Key Points
Terminal galactose exposure on VWF is significantly increased in a subgroup of patients with low VWF compared with controls.
Enhanced clearance of aberrantly glycosylated VWF contributes to the etiology underlying low VWF.

Abstract
Glycan determinants on von Willebrand factor (VWF) play critical roles in regulating its susceptibility to proteolysis and clearance. Abnormal glycosylation has been shown to cause von Willebrand disease (VWD) in a number of different mouse models. However, because of the significant technical challenges associated with accurate assessment of VWF glycan composition, the importance of carbohydrates in human VWD pathogenesis remains largely unexplored. To address this, we developed a novel lectin-binding panel to enable human VWF glycan characterization. This methodology was then used to study glycan expression in a cohort of 110 patients with low VWF compared with O blood group-matched healthy controls. Interestingly, significant interindividual heterogeneity in VWF glycan expression was seen in the healthy control population. This variation included terminal sialylation and ABO(H) blood group expression on VWF. Importantly, we also observed evidence of aberrant glycosylation in a subgroup of patients with low VWF. In particular, terminal α(2-6)-linked sialylation was reduced in patients with low VWF, with a secondary increase in galactose (Gal) exposure. Furthermore, an inverse correlation between Gal exposure and estimated VWF half-life was observed in those patients with enhanced VWF clearance. Together, these findings support the hypothesis that loss of terminal sialylation contributes to the pathophysiology underpinning low VWF in at least a subgroup of patients by promoting enhanced clearance. In addition, alterations in VWF carbohydrate expression are likely to contribute to quantitative and qualitative variations in VWF levels in the normal population. This trial was registered at www.clinicaltrials.gov as #NCT03167320.
Introduction
von Willebrand disease (VWD) represents the most common inherited bleeding disorder and is caused by either a quantitative or qualitative deficiency in von Willebrand factor (VWF).1,2 International consensus guidelines recommend that patients with partial quantitative deficiencies of VWF should be considered in 2 distinct subsets.3,4 First, patients with significant reductions in plasma VWF levels (<30 IU/dL) and a bleeding phenotype should be classified as type 1 VWD. The majority of these patients have dominant-negative VWF gene missense mutations. Second, patients with intermediate plasma VWF:Ag levels (30-50 IU/dL) should be considered a separate entity and diagnosed as having low VWF levels.5 Importantly, VWF mutations are significantly less common in this cohort of patients, with milder reductions in plasma VWF levels.3,4 Furthermore, linkage studies have demonstrated that in many families with low VWF, inheritance is independent of the VWF locus on chromosome 12.6-8 Together, these data support the hypothesis that as yet unidentified modifier loci contribute to the pathobiology underlying low VWF levels.
Before its secretion from endothelial cells, VWF undergoes complex posttranslational modifications including significant glycosylation.9 As a consequence, each VWF monomer contains 12 N- and 10 O-linked glycan structures that together account for 20% of the final monomeric mass. The N- and O-glycans of human VWF demonstrate significant heterogeneity.10-12 However, sialylated bi-antennary complex-type chains account for approximately 80% of the N-glycan structures expressed on VWF.11 In contrast, disialyl core 1 (T-antigen) structures account for approximately 70% of the total O-linked glycome on VWF.12 Importantly, covalently linked ABO(H) blood group carbohydrate determinants have also been described as terminal sugar residues on a proportion of both the N- and O-linked glycans of VWF.11,12
Recent studies have demonstrated that glycan determinants play critical roles in regulating multiple aspects of VWF biology, including its biosynthesis, proteolysis, and clearance.12-22 For example, site-directed mutagenesis studies have highlighted that the N-linked glycan chains at N99, N857, N2400, and N2790 influence VWF synthesis and secretion within endothelial cells.19 Furthermore, glycosylation, notably at N1515 and N1574, has also been shown to significantly influence VWF clearance.18,20,21 Although ABO(H) blood group determinants are only expressed on 13% of the N-glycans and 1% of the O-glycans, nevertheless, ABO has a significant effect on VWF clearance, such that plasma VWF levels are 20% to 30% lower in blood group O compared with non-O individuals.11,12,23-25 Terminal sialylation has also been shown to play a major role in regulating VWF clearance. Enzymatic removal of sialic acid residues from VWF ex vivo markedly reduces plasma half-life.16,18 Moreover, genetic inactivation of a specific sialyltransferase (ST3Gal-IV) in mice causes reduced plasma VWF levels as a result of significantly enhanced clearance.26
The biological importance of glycan determinants in regulating VWF clearance is perhaps best exemplified by the RIIIS/J mouse.13,27,28 This murine strain has plasma VWF levels that are markedly reduced compared with other strains.27 These reduced VWF levels were shown to result from enhanced clearance secondary to aberrant VWF glycosylation. Thus, the RIIIS/J mouse provides naturally occurring proof of principle that abnormalities of VWF glycosylation independent of the VWF gene can cause quantitative VWD through enhanced VWF clearance.13,27,28 Alteration in VWF glycosylation, and/or variation in plasma VWF clearance, both therefore constitute biologically plausible candidate mechanisms through which VWF-independent modifiers could contribute to low VWF levels. In this context, it is perhaps surprising that the importance of carbohydrates in the pathogenesis of human VWD remains poorly explored, largely because of the significant technical challenges associated with accurate determination of VWF glycan composition. Two previous studies reported increased binding of the lectin Ricinus communis agglