Abstract
Our knowledge about the genetics of myelodysplastic syndromes (MDS) and related myeloid disorders has been dramatically improved during the past decade, in which revolutionized sequencing technologies have played a major role. Through intensive efforts of sequencing of a large number of MDS genomes, a comprehensive registry of driver mutations recurrently found in a recognizable fraction of MDS patients has been revealed, and ongoing efforts are being made to clarify their impacts on clinical phenotype and prognosis, as well as their role in the pathogenesis of MDS. Among major mutational targets in MDS are the molecules involved in DNA methylations, chromatin modification, RNA splicing, transcription, signal transduction, cohesin regulation, and DNA repair. Showing substantial overlaps with driver mutations seen in acute myeloid leukemia (AML), as well as age-related clonal hematopoiesis in healthy individuals, these mutations are presumed to have a common clonal origin. Mutations are thought to be acquired and positively selected in a well-organized manner to allow for expansion of the initiating clone to compromise normal hematopoiesis, ultimately giving rise to MDS and subsequent transformation to AML in many patients. Significant correlations between mutations suggest the presence of functional interactions between mutations, which dictate disease progression. Mutations are frequently associated with specific disease phenotype, drug response, and clinical outcomes, and thus, it is essential to be familiar with MDS genetics for better management of patients. This review aims to provide a brief overview of the recent progresses in MDS genetics.
Introduction
Myelodysplastic syndromes (MDS) comprises a heterogeneous group of myeloid neoplasms, which are characterized in common by manifestations of bone marrow failure with abnormal cell morphology and a high propensity to acute myeloid leukemia (AML).1,2 Now, it is well recognized that MDS is, like other cancers, shaped by recursive rounds of positive selections, where gene mutations and other genetic alterations play central roles.3-5 A patient’s bone marrow milieus as well as extrinsic factors, such as autoimmunity and chemoradiotherapy, are likely also engaged in this process.6-10 It is central to the understanding of the pathogenesis and disease phenotypes of MDS to decipher the mutations that are involved in the positive selection(“driver” mutations) and the mechanisms by which those mutations are positively selected. Although the latter requires functional evaluations of each driver mutation, which are often complicated, the detection of driver mutations is reliably accomplished by interrogating genetic evidence of positive selection, ie, significantly higher mutation frequency than expected only by chance (ie, recurrence). In fact, through revolutionized sequencing technologies or next-generation sequencing (NGS), an almost complete registry of those driver mutations engaged in positive selections of MDS has been revealed during the past decade, together with those in other cancers. Many driver alterations in MDS are shared by primary AML but still show several distinct features of their own, which are thought to make up the unique pathophysiology of this heterogeneous group of myeloid neoplasms.11 Most importantly, a patient’s genetic profile critically impacts the clinical phenotype, prognosis, and response to therapy, underscoring the importance of the updated knowledge about MDS genetics for predicting a patient’s clinical course as well as optimizing therapy and management.
The purpose of this review is to overview the recent advances in the genetics of MDS and related disorders, including chronic myeloid leukemia and other myelodysplastic/myeloproliferative neoplasms, largely focusing on somatic mutations. Although recent studies suggest that germline mutations could have a much larger impact on the development of myeloid malignancies than previously expected,12 the issues of germline predisposition are not included here but will be reviewed in other articles of this review series.
Driver alterations in MDS and related disorders
Chromosomal and copy-number abnormalities (CNAs)
Early genetic studies on MDS mostly focused on cytogenetical abnormalities detected by conventional karyotyping, which are found in ∼50% of MDS cases.13,14 In contrast to the case with AML, in which balanced abnormalities, such as t(8;21)(q22;q22), t(15;17)(q22;q21), inv(16)(p13q22)/t(16;16)(p13;q22), and 11q23-involved translocations,14 are predominant, the majority of abnormalities in MDS are unbalanced changes, which result in CNAs, ie, gains or losses of chromosomal materials.15 The most frequent among these are −7/del(7q) and −5/del(5q), followed by +8, dup(1q), del(20q), del(11q), del(12p)/t(12p), del(17p)/iso(17q), del(18q), +21q gains, del(13q), and +der(1;7)(q10;p10) (Figure 1A).14 Many of these lesions often cooccur as a part of complex abnormalities, designated as complex karyotypes (CKs). Involving multiple (≥3) chromosomes or chromosomal arms/segments, CKs are frequently accompanied by TP53 mutation (∼40% to 50%), generally predicting poor clinical outcomes, particularly when −5/del(5q) and del(17p) are involved.16-18 Rare cases (less than ∼2% to 3%) show recurrent reciprocal translocations, including t(3;21)(q26;q22), inv(3)(q21q26), t(3;3)(q21;q26), t(1;3)(q36;q21), and t(6;9)(q22;q34), which are associated with unique clinicopathological features and have high diagnostic values for MDS, even without clear morphologic evidence of myelodysplasia.19-21 CNAs have also been investigated using single-nucleotide polymorphism (SNP) array–based copy-number detection.22 The SNP array–based platforms can detect not only CNAs but also copy-neutral loss of heterozygosity (LOH) or uniparental disomy (UPD), which are commonly seen in chromosomes 1p, 4q, 7q, 11q, 13q, 14q, 17p.23 Extremely high resolution of SNP array-based analysis allows for identification of sub-chromosomal/focal lesions, as small as 20 kb in size, leading to the identification of relevant driver genes they involve, such as MPL and NRAS (1pUPD), TET2 (4qLOH), CUX1 and EZH2 (7qLOH), CBL (11qLOH), ETV6 (del(12p)), FLT3 (13qUPD), TP53 (17pLOH), and RUNX1 (21qLOH).23-26
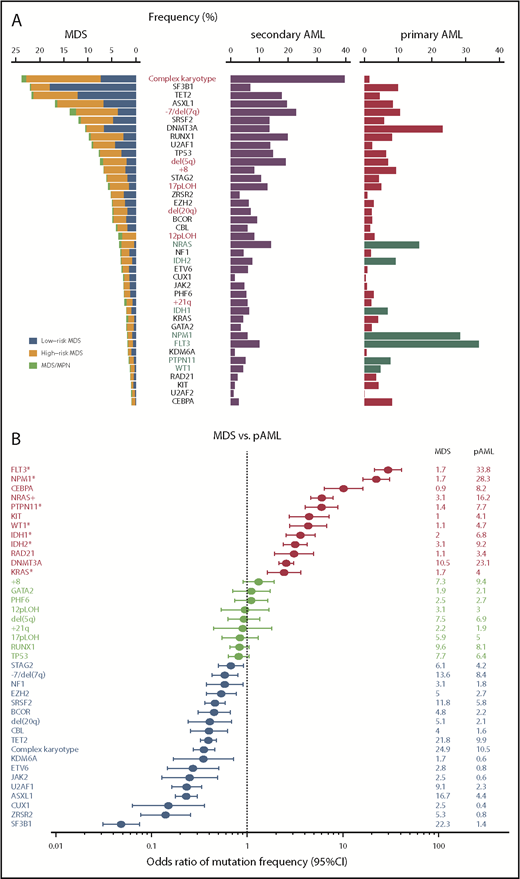
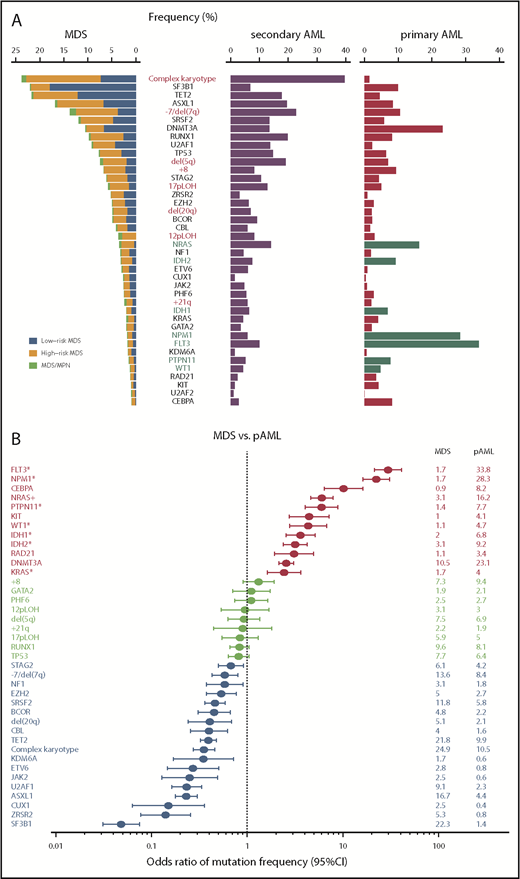
Common driver alterations in MDS and other myeloid neoplasms. (A) Frequencies of major driver mutations and CNAs are plotted, combining data from 7 publications,11,18,35-38,40 which include 1449 low- and 966 high-risk MDS, 46 MDS/MPN, 549 sAML, and 1540 primary AML (pAML) cases. Green bars indicate type I genes.36 (B) Odds ratios and their 95% confidence intervals (CIs) of frequencies of major driver mutations and CNAs between MDS (including MDS/MPN) and pAML are shown in forest plots.
Common driver alterations in MDS and other myeloid neoplasms. (A) Frequencies of major driver mutations and CNAs are plotted, combining data from 7 publications,11,18,35-38,40 which include 1449 low- and 966 high-risk MDS, 46 MDS/MPN, 549 sAML, and 1540 primary AML (pAML) cases. Green bars indicate type I genes.36 (B) Odds ratios and their 95% confidence intervals (CIs) of frequencies of major driver mutations and CNAs between MDS (including MDS/MPN) and pAML are shown in forest plots.
