ex could be detected. Contrarily, the sctb also leads to the depletion of PD-L1+ cells in vitro as well as body weight loss and leukemia-unrelated PD-1 upregulation in vivo. Therefore, we consider the new CiTE format a promising therapeutic approach to treat patients with AML with high efficacy and minimize the risk to induce irAEs that are associated with systemic immune checkpoint blockade.
Methods
Expression and purification
PD-1ex was amplified from human muscle complementary DNA (cDNA; PDCD1 gene). The αPD-L1 scFv was published before (YW243.55.S70, atezolizumab-derived) with variable light and variable heavy chains connected by a (G4S4)4 linker.28 The OKT3-based αCD3 scFv and hP67.6-derived αCD33 scFv were obtained from published sequences.29,30 Coding sequences for CiTE, sctb, and controls were cloned into the expression vector pSecTag2/HygroC (Thermo Fisher Scientific, Waltham, MA) containing a His6-tag. As control, PD-1ex was fused to a C-terminal human IgG1-Fc. These molecules and the specificity control27 were expressed in FreeStyle 293-F or Expi293F cells (Thermo Fisher Scientific). The αPD-L1 scFv was cloned into the pAK40031 vector and expressed in Escherichia coli BL21 cells (NEB, Ipswich, MA). Proteins were purified by nickel affinity and size exclusion chromatography (SEC) using Superdex 200 increase 10/300 or Superdex 75 10/300 columns (GE Healthcare, Little Chalfont, UK) in 20 mM histidine and 300 mM NaCl (pH 6.5). Proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analytical SEC (Superdex 200 increase 5/150, GE Healthcare). For mouse studies, proteins were prepared in 1× DPBS (Thermo Fisher Scientific) and endotoxin levels were confirmed to be <5 EU/kg per day.32 Stability was measured by fluorescence-based thermal shift (ThermoFluor) assay using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA).33
Cell lines
All cell lines were cultivated at standard conditions. Flp-In T-REx 293 cells (Thermo Fisher Scientific) were modified for expression of human PD-L1 and CD33 (HEK:PD-L1 and HEK:CD33:PD-L1), which could be enhanced by tetracycline induction (HEK:PD-L1_ind.). MOLM-13, OCI-AML3, BA/F3, and Jurkat cells were purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (Leibniz-Institut DSMZ, Braunschweig, Germany). Stable PD-L1–expressing cells were generated by retroviral transduction with cDNA of human PD-L1 (MOLM-13:PD-L1 and OCI-AML3:PD-L1) or murine PD-L1 (Panc02-OVA:mPD-L1), BA/F3:CD33:PD-L1 cells by further transduction with cDNA of human CD33.34
Patient and healthy donor material
After written informed consent in accordance with the Declaration of Helsinki and approval by the Institutional Review Board of the Ludwig-Maximilians-Universität (Munich, Germany), peripheral blood or bone marrow (BM) samples were collected from healthy donors (HDs) and AML patients. At initial diagnosis or relapse, samples were analyzed at the Laboratory for Leukemia Diagnostics of the Klinikum der Universität München as described previously.35-37 Patient characteristics are summarized in supplemental Table 2, available on the Blood Web site.
Flow cytometry
Flow cytometry measurements were performed on a Guava easyCyte 6HT instrument (Merck Millipore, Burlington, MA) and analyzed using GuavaSoft, version 3.1.1 (Merck Millipore) or on an LSR II flow cytometer (BD Biosciences, Franklin Lakes, NJ) and data were evaluated using FlowJo, version 9.6 (Tree Star Inc., Ashland, OR). Commercial antibodies are listed in the supplemental Methods. Surface antigen density of cell lines was evaluated with QIFIKIT (Agilent Dako, Santa Clara, CA). Apparent dissociation constants were determined by calibrated flow cytometry as described.38 A total of 3.0 to 3.4 μm Rainbow Calibration particles (BioLegend, San Diego, CA) served as calibration control. Data points were normalized to the maximum mean fluorescence intensity and fitted to a 1-site specific binding model.
T-cell activation and cytotoxicity assays
HD T cells were incubated with target cell lines at an effector to target cell (E:T) ratio of 2:1, 1:3, or without targets in the presence of CiTE, sctb, and control molecules. Assays were performed in RPMI1640 + GlutaMAX supplemented with 10% fetal calf serum and penicillin/streptomycin (100 U/mL) (Thermo Fisher Scientific). BA/F3 medium included 10% WEHI-3B supernatant and 2.5 µg/mL αCD28 mAB (BD Pharmingen). Bead-immobilized αCD3/αCD28 antibodies (Thermo Fisher Scientific) served as positive control. After 96 hours, T-cell activation was assessed by flow cytometry quantifying the CD2+CD69+, CD2+CD25+, or CD2+PD-1+ population. For cytotoxicity readout, MOLM-13– and BA/F3-derived cells were directly applied, OCI-AML3:PD-L1 and OCI-AML3 were labeled with PKH67 (Sigma-Aldrich, St. Louis, MO). After 72 hours, total target cell numbers were assessed by flow cytometry as live CD2−CD33+ or CD2−PKH67+ population, respectively, and normalized to negative control. Data were transformed with a 4-parameter nonlinear fit model. Interferon-γ (IFN-γ) and Granzyme B release were determined after 72 hours by Cytometric Bead Array (Human IFN-γ/Granzyme B Flex Set, BD Biosciences).
HEK:PD-L1 and HEK:CD33:PD-L1 cells were labeled with 15 µM Calcein AM (Thermo Fisher Scientific). Preactivated T cells derived from an 18-day ex vivo peripheral blood mononuclear cell expansion were incubated with a 1:1 mixture of unlabeled HEK:PD-L1 and labeled HEK:CD33:PD-L1 cells and vice versa at a total E:T ratio of 2:1 and increasing concentrations of molecules.39 The 2.5% Triton X-100 served as maximum lysis. Fluorescence intensity was measured using an Infinite M100 plate reader (TECAN, Männedorf, Switzerland) and specific lysis was calculated and analyzed with a 4-parameter nonlinear fit model.
Specific lysis [%] = (fluorescence(sample) – fluorescence(spontaneous lysis))/(fluorescence(maximum lysis) – fluorescence(background)) × 100
Ex vivo redirected lysis assay of cocultured AML patient cells
Redirected lysis assays of AML patient samples were performed in α-MEM (Thermo Fisher Scientific) supplemented with 12.5% fetal calf serum, 12.5% horse serum, 1% penicillin/streptomycin/glutamine (Invitrogen, Carlsbad, CA), and a distinct cytokine cocktail on irradiated MS-5 cells as described elsewhere.10,13,40-42 HD T and AML cells were incubated at an E:T ratio of 1:4 and addition of 10 nM of molecules or 10 µg/mL αPD-L1 mAB (eBioscience Thermo Fisher Scientific). Cell populations were assessed by flow cytometry. Cytotoxicity and T-cell proliferation were evaluated as described previously.10,13
Murine AML xenograft studies
Female non-obese diabetic severe combined immunodeficiency γ (NSG) mice 170 to 265 days of age were housed under pathogen-free conditions at the research animal facility of the Helmholtz Zentrum München, Munich, Germany. Animal experiments were approved by regional regulating authorities (Regierung von Oberbayern) and conducted as described in a published protocol.43 At day 0, 2 × 104 MOLM-13:PD-L1 cells were injected IV. At day 3, 107 in vitro preactivated T cells were transferred intraperitoneally and mice were randomized into 5 groups: 3 treatment groups containing 6 mice each, a specificity control group of 4 mice, and a 1× DPBS control group of 5 mice. At day 4, 1.7 pmol of molecules/g body weight or 1× DPBS were daily IV injected until day 12. At day 13, mice were euthanized, spleens were removed, and BM was obtained from femora of hind legs.
Plotting and statistical analysis
Statistical evaluation was performed using GraphPad Prism version 6.07 (GraphPad Software Inc., San Diego, CA) applying unpaired Student t test with Welch correction for cell line–based assays with the same T-cell donors, Wilcoxon test for different HDs and patient samples, and Mann-Whitney U test for mouse xenograft experiments. If P < .05, results were considered statistically significant.
Results
Generation and characterization of the CiTE antibody
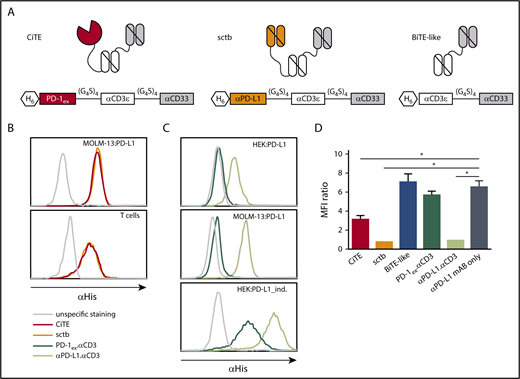
To combine specific T-cell redirection to AML cells with a target cell–restricted PD-1/PD-L1 blockade, we generated a CiTE antibody by fusing PD-1ex to an αCD3.αCD33 BiTE-like molecule. The CiTE was compared with a sctb,27 in which PD-1ex was replaced by a high-affinity αPD-L1 scFv. The BiTE-like molecule αCD3.αCD33, PD-1ex.αCD3 and αPD-L1.αCD3, as well as a nontargeting molecule served as controls (Figure 1A; supplemental Figure 1A). Purified proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analytical SEC (supplemental Figure 1B-C) and protein stability was assessed by fluorescence-based thermal shift assay (supplemental Figure 1D).
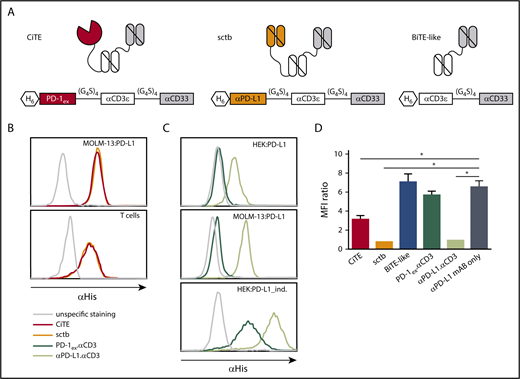
Schematic drawing and binding characteristics of CiTE, sctb, and control molecules. (A) Modular composition of CiTE, sctb, and BiTE-like molecule. (B) Binding analysis of CiTE and sctb to MOLM-13:PD-L1 cells and HD T cells at 15 ng/µL concentration by flow cytometry. (C) Binding analysis of PD-1ex (in PD-1ex.αCD3) and αPD-L1 scFv (in αPD-L1.αCD3) to HEK:PD-L1, MOLM-13:PD-L1, and HEK:PD-L1_ind. cells at 1.5 ng/µL concentration. Gray line indicates unspecific staining by the secondary antibody. Histograms show 1 of 3 experiments with similar results. (D) Binding of αPD-L1 mAB (clone MIH1) to MOLM-13:PD-L1 cells in the presence or absence of CiTE, sctb, or controls at 150 nM concentration as measured by flow cytometry. Mean values show n = 3 independent experiments with standard error of the mean (SEM) as error bars. For statistical analysis, unpaired Student t test with Welch correction was applied. *P < .05, **P < .01, ***P < .001. MFI, mean fluorescence intensity.
Schematic drawing and binding characteristics of CiTE, sctb, and control molecules. (A) Modular composition of CiTE, sctb, and BiTE-like molecule. (B) Binding analysis of CiTE and sctb to MOLM-13:PD-L1 cells and HD T cells at 15 ng/µL concentration by flow cytometry. (C) Binding analysis of PD-1ex (in PD-1ex.αCD3) and αPD-L1 scFv (in αPD-L1.αCD3) to HEK:PD-L1, MOLM-13:PD-L1, and HEK:PD-L1_ind. cells at 1.5 ng/µL concentration. Gray line indicates unspecific staining by the secondary antibody. Histograms show 1 of 3 experiments with similar results. (D) Binding of αPD-L1 mAB (clone MIH1) to MOLM-13:PD-L1 cells in the presence or absence of CiTE, sctb, or controls at 150 nM concentration as measured by flow cytometry. Mean values show n = 3 independent experiments with standard error of the mean (SEM) as error bars. For statistical analysis, unpaired Student t test with Welch correction was applied. *P < .05, **P < .01, ***P < .001. MFI, mean fluorescence intensity.
The binding properties and apparent dissociation constants of CiTE and sctb to antigen-presenting cells were analyzed by flow cytometry (supplemental Figure 2). When investigating CiTE and sctb as whole molecules, both bound similarly to CD33+PD-L1+ AML cell lines and HD T cells (Figure 1B). Because the unique feature of the CiTE format is the weak PD-1ex affinity to PD-L1, we evaluated the binding abilities of PD-1ex and the αPD-L1 scFv independently. To this end, MOLM-13 and tetracycline-inducible HEK293 cells both stably expressing PD-L1 (MOLM-13:PD-L1 and HEK:PD-L1) were quantified for their PD-L1 surface antigen density (supplemental Table 1A). As expected, our results showed weak physiological binding of PD-1ex (described in the low micromolar range)44,45 and comparably strong binding of the αPD-L1 scFv (Figure 1C).
Consequently, CiTE-mediated checkpoint inhibition on AML cells depends on avidity contribution of the CD33 targeting module. We performed a blocking assay with a labeled αPD-L1 mAB that interferes with the binding of the checkpoint modules. Despite the weak interaction of PD-1ex in comparison with the αPD-L1 scFv, the CiTE was able to block subsequent binding of the αPD-L1 mAB on CD33+PD-L1+ AML cells (Figure 1D). However, it was not as efficient as the sctb and the high-affinity αPD-L1.αCD3 control, which were able to completely occupy accessible PD-L1 surface molecules. In line with the binding studies, the low-affinity PD-1ex.αCD3 control was displaced by the αPD-L1 mAB. Thus, PD-1ex only interacts with its ligand on AML cells when it is covalently linked to a high-affinity leukemia-targeting arm.
CiTE-mediated activation of resting T cells
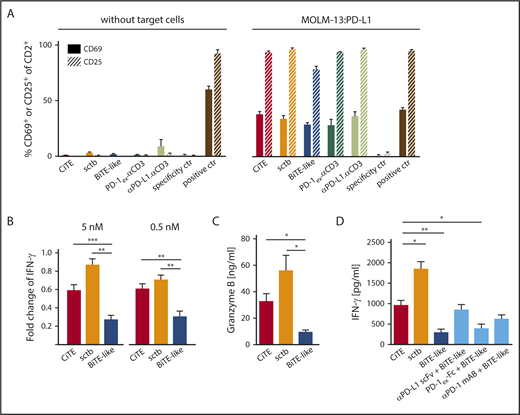
In vitro, BiTE-mediated T-cell activation strictly depends on the crosslink to target cells.46 To assess T-cell activation caused by sole CD3 engagement, we incubated HD T cells with CiTE and sctb in the absence or presence of MOLM-13:PD-L1 cells (Figure 2A). As expected, none of the molecules induced expression of CD25 and CD69 without target cells, whereas T cells significantly upregulated both markers in the presence of MOLM-13:PD-L1 cells. As a further hallmark of T-cell activation, we quantified the IFN-γ and Granzyme B release (Figure 2B-D). On CD33+PD-L1+ cells, both CiTE and sctb led to a significant increase in IFN-γ and Granzyme B levels compared with the BiTE-like molecule. We also observed an upregulation of PD-1 upon T-cell activation (supplemental Figure 3).
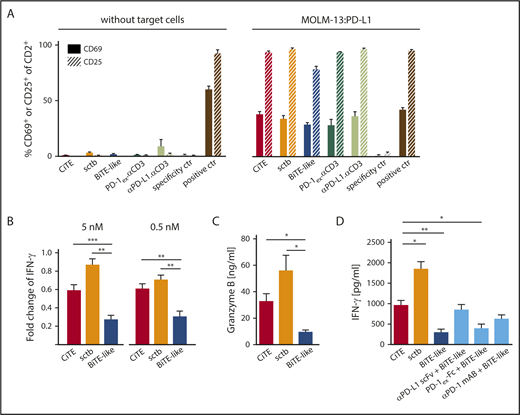
CiTE-mediated T-cell activation depends on crosslink to target cells and is enhanced compared with BiTE-like molecule. (A) CiTE- and sctb-induced upregulation of CD69 and CD25 on HD T cells in comparison with controls as analyzed by flow cytometry in the absence or presence of MOLM-13:PD-L1 cells after 96 hours and E:T ratio of 2:1. Bar charts show mean values of n = 3 independent experiments at 5 nM concentration with SEM as error bars. (B) Fold change of IFN-γ release in the presence of MOLM-13:PD-L1 compared with MOLM-13 cells at 5 and 0.5 nM protein concentrations. (C) Granzyme B release in the presence of MOLM-13:PD-L1 cells at 0.5 nM protein concentration. (D) IFN-γ release in the presence of MOLM-13:PD-L1 cells upon addition of CiTE, sctb, or BiTE-like molecule at 5 nM protein concentration and combination of BiTE-like molecule with PD-1/PD-L1 blocking agents at equimolar concentration. Cytokine and Granzyme B release were measured after 72 hours at an E:T ratio of 2:1 using nonstimulated HD T cells. Bar charts represent mean values of (B,D) n = 4-5 or (C) n = 4-6 independent experiments with SEM as error bars. For statistical analysis, unpaired Student t test with Welch correction was applied. *P < .05, **P < .01, ***P < .001.