Somatic mutations
Until the early 2000s, only a handful of genes, such as NRAS,27 TP53,28 RUNX1,29 and ATRX,30 were known to be recurrently mutated in MDS. Since then, assisted by SNP array karyotyping and high-throughput capillary sequencing, a number of additional mutational targets, including NPM1, TET2, CBL, EZH2, FLT3, KRAS, MPL, ASXL1, PTPN11, KRAS, and KIT, were identified in MDS until 2010.24-26,31-34 However, during the following decade, our knowledge about MDS genomes has been fully revised through a comprehensive detection of somatic mutations in MDS and related myeloid neoplasms35-38 based on advanced sequencing technologies.39,40
Including both driver and nondriver (“passenger”) mutations, MDS patients carry a median of 9 somatic mutations within the coding sequence36,38,41,42 or ∼1500 mutations across the entire genome (Yasuhito Nannya and the MDS Whole Genome Project, unpublished observation), which is substantially lower than the numbers typically seen in solid cancers. Mutation numbers vary depending on disease subtype, with 6 per exome in low-risk MDS, 9 in MDS with excess blasts (MDS-EB), 12 in chronic myelomonocytic leukemia (CMML), 8 MDS/myeloproliferative neoplasms (MPN) unclassified, and 13 in secondary AML (sAML).36 Until now, >30 driver genes that are involved in the pathogenesis of MDS have been identified (Figure 1A).35,37 While a typical MDS patient harbors a median of 2 or 3 driver mutations, high-risk MDS (MDS-EB and MDS with multilineage dysplasia) and CMML tend to show higher numbers of driver mutations than lower-risk MDS, including MDS with single-lineage dysplasia with or without increased sideroblasts (RS) and MDS with isolated del(5q). The target driver genes are categorized into several discrete functional pathways, including RNA splicing, DNA methylation, transcription, chromatin modification, signal transduction, DNA repair, and other pathways and molecules (Table 1).35,37 Combined with CNAs and chromosomal abnormalities, >78% to 90% of MDS patients had ≥1 known recurrent genetic abnormalities.35,37 Among these, most frequently observed are those affecting epigenetic regulation and RNA splicing, including DNA methylation (TET2, DNMT3A, and IDH1/IDH2)24,33,43 and chromatin/histone modification (MLL2, EZH2 and other polycomb repressive complex 2 (PRC2) components, ARID2 and ASXL1),25,44 as well as RNA splicing (SF3B1, SRSF2, U2AF1, U2AF2, ZXRSR2, SF1, and SF3A1).41,42,45 Other pathways or molecules commonly affected include p53, cohesin and associated proteins (STAG2, RAD21, SMC1A, SMC3, NIPBL, PDS5b, and CTCF),46 transcription factors (RUNX1, ETV6, GATA32, and IRF1), the RAS pathway (NRAS, KRAS, PTPN11, NF1, and CBL),26 other signaling molecules (JAK2, KIT, MPL, GNB1, and FLT3), and several components of the DNA repair machinery (BRCC3, FANCL, and ATM).35,37 Mutational targets are largely similar between MDS and primary AML (Figure 1A), suggesting common pathogenesis in different myeloid neoplasms. However, the frequencies of these mutations are significantly different between MDS and primary AML; there is an overrepresentation of mutations in receptor tyrosine kinase (FLT3 and KIT) and RAS pathway genes, as well as CEBPA and IDH1/IDH2 mutations in AML, while mutations in splicing factors (SFs) and epigenetic regulators, as well as CNAs, are more prevalent in MDS (Figure 1B). Mutations in several genes, such as DDX41, RUNX1, GATA2, and TP53, may be present in the germline and responsible for predisposition to AML and MDS (Table 1). This raises a challenging issue of how to discriminate pathogenic variants vs nonpathogenic polymorphisms for the correct diagnosis of familial cases, which will be discussed in another article in this review series.
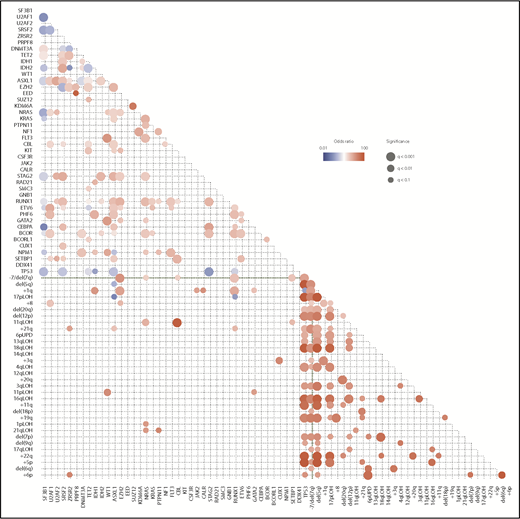
When multiple driver genes are affected, their combination is not totally random, but some combinations of driver mutations cooccur more frequently than expected by chance, while others are observed in a mutually exclusive manner, suggesting the presence of functional interactions between these mutations that are involved in positive and negative selections (Figure 2). For example, SF mutations are almost mutually exclusive of each other, and so are mutations in the components of the cohesin complex, likely due to synthetic lethality of multiple mutations in these pathways and complexes. By contrast, STAG2 mutations are significantly associated with mutations in RUNX1, SRSF2, ASXL1, EZH2, BCOR, and IDH2. As seen from the paucity of significant correlations in the lower left rectangle in Figure 2, positive and negative correlations tend to be separately compartmentalized within gene mutations and chromosomal CNAs. Major exceptions are significant positive correlations observed between TP53 mutations and CKs and between 1p gains, +8, 11p LOH, 11q LOH, 17p LOH, and +21 and corresponding mutations (Figure 2).