CiTE-mediated T-cell activation depends on crosslink to target cells and is enhanced compared with BiTE-like molecule. (A) CiTE- and sctb-induced upregulation of CD69 and CD25 on HD T cells in comparison with controls as analyzed by flow cytometry in the absence or presence of MOLM-13:PD-L1 cells after 96 hours and E:T ratio of 2:1. Bar charts show mean values of n = 3 independent experiments at 5 nM concentration with SEM as error bars. (B) Fold change of IFN-γ release in the presence of MOLM-13:PD-L1 compared with MOLM-13 cells at 5 and 0.5 nM protein concentrations. (C) Granzyme B release in the presence of MOLM-13:PD-L1 cells at 0.5 nM protein concentration. (D) IFN-γ release in the presence of MOLM-13:PD-L1 cells upon addition of CiTE, sctb, or BiTE-like molecule at 5 nM protein concentration and combination of BiTE-like molecule with PD-1/PD-L1 blocking agents at equimolar concentration. Cytokine and Granzyme B release were measured after 72 hours at an E:T ratio of 2:1 using nonstimulated HD T cells. Bar charts represent mean values of (B,D) n = 4-5 or (C) n = 4-6 independent experiments with SEM as error bars. For statistical analysis, unpaired Student t test with Welch correction was applied. *P < .05, **P < .01, ***P < .001.
To effectively counteract adaptive immune resistance caused by PD-1/PD-L1 signaling, current clinical trials investigate combination therapies of targeting agents with checkpoint inhibitors.47,48 Thus, INF-γ levels were measured upon T-cell activation by CiTE, BiTE-like molecule, or combinations of BiTE-like and checkpoint inhibitors. Strikingly, the CiTE induced similar cytokine levels compared with high-affinity blocking agents plus BiTE-like molecule, whereas the equimolar addition of PD-1ex-Fc (low-affinity blocking module) triggered a weaker IFN-γ release (Figure 2D). We conclude that the fusion of PD-1ex to a BiTE-like scaffold leads to similar T-cell activation as combination approaches, but with the advantage of local restriction to CD33+ cells. We hypothesize that this effect is due to a synergy of avidity-dependent binding and PD-1/PD-L1 checkpoint blockade.
CiTE-mediated cytotoxicity is limited to CD33+ cells
With the CiTE format, we provide a molecule that targets CD33+ leukemic cells with high affinity and locally blocks PD-L1 because of the low affinity of PD-1ex. Furthermore, we expect the CiTE to address CD33+PD-L1+ cells more efficiently than CD33+PD-L1− cells because of avidity-dependent binding of the αCD33 scFv and PD-1ex. To test this hypothesis, the molecules were incubated with nonstimulated HD T cells and MOLM-13 or MOLM-13:PD-L1 cells, expressing high levels of CD33 (Figure 3; supplemental Figure 5; supplemental Table 1). Both CiTE and sctb induced specific lysis of both cell lines, yet, PD-L1 expression on AML cells increased the efficacy of target cell depletion (Figure 3A,C). Also, T-cell proliferation was triggered more strongly on CD33+PD-L1+ target cells (supplemental Figure 4). Interestingly, both molecules revealed similar 50% effective concentration (EC50) values despite their different affinities for PD-L1. Consistent with the previous characterization, the low-affinity PD-1ex.αCD3 control had a low impact on cytotoxicity, whereas the high-affinity αPD-L1.αCD3 control led to target cell lysis when PD-L1 was expressed. Because CD33 levels on AML cells exhibit a high inter- and intrapatient heterogeneity,10 the results were confirmed with OCI-AML3 and OCI-AML3:PD-L1 cells, which express low CD33 levels (Figure 3B,D; supplemental Table 1). The advantage of the bifunctional CiTE and sctb in comparison with the standard BiTE-like molecule was further investigated by T cell–induced cytotoxicity assays using MOLM13:PD-L1 or BA/F3:CD33:PD-L1 target cells at an E:T ratio of 1:3 (Figure 3E-F). In contrast to the BiTE-like molecule, both CiTE and sctb significantly enhanced target cell lysis. Collectively, CiTE- and sctb-mediated cytolysis is strongest against CD33+PD-L1+ double-positive cells, independent of the absolute affinity of the checkpoint blocking module, and the fused PD-L1 blocking module increases lysis of double-positive cells.

CiTE and sctb enhance lysis of CD33+PD-L1+target cells. (A) Dose-dependent lysis of MOLM-13 vs MOLM-13:PD-L1 cells or (B) OCI-AML3 vs OCI-AML3:PD-L1 cells by nonstimulated HD T cells. Calculated EC50 values for CiTE and sctb on MOLM-13 cells were 79.1 and 81.6 pM, on MOLM-13:PD-L1 cells were 26.9 and 13.2 pM, on OCI-AML3 cells were 20.8 and 27.4 pM, and on OCI-AML3:PD-L1 cells were 2.3 and 1.9 pM, respectively. (C) Lysis of MOLM-13 vs MOLM-13:PD-L1 cells at 50 pM protein concentration and (D) OCI-AML3 vs OCI-AML3:PD-L1 cells at 5 pM concentration at an E:T ratio of 2:1 after 72 hours. Lysis of (E) MOLM-13:PD-L1 cells and (F) BA/F3:CD33:PD-L1 cells at 0.5 nM concentration, E:T ratio of 1:3, and culture time of 72 hours. Graphs display mean values of (A-D) n = 4-5, (E) n = 7, or (F) n = 6 independent experiments with SEM as error bars. For statistical analysis, unpaired Student t test with Welch correction (C-D) or Wilcoxon test (E-F) was applied. *P < .05, **P < .01, ***P < .001.
CiTE and sctb enhance lysis of CD33+PD-L1+target cells. (A) Dose-dependent lysis of MOLM-13 vs MOLM-13:PD-L1 cells or (B) OCI-AML3 vs OCI-AML3:PD-L1 cells by nonstimulated HD T cells. Calculated EC50 values for CiTE and sctb on MOLM-13 cells were 79.1 and 81.6 pM, on MOLM-13:PD-L1 cells were 26.9 and 13.2 pM, on OCI-AML3 cells were 20.8 and 27.4 pM, and on OCI-AML3:PD-L1 cells were 2.3 and 1.9 pM, respectively. (C) Lysis of MOLM-13 vs MOLM-13:PD-L1 cells at 50 pM protein concentration and (D) OCI-AML3 vs OCI-AML3:PD-L1 cells at 5 pM concentration at an E:T ratio of 2:1 after 72 hours. Lysis of (E) MOLM-13:PD-L1 cells and (F) BA/F3:CD33:PD-L1 cells at 0.5 nM concentration, E:T ratio of 1:3, and culture time of 72 hours. Graphs display mean values of (A-D) n = 4-5, (E) n = 7, or (F) n = 6 independent experiments with SEM as error bars. For statistical analysis, unpaired Student t test with Welch correction (C-D) or Wilcoxon test (E-F) was applied. *P < .05, **P < .01, ***P < .001.
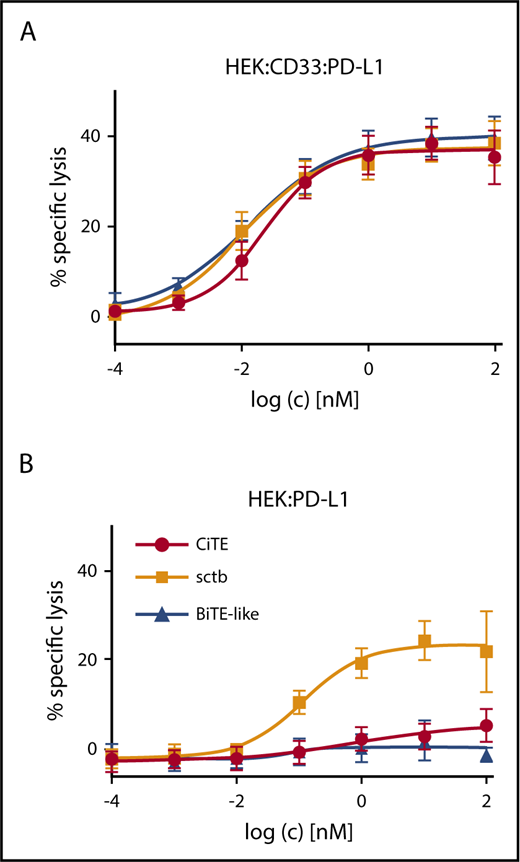
We next evaluated whether the CiTE molecule is able to induce elimination of CD33+PD-L1+ cells selectively in the presence of PD-L1+ cells. To this end, preferential lysis was analyzed in a mixed target cell population (Figure 4; supplemental Figure 6). Although the CiTE triggered preferential lysis of CD33+PD-L1+ cells, molecules with high affinity to PD-L1 revealed dose-dependent elimination of both CD33+PD-L1+ and PD-L1+ cell lines. This indicates that the low-affinity PD-1ex module is not sufficient to redirect T cells to PD-L1+ non-AML cells, which might provide an important safety feature for the CiTE platform.

CiTE induces preferential lysis of CD33+PD-L1+cells and has no activity against CD33-PD-L1+cells. Dose-dependent preferential lysis of (A) HEK:CD33:PD-L1 over (B) HEK:PD-L1 cells induced by CiTE, sctb, or BiTE-like molecule and vice versa. Preactivated HD T cells were incubated with target cells and CiTE, sctb, or BiTE-like molecule for 4 hours at a total E:T ratio of 2:1. EC50 values for CiTE, sctb, and BiTE-like molecule were 22.8, 10.4, and 13.5 pM for HEK:CD33:PD-L1 cells. For HEK:PD-L1 cells, the EC50 value for the sctb was 114.4 pM. Graphs represent mean values of n = 4 independent experiments with SEM as error bars.
CiTE induces preferential lysis of CD33+PD-L1+cells and has no activity against CD33-PD-L1+cells. Dose-dependent preferential lysis of (A) HEK:CD33:PD-L1 over (B) HEK:PD-L1 cells induced by CiTE, sctb, or BiTE-like molecule and vice versa. Preactivated HD T cells were incubated with target cells and CiTE, sctb, or BiTE-like molecule for 4 hours at a total E:T ratio of 2:1. EC50 values for CiTE, sctb, and BiTE-like molecule were 22.8, 10.4, and 13.5 pM for HEK:CD33:PD-L1 cells. For HEK:PD-L1 cells, the EC50 value for the sctb was 114.4 pM. Graphs represent mean values of n = 4 independent experiments with SEM as error bars.