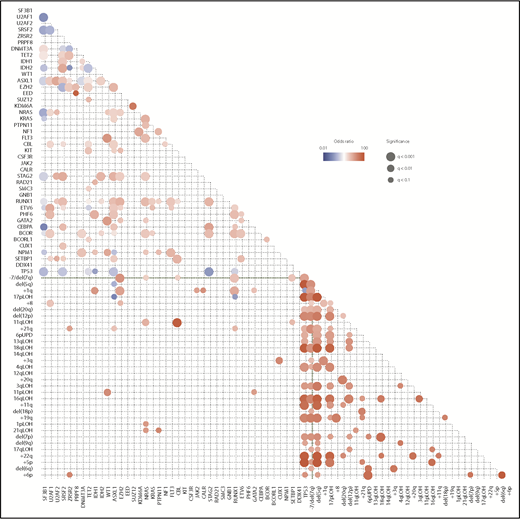
Correlations between driver mutations and CNAs in MDS. Significantly cooccurring and mutually exclusive alterations are shown in red and blue circles, respectively, on the basis of data from 6 published studies,11,18,35-38 where effect size and q-values are indicated by the size of circles and color gradient.
Correlations between driver mutations and CNAs in MDS. Significantly cooccurring and mutually exclusive alterations are shown in red and blue circles, respectively, on the basis of data from 6 published studies,11,18,35-38 where effect size and q-values are indicated by the size of circles and color gradient.
Genotype–phenotype correlations and impact on clinical outcomes
Mutations and CNAs significantly correlate with disease phenotype and clinical outcome. Such correlations have long been recognized for a number of cytogenetic abnormalities and were first documented for isolated del(5q), which is associated with unique phenotypes of refractory anemia showing normal to increased platelet counts with micromegakaryocytes.47 Other examples are 3q26- and 1p36-involved translocations. Affecting MECOM (EVI-1) and PRDM16, respectively, these translocations are associated with prominent dysmegakaryopoiesis and frequent progression to AML, invariably predicting a very poor prognosis.40,48 Clinical effects of gene mutations have also been investigated in large cohorts of MDS patients.35,37,49 The number of driver mutations significantly correlates with overall survival.35,37 Moreover, mutations in particular drivers are linked to characteristic hematological pictures and predict clinical outcomes, independently of established risk factors, such as the International Prognostic Scoring System or the Revised International Prognostic Scoring System. For example, found in the majority of MDS-RS cases, SF3B1 mutations are strongly associated with increased bone marrow RS19 and generally predict a favorable prognosis.41,50 Because of this strong association, the World Health Organization authors recommend the diagnosis of MDS-RS with ≥5% (otherwise ≥15%) of RS in the presence of SF3B1 mutation.19 SF3B1 mutations are mutually exclusive of not only other SF mutations but also most other mutations/CNAs, except for DNMT3A, TET2, JAK2, and del(5q).35,37 Of interest, JAK2 mutations and, to a lesser extent, those in MPL and CALR are frequently found in MDS/MPN-RS and thrombocytosis, which is a distinct MDS/MPN overlap entity characterized by MDS-RS with an elevated platelet count (≥450 × 109/L).19,51 It is also of note that generally predicting a favorable prognosis, SF3B1 mutation is found in the majority of cases with EVI1 (MECOM)–involved rearrangements, which are uniformly associated with very poor clinical outcomes, as mentioned above.52,53 On the basis of these unique genetic, morphological, and clinical features, it is suggested that SF3B1 mutations define a unique subset of myeloid neoplasms.51 In contrast to generally favorable prognosis in SF3B1-mutated cases, patients with other SF mutations, including U2AF1 and SRSF2, have poor overall survival.45,54-56 SRSF2 mutations are among the most predominant mutations in CMML, in which mutated SRSF2 was found in approximately half of these patients, frequently cooccurring with TET2 and ASXL1 mutations.57-59
TP53 mutations account for approximately one-fourth of high-risk MDS and also characterize ∼10% of primary AML.60 Mutations are especially common in therapy-related MDS/AML, where as many as 50% of the cases have TP53 mutations.61 Most (∼90%) TP53-mutated patients, particularly those with biallelic TP53 lesions, exhibit CK.18 Extensive CNAs are also tightly associated with TP53 mutation in many solid cancers, as well as other blood cancers, such as malignant lymphomas,62 suggesting a causative role of TP53 inactivation in the acquisition of CK, where TP53 mutation might allow abnormal aneuploid cells to survive without undergoing p53-mediated apoptosis or cell-cycle arrest at the G2 checkpoint. While CKs are also seen in TP53-unmutated cases, those in TP53-mutated cases can be discriminated from other CKs by a very high frequency of del(5q) and 17qLOH, which are rarely seen in TP53-unmutated cases.18 Of note, del(5q) may exist as an isolated cytogenetic lesion, which is associated with a unique hematological pictures (isolated del(5q)), but is more commonly seen as a part of CKs among TP53-mutated cases.18,63 In contrast to frequent associations with CKs, fewer numbers of other driver mutations are present in TP53-mutated MDS cases than TP53-unmutated cases.18,37 Regardless of their clinical/pathological diagnosis, patients with TP53-mutated myeloid neoplasms uniformly show a poor prognosis.18,64 It appears that the strong negative impact of a TP53 mutation on prognosis cannot be overcome, even with allogenic stem-cell transplantation.18,65,66 Recently, a dramatic clinical effect of decitabine has been reported, where 21 out of 21 AML/MDS patients with TP53 mutations achieved complete molecular remission after treatment with decitabine.67 Although the response was invariably transient and most patients relapsed within 6 months after achieving complete remission, an initial response to decitabine, if obtained, might be promising and could be used to overcome this most dismal subtype of myeloid neoplasm (eg, in combination with stem cell transplantation).