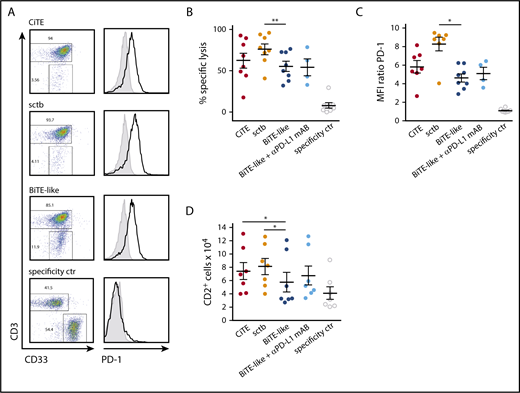
CiTE and sctb increase specific cytotoxicity against patient-derived AML cells and enhance T-cell proliferation
In ALL, relapse after blinatumomab treatment was suggested to originate from PD-L1 expressing leukemic cells, which are resistant to T cell–mediated cytotoxicity.14 A similar mechanism was identified in AML, where AMG 330-induced T-cell activation was accompanied by PD-L1 upregulation on patient-derived AML cells as well as PD-1 expression on T cells ex vivo.13 Also, CiTE-mediated T-cell activation led to the upregulation of PD-L1 on primary AML patient cells (supplemental Figure 7A). In 7 of 8 patients, the CiTE was able to induce equal or enhanced redirected lysis of target cells compared with the BiTE-like molecule (62 ± 9% compared with 55 ± 6%), and the sctb triggered similar or higher lysis in all patients (76 ± 6%) (Figure 5A-B; supplemental Figure 7B). An increase in T-cell proliferation was induced by the CiTE and sctb in contrast to the BiTE-like molecule through prolongation of coculture time to 6 to 7 days (Figure 5D). Furthermore, elevated T-cell activity was demonstrated by virtue of PD-1 expression as well as IFN-γ release (Figure 5C; supplemental Figure 7C). Interestingly, addition of a PD-L1 blocking mAB to the BiTE-like molecule had a lower impact on cytotoxicity and T-cell proliferation than the sctb. Thus, we hypothesize that CiTE and sctb are able to efficiently counteract PD-L1–mediated resistance mechanisms and to induce specific lysis of AML cells by a synergy of avidity-dependent binding and checkpoint blockade.
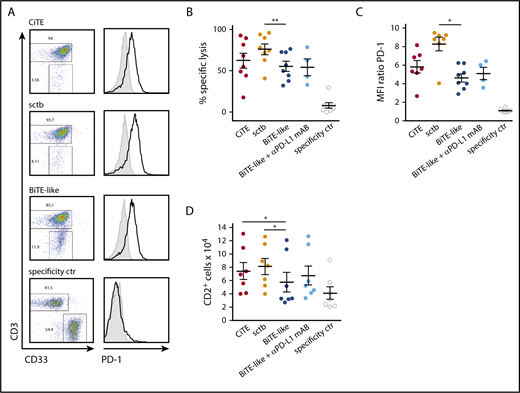
CiTE and sctb enhance lysis of patient-derived primary AML cells and increase T-cell proliferation. (A) Cytotoxic lysis of primary AML cells from an exemplary patient and PD-1 expression on T cells after 3 to 4 days of cocultivation. (B) Mean cytotoxic lysis of primary AML cells induced by respective molecules after 3 to 4 days. (C) PD-1 expression on T cells after 3 to 4 days. (D) T-cell proliferation after 6 to 7 days measured by flow cytometry. CiTE, sctb, BiTE-like molecule, and specificity control were applied at 10 nM; αPD-L1 mAB at 10 µg/mL concentration. Assays were performed at an initial E:T ratio of 1:4. Error bars display SEM. For statistical analysis, Wilcoxon test was applied. *P < .05, **P < .01, ***P < .001.
CiTE and sctb enhance lysis of patient-derived primary AML cells and increase T-cell proliferation. (A) Cytotoxic lysis of primary AML cells from an exemplary patient and PD-1 expression on T cells after 3 to 4 days of cocultivation. (B) Mean cytotoxic lysis of primary AML cells induced by respective molecules after 3 to 4 days. (C) PD-1 expression on T cells after 3 to 4 days. (D) T-cell proliferation after 6 to 7 days measured by flow cytometry. CiTE, sctb, BiTE-like molecule, and specificity control were applied at 10 nM; αPD-L1 mAB at 10 µg/mL concentration. Assays were performed at an initial E:T ratio of 1:4. Error bars display SEM. For statistical analysis, Wilcoxon test was applied. *P < .05, **P < .01, ***P < .001.