Evolution of MDS clones
In most cases, it is impossible to track an entire history of clonal evolution of MDS back to its cell of origin. However, several observations provide insight into how driver mutations shape MDS development: enrichment of mutations in particular MDS stages and clonal hematopoiesis (CH).
Enrichment of mutations in different stage of MDS
One of the cardinal features of MDS is disease progression over years with an increasing blast count during its clinical courses, which is dictated by the emergence of new clones that acquired additional mutations and other genetic alterations.38 In accordance with this, substantial differences in mutational profiles are seen across different disease stages. Mutations in some genes are significantly enriched in sAML compared with high-risk MDS, including FLT3, NPM1, NRAS, PTPN11, WT1, IDH1, and IDH2 (type I genes), while other genes are more commonly mutated in high-risk compared with low-risk MDS, such as GATA2, RUNX1, TET2, ZRSR2, TP53, STAG2, and ASXL1 (type II genes).36 It is of note that all type I genes are among the most frequently mutated genes in primary AML11 and highly enriched in primary AML compared with MDS (Figure 1A-B).11,36 In addition, patients with type I mutations show a significantly faster leukemic transformation than those without type I mutations. Significantly cooccurring with ≥1 type II mutation, type I mutations have significantly smaller allele frequencies than type II mutations, suggesting the ordered acquisition of type II followed by type I mutations during disease progression. In the analysis of serially collected samples, type I mutations tend to be more newly acquired than lost. Taken together, these observations suggest critical roles of type I mutations in AML phenotypes and transformation to sAML.
CH
Recently, it has been reported that common driver mutations or CNAs found in MDS and AML may also be detected in apparently healthy individuals with normal blood counts, particularly in the elderly, suggesting the presence of CH,3,68-70 also termed CH with indeterminate potential (CHIP) or age-related CH. While CHIP and age-related CH are frequently used synonymously, CHIP is defined in a more practical manner, taking into consideration the limitation of typical NGS platforms to detect somatic variants (ie, ≥2% of variant allele frequencies).71 In most cases, CH is accompanied by solitary mutations affecting genes most frequently mutated in MDS/AML, such as DNMT3A, TET2, ASXL1, JAK2, SF3B1, SRSF2, TP53, CBL, U2AF1, and IDH2 (Figure 1A) with an exception of PPM1D, which is rarely mutated primary MDS and AML. CH is also detected by CNA, which also show significant overlaps with those commonly seen in MDS/AML, such as del(20q), del(13q), 14qUPD, del(11q)/11qUPD, and +8.69,70,72 Importantly, the presence of CH confers a 10 (for mutations3,68 ) to 30 (for CNAs69,70 ) times higher risk for the development of hematological malignancies, including MDS and AML. The risk is even higher in cytopenic patients with CH.73 These observations suggest that MDS might be initiated by CH-related mutations or chromosomal lesions. However, the absolute risk of AML/MDS development remains small (∼1% per year on average). Moreover, the risk of developing AML/MDS seems to differ substantially depending on associated mutations.74 CH with mutations in TP53 and SFs, particularly U2AF1 and SRSF2, is associated with a higher risk of the subsequent development of AML.75 By contrast, DNMT3A and TET2 mutations are most prevalent among CH but seem to confer only a moderate risk of AML development.74,76
CH is more frequently seen among patients with aplastic anemia (AA) successfully treated with immunosuppressive therapy77 and lymphoma patients who received cytotoxic agents.78 However, mutations and CNAs positively selected in these conditions substantially differ from those of healthy individuals and differentially affect the risk of MDS/AML, suggesting a role of autoimmunity and chemotherapy in the positive selection of clones, respectively.77,78 In AA, PIGA and BCOR/BCORL1 mutations as well as 6pUPD are overrepresented, while mutations in DNMT3A and ASXL1, but not TET2, are also common in AA and associated with an elevated risk of progression to MDS/AML. In CH seen in postchemotherapy lymphoma patients, PPMID mutations are most frequently selected, followed by DNMT3A, TET2, and TP53 mutations. It has not fully been investigated whether the clones in CH represent the subsequent AML/MDS clones, although it has been demonstrated that TP53-mutated clones in therapy-related AML can be detected before therapy.61 More studies are needed to elucidate the precise roles of CH in leukemogenesis.76
Functional implications of major genes and pathways mutated in MDS
In contrast to rapidly expanded knowledge about driver mutations in MDS, the mechanisms by which those mutations promote positive selection for the development of MDS have not fully been elucidated. A number of novel mutational targets have been identified through unbiased sequencing analysis, many of which involve previously unknown pathways or genes in cancer development, such as SFs and cohesin. Intensive studies on the functional aspects of these newly identified driver alterations are ongoing, although it is much more complicated than the discovery of mutations by itself. In the remaining part of this review, some functional aspects of newly identified mutations are discussed.
Mutations in genes involved in DNA methylation and chromatin modification
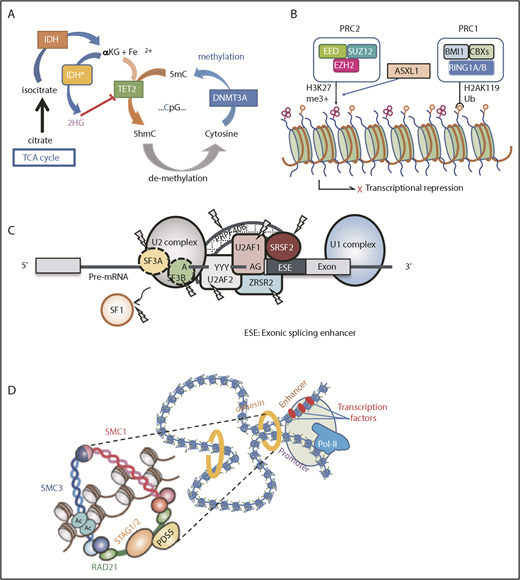
This group of genes is mutated as many as 60% to 70% of MDS cases in MDS, in which TET2, DNMT3A, and ASXL1 are the predominant targets.24,33,79 Together with TP53 and SF mutations, these mutations represent the most common targets of mutations in age-related CH, suggesting their role in the early development of MDS as founder mutations. DNMT3A encodes an enzyme involved in de novo DNA methylation of cytosine to methylated cytosine at CpG residues (Figure 3A).80,81 By contrast, in the presence of Fe2+ and α-ketoglutarate (αKG), TET2 catalyzes the conversion of methylated cytosine to 5′-hydroxymethylcytosine, the initial step of DNA demethylation, which is ultimately oxidized to cytosine.24,82 IDH1 and IDH2 are also common mutational targets in MDS and AML.35,43 Both genes encode 2 different isoforms of isocitrate dehydrogenase, which normally catalyze a conversion of isocitrate to αKG in the cytoplasm and mitochondria, respectively.83 However, when mutated, both isoforms no longer catalyze this reaction but convert αKG to an oncometabolite, 2-hydroxyglutalate, which in turn inhibits many oxidation reactions that require αKG as a substrate, including TET2 and a number of histone demethylases, such as KDM1A, KDM4 (A-E), and KDM6A (UTX).84,85 Another group of genes commonly mutated in MDS includes a number of genes involved in chromatin/histone modification, including several components of PRC2 and a polycomb-related protein, ASXL1. PRC2 is a large protein complex comprising EZH2, EED, JARID2, SUZ12, and other components and is involved in the repression of key genes in hematopoiesis, including the posterior HOXA clusters, via trimethylation of histone H3K27 (Figure 3B).86 Thus, loss-of-function mutations of PRC2 components are thought to deregulate the normal program of hematopoiesis through derepression of key genes in hematopoietic stem/progenitor cells, contributing to positive selection.25,87,88 Physically interacting with PRC2, ASXL1 is required for PRC2 activity, and when mutated and truncated, it inhibits PRC2-mediated trimethylation of H3K27.89,90
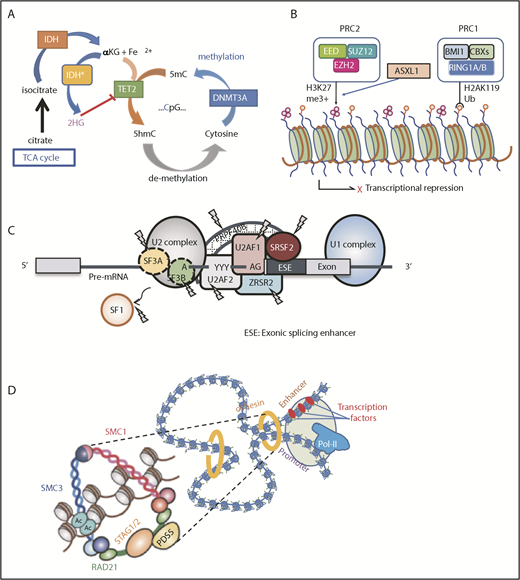
Major functional pathways newly identified to be affected by somatic mutations. Major functional pathways newly identified as somatic alterations include DNA methylation (A), PRC2 and related genes (B), SFs (C), and the cohesion complex (D). For each panel, major targets of mutations are indicated with functional implications. 5hmc, 5′-hydroxymethylcytosine; 5mc, methylated cytosine; TCA, tricarboxylic acid.
Major functional pathways newly identified to be affected by somatic mutations. Major functional pathways newly identified as somatic alterations include DNA methylation (A), PRC2 and related genes (B), SFs (C), and the cohesion complex (D). For each panel, major targets of mutations are indicated with functional implications. 5hmc, 5′-hydroxymethylcytosine; 5mc, methylated cytosine; TCA, tricarboxylic acid.