CiTE induces leukemia eradication in vivo without on-target off-leukemia events
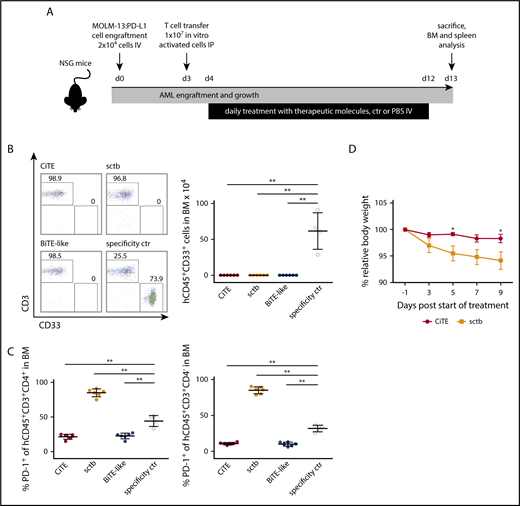
Because T cell–based immunotherapies such as BiTEs, chimeric antigen receptor T cells and hematopoietic stem cell transplantation rely on T-cell activation, the induced proinflammatory response will consistently evoke PD-L1 upregulation on AML cells.13,49-52 To mimic this physiological situation in vivo, we engrafted MOLM-13:PD-L1 cells into NSG mice followed by transfer of in vitro activated human HD T cells (Figure 6A). Evaluation of the residual hCD45+CD33+ AML population in the BM after 9 days of treatment revealed complete eradication of leukemic cells in all 3 treatment groups (Figure 6B). In contrast, the control cohort showed 1% to 3% AML cells in the BM, which resembles minimal residual disease criteria of <5% myeloblasts in humans (supplemental Figure 8A).53
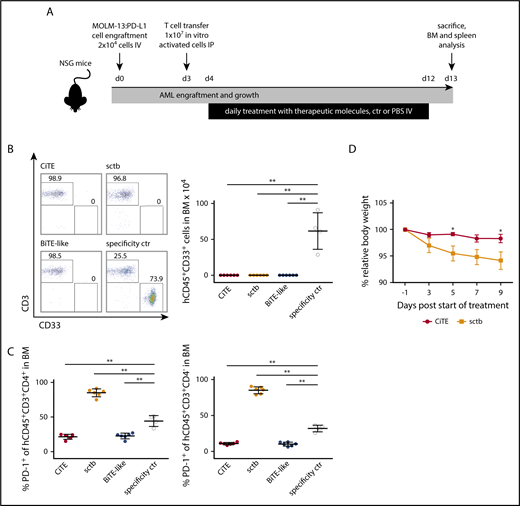
CiTE leads to eradication of AML cells in a murine NSG xenograft model without indication of enhanced PD-1 upregulation or body weight loss. (A) Experimental design of mouse studies. (B) Remaining engrafted MOLM-13:PD-L1 (live hCD45+CD33+) cells in BM of exemplary mice per cohort (left) and as mean (right) after 13 days. (C) PD-1 upregulation on human CD45+CD3+CD4+ and CD45+CD3+CD4− T cells. (D) Relative body weight of mice as scored during treatment with CiTE and sctb. Cohorts contained 4 to 6 mice. Error bars in (B-C) indicate standard deviation, in (D) SEM. For statistical analysis, Mann-Whitney U test was applied. *P < .05, **P < .01, ***P < .001. IP, intraperitoneally.
CiTE leads to eradication of AML cells in a murine NSG xenograft model without indication of enhanced PD-1 upregulation or body weight loss. (A) Experimental design of mouse studies. (B) Remaining engrafted MOLM-13:PD-L1 (live hCD45+CD33+) cells in BM of exemplary mice per cohort (left) and as mean (right) after 13 days. (C) PD-1 upregulation on human CD45+CD3+CD4+ and CD45+CD3+CD4− T cells. (D) Relative body weight of mice as scored during treatment with CiTE and sctb. Cohorts contained 4 to 6 mice. Error bars in (B-C) indicate standard deviation, in (D) SEM. For statistical analysis, Mann-Whitney U test was applied. *P < .05, **P < .01, ***P < .001. IP, intraperitoneally.
Besides efficient eradication of AML cells, the main purpose of the CiTE antibody is to avoid irAEs that originate from systemic binding to PD-L1+ tissue. To investigate potential targeting of non-AML cells, we took advantage of the cross-reactivity of both PD-L1 checkpoint blocking modules to murine PD-L1, which bound murine PD-L1 with comparable affinities than human PD-L1 (supplemental Figure 8B). Mice treated with the high-affinity sctb lost body weight compared with the other treatment groups (Figure 6D; supplemental Figure 8C). PD-1 was significantly upregulated on CD4+ and CD4− T cells in the BM (Figure 6C); a similar T-cell phenotype was noted when splenic T cells were analyzed (data not shown). We hypothesize that this observation is due to sctb-mediated T-cell redirection to PD-L1+ murine cells and represents on-target off-leukemia events. Most importantly, no such effects were observed for the CiTE antibody. These findings demonstrate that the CiTE efficiently induces specific AML eradication in vivo without affecting the body weight as indication for systemic PD-L1 targeting. Thus, we consider the new CiTE format as favorable postremission approach in AML, which is particularly suited to counteract PD-L1–mediated adaptive immune resistance.
Discussion
The BiTE technology is a successful immunotherapeutic approach in ALL, and with AMG 330, a first T-cell engager recently entered the clinics for AML treatment. However, it has been shown that BiTE-mediated T-cell activation and the associated release of proinflammatory cytokines trigger the upregulation of the inhibitory ligand PD-L1 on AML and ALL cells.13,14 As reflected in ex vivo experiments using human patient samples, the combination of AMG 330 and PD-1/PD-L1 inhibitors might abrogate this axis and restore T-cell activity.13 Yet, PD-1 and PD-L1 blocking mABs that have hitherto been approved by regulatory authorities are limited by their risk to induce irAEs. Although adverse events are often successfully managed, they can develop into a severe state and require therapy interruption or discontinuation; thus, new approaches are urgently needed.
The presented CiTE format is able to combine T-cell redirection with a restricted PD-1/PD-L1 blockade to the surface of AML cells and thereby to sustain immune tolerance against healthy tissue. This is achieved by fusing the extracellular domain of human PD-1, which naturally holds a low affinity to PD-L1, to a BiTE-like scaffold. PD-1ex is not sufficient to bind PD-L1 alone, but only linked to a high-affinity leukemia-targeting module. As a consequence, the CiTE exclusively induces lysis of CD33+PD-L1+ cells in vitro, whereas PD-L1+ non-AML cells are not affected. In vivo, the CiTE did not lead to on-target off-leukemia events indicated by body weight loss and leukemia-unrelated T-cell activation. Thus, the bifunctional format displays a promising therapeutic strategy to lower irAEs compared with high-affinity PD-1 and PD-L1 blocking agents.
Because CD3-addressing approaches by T-cell–engaging molecules are effective at very low protein concentrations (picomolar or even subpicomolar),9,54,55 an obvious question is whether fusing checkpoint ligands to CD3-binding modules in a single molecule would be sufficient to block PD-1/PD-L1 interactions. However, T-cell receptor and PD-1 are suggested to be closely associated in the immunological synapse.56 Consequently, a locally restricted full or even partial inhibition of PD-1/PD-L1 interactions at the T-cell receptor could lead to a more efficient T-cell activation even at low antibody concentrations.
Stimulation of CD3ε on T cells with monoclonal antibodies was shown to induce T-cell activation.46,57 Accordingly, patients treated with muromonab (Orthoclone OKT3) frequently experience cytokine release syndrome.58,59 In contrast, monovalent CD3 stimulation by the CiTE does not per se trigger upregulation of T-cell activation markers such as CD69 or CD25 in vitro. In concordance to preclinical studies of BiTE antibodies,46 T cells are exclusively activated by crosslinking to leukemic cells that express the targeted CD33. Nevertheless, blinatumomab does induce cytokine release syndrome in some patients.60 Intensive investigations in animal models are therefore indispensable.
Similar to BiTE molecules,9,54,55 the CiTE is able to induce redirected lysis of cancer cells at very low concentrations with EC50 values in the low picomolar range. Because of avidity-dependent binding, the targeting efficacy of CD33+ AML cells that express PD-L1 is increased. This might provide the possibility to preferentially address double-positive cells, which is especially important because CD33 is also expressed on CD34+CD38− hematopoietic stem cells and healthy myeloid cells, and the general depletion of CD33+ cells by CD33 monotargeting agents such as gemtuzumab ozogamicin consequentially results in neutropenia.10,61-66
Because the upregulation of immune checkpoints in response to T-cell activation is a general mechanism of adaptive immune resistance, combination therapies of targeting agents, chemotherapies, or kinase inhibitors with blocking mAB are under intensive investigation.16,47,48,67 We were able to demonstrate that the CiTE molecule, despite the low-affinity PD-1ex domain, induces similar IFN-γ levels in comparison with the combination of the BiTE-like molecule and PD-1 or PD-L1 inhibitors. However, most importantly and in contrast to high-affinity PD-L1 binders applied in combination therapies, the CiTE preferentially and highly selectively eliminated CD33+PD-L1+ double-positive target cells. This is expected to translate into a decreased incidence of irAEs, as was observed in our xenograft mouse model.
Collectively, we showed that the CiTE antibody reveals a high potency to activate resting T cells and to induce efficient cytotoxicity against AML cells. It features a high specificity for CD33+PD-L1+ target cells in vitro and does not show adverse events in vivo. Because of its beneficial performance compared with the BiTE format, we consider the CiTE a promising candidate to reverse immune resistance in AML. Future studies will have to examine efficacy and tolerance in more advanced in vivo models before applying the CiTE format into a clinical setting.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Alexandra Schele and Constanze Heise for excellent technical assistance and Stefanie Schneider for contributing to the characterization of patient samples. The authors are grateful to Matthias Peipp for providing the αHer2 scFv sequence and Claudia C. Roskopf for sharing the expression vector for αHer2.αCD3.αHer2.
This work is funded by the Deutsche Forschungsgemeinschaft (DFG) CRC1243 and the Bavarian Elite Graduate Training Network “i-Target” (S.K., M.S., and K.-P.H.) and by the m4-Award of the Bavarian Ministry of Economic Affairs (N.C.F., K.-P.H., and M.S.). K.-P.H. and S.K. are further supported by the Marie-Sklodowska-Curie Training Network “Immutrain” funded by the H2020 Program of the European Union. M.H. was supported by a DFG fellowship through the Research Training Group 1202 “Oligonucleotides in cell biology and therapy.” S.K. is supported by the Else-Kröner Fresenius-Stiftung, the German Cancer Aid and the European Research Council.
M.H. is a doctoral candidate at Ludwig-Maximilians-Universität München; this work is submitted in partial fulfillment of the requirement for that degree.
Authorship
Contribution: M.H., N.C.F., M.S., and K.-P.H. designed the experiments and interpreted the data. M.H. generated and characterized the molecules and performed cell line–based assays. F.R. and S.K. contributed to cell line generation. C.K. and B.B. performed the evaluation on acute myeloid leukemia patient samples and analyzed the results. A.O.W. and A.M. contributed to the cell line–based assays and patient sample evaluation. K.D. performed the in vivo studies and evaluated the data. K.H.M., K.S., and M.S. contributed to patient characterization including cytogenetic and molecular data. R.M. wrote the animal test proposal and supervised animal experiments. K.-P.H. and M.S. supervised the project. M.H. and N.C.F. wrote the manuscript with input from all authors.
Conflict of interest disclosure: The authors declare no competing financial interests.
Correspondence: Marion Subklewe, Department of Medicine III, University Hospital, Ludwig-Maximilians-Universität München, Marchioninistr 15, 81377 Munich, Germany; e-mail: marion.subklewe@med.uni-muenchen.de; Karl-Peter Hopfner, Gene Center and Department of Biochemistry, Ludwig-Maximilians-Universität München, Feodor-Lynen-Str 25, 81377 Munich, Germany; e-mail: hopfner@genzentrum.lmu.de; and Nadia C. Fenn, Gene Center and Department of Biochemistry, Ludwig-Maximilians-Universität München, Feodor-Lynen-Str 25, 81377 Munich, Germany; e-mail: nfenn@genzentrum.lmu.de.