SF mutations
Another class of the major mutational targets in MDS is SFs (Figure 3C), of which the most frequently affected are those involved in the U2RNP complex,91 including SF3B1, SRSF2, U2AF1, and ZRSR2 and, less commonly, U2AF2, SF1, and SF3A1. Some of these genes (SF3B1, U2AF1, and SRSF2) show discrete mutational hotspots without any frameshift or nonsense alterations, suggesting neomorphic functions of these mutated SFs.41,42,45 By contrast, encoded on the X chromosome, ZRSR2 is inactivated by nonsense or frameshift mutations in most cases, particularly in males; mutations are widely distributed across its entire coding sequence, causing a truncation of the protein to abolish gene function.42 Except in the case with ZRSR2, all SF mutations are heterozygous and rarely cooccur, suggesting that loss of multiple alleles of these vital genes would not be compatible with cell viability and leukemic growth.92 In terms of therapeutics, it is argued that this might be potentially used to specifically kill SF-mutated cells by inducing synthetic lethality using SF inhibitors.93
All SF mutations cause widespread changes in RNA splicing, including alternative 3′- or 5′-splice site usage, enhanced or reduced intron retention, and inclusion or exclusion of cassette exons,94 although patterns of splicing alterations differ substantially depending on the type of SF affected.95-97 While the roles of these SF mutations in the pathogenesis of MDS remains to be fully elucidated, several genes have been reported as functional targets of mutated SFs in MDS. For example, mutated-SF3B1 promotes alternative 3′-splice site usage in ABCB7 and other genes involved in mitochondrial iron transportation and leads to their premature truncation and reduced gene expression, which is implicated in the formation of RS caused by abnormal iron deposition around mitochondria.98 Other putative targets of mutant SFs implicated in MDS pathogenesis include MAP3K7, NF1, and PDS5A for SF3B1 mutation and EZH2, CASP8, CDK10 for SRSF2 mutation.96-98 Despite a number of targets of missplicing unique to each SF mutation, currently, few common targets of missplicing of different SFs are known to explain common MDS phenotypes.
Cohesin mutations
Cohesin is a multimeric protein complex comprising RAD21, SMC1A, SMC3, and a STAG protein (STAG1-3).99 Taking a ring-like structure, these molecules are recruited on the chromatin in concert with cohesin-associated molecules, such as NIPBL and ESCO proteins, as well as CTCF, to participate in the regulation of gene expression through the formation of large-scale chromatin structures (Figure 3D). In MDS and other myeloid malignancies, ∼10% to 15% of the cases harbor mutations in cohesin and related molecules.46 Cohesin mutations are largely mutually exclusive, suggesting synthetic lethality of mutations in multiple cohesion components. Recent studies showed that cohesin-deficient cells had altered chromatin structures that allowed for accessibility to a number of transcription factors, including RUNX1, GATA2, and ERG, suggesting functional interaction of cohesion mutations and these transcription factors.100
Del(5q) and TP53 mutations with CK
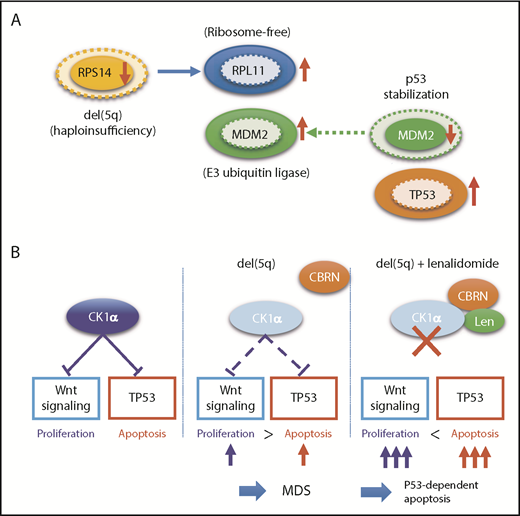
The molecular pathogenesis of del(5q) has been extensively studied, and important roles of enhanced p53-mediated apoptosis have been highlighted, together with abnormally activated Wnt/β-catenin signaling and deregulated inflammatory signaling. Del(5q) causes haploinsufficiency of several genes within the critical 5q deletion, including RPS14, CSNK1A1, APC, DDX41, and miR-145/miR-146a, of which RPS14, CSNK1A1, and miR-145 are included in the critical 5q deletion. RPS14 encodes a small subunit of ribosomal components. RPS14 haploinsufficiency induces an enhanced translation of other ribosomal components, such as RPL11, which bind additional MDM2 molecules, leading to the reduction of the p53-bound MDM2 to stabilize p53 through reduced degradation in the proteasome (Figure 4A). Reduced RPS14 expression is also implicated in a block in erythroid differentiation via a marked elevation of S100A8 and S100A9.10 Encoding casein kinase 1α, CSNK1A1 negatively regulates both Wnt/β-catenin signaling and p53 activity. Thus, haploinsufficient CSNK1A1 results in an enhanced apoptosis of bone marrow progenitors through activated p53 activity, while it also leads to derepression of Wnt/β-catenin signaling, through which promoting proliferation of hematopoietic progenitors (Figure 4B).101-103 These mechanisms not only explain del(5q) pathophysiology characterized by ineffective blood cell production in the face of hyperplastic bone marrow but also provide a basis for frequent evolution of TP53-mutated clones in del(5q) patients, in whom enhanced apoptotic activity should critically depend on intact p53 activity. Surprisingly, the haploinsufficient CSNK1A1-mediated del(5q) pathogenesis also provides a unique mechanism of the pharmacological effect of lenalidomide for del(5q)-positive MDS, which is based on the specific binding of lenalidomide to human, but not mouse, CRBN, a Cul4-based E3 ubiquitin ligase.104 When it binds to CRBN, lenalidomide changes the substrate specificity of CRBN, allowing lenalidomide-bound CRBN to recruit CSNK1A1 as a neosubstrate for ubiquitination and subsequent degradation in proteasome. This causes a further reduction of haploinsufficient CSNK1A1 levels in del(5q) cells, leading to p53-dependent apoptosis of del(5q) cells (Figure 4B).104,105
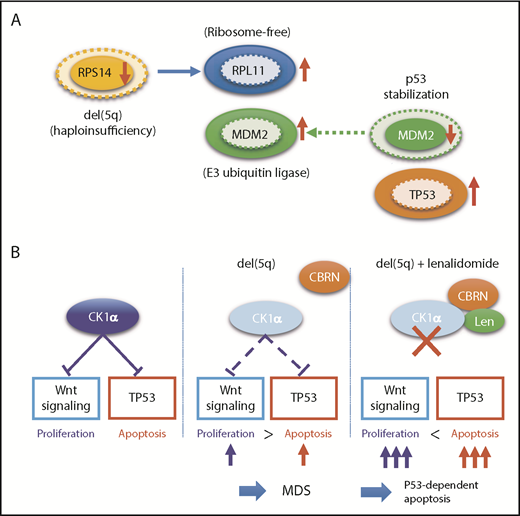
Functional interaction between TP53 and del(5q). (A) Effects of RPS14 haploinsufficiency on TP53 protein levels. In this model, haploinsufficient RPS14 results in an increase in ribosome-free RPL11, which by competing with TP53 for binding MDM2, an E3 ubiquitin ligase that also targets p53 for destruction, inhibits TP53 destruction in the proteasome, leading to an elevated TP53 level and an enhanced TP53-mediated apoptosis of erythroid progenitors.101,102 Protein levels corresponding to diploid and haploid status of RPS14 are indicated by solid and broken lines, respectively. (B) Effects of haploinsufficiency of CSKN2A on Wnt signaling and TP53 activation and their implication in the mechanism behind the high response of del(5q) clones to lenalidomide. Haploinsufficiency of RPS14 and CSKN2A induced TP53 activation, leading to enhanced apoptosis of del(5q) clones, while haploinsufficient CSKN2A results in the activation of Wnt signaling and cell proliferation. Apoptosis of del(5q) clones, particularly that induced by lenalidomide, critically depends on intact TP53, which explains the strong correlation between del(5q) and mutated TP53.
Functional interaction between TP53 and del(5q). (A) Effects of RPS14 haploinsufficiency on TP53 protein levels. In this model, haploinsufficient RPS14 results in an increase in ribosome-free RPL11, which by competing with TP53 for binding MDM2, an E3 ubiquitin ligase that also targets p53 for destruction, inhibits TP53 destruction in the proteasome, leading to an elevated TP53 level and an enhanced TP53-mediated apoptosis of erythroid progenitors.101,102 Protein levels corresponding to diploid and haploid status of RPS14 are indicated by solid and broken lines, respectively. (B) Effects of haploinsufficiency of CSKN2A on Wnt signaling and TP53 activation and their implication in the mechanism behind the high response of del(5q) clones to lenalidomide. Haploinsufficiency of RPS14 and CSKN2A induced TP53 activation, leading to enhanced apoptosis of del(5q) clones, while haploinsufficient CSKN2A results in the activation of Wnt signaling and cell proliferation. Apoptosis of del(5q) clones, particularly that induced by lenalidomide, critically depends on intact TP53, which explains the strong correlation between del(5q) and mutated TP53.
Summary and future directions
Advanced sequencing technologies have clarified a comprehensive registry of driver mutations in MDS, their frequency and mutual correlations, and their impact on disease phenotypes and prognosis in large cohorts of MDS patients. Now, unified efforts are underway by the International Working Group for the Prognosis of MDS, in which a huge number of MDS patients are being investigated for major driver mutations to confirm and further extend these initial findings. These studies should provide a solid basis for novel molecular classification of MDS, which will help make accurate diagnosis and prognostication, as well as a better choice of therapy, to ultimately improve patient survival. Finally, despite the large number of driver mutations that have been newly detected with advanced genomics, the functional roles of these mutations in MDS pathogenesis remain to be elucidated for many drivers. For example, SF mutations unequivocally play critical roles in MDS pathogenesis, but no functional evidence has been obtained supporting that SF-mutated hematopoietic cells achieve a clonal dominance; SF-mutated stem cells show a compromised competitive repopulation potential over normal stem cells in mouse models.96,98,106-108 The importance of understanding the functional and molecular mechanisms of the driver mutations identified through genetic analyses cannot be overstated.
Acknowledgments
The author thanks Hideki Makishima and Testuichi Yoshizato for helping to generate figures.
This work was supported by the Project for Development of Innovative Research on Cancer Therapeutics (P-DIRECT, 16cm0106501h0001), Practical Research for Innovative Cancer Control (15Ack0106014h0002 and 16ck0106073h0003), and the Project for Cancer Research and Therapeutic Evolution (P-CREATE, 16cm0106501h0001) from the Japan Agency for Medical Research and Development, Japan Society for the Promotion of Science KAKENHI (15H05668 and 15H05909 (S.O.).
Authorship
Contribution: S.O. is responsible for designing and writing the contents of the manuscript.
Conflict-of-interest disclosure: The author declares no competing financial interests.
Correspondence: Seishi Ogawa, Department of Pathology and Tumor Biology, Graduate School of Medicine, Kyoto University, Yoshida-Konoe-cho, Sakyo-ku, Kyoto 606-8501, Japan: e-mail: sogawa-tky@umin.ac.jp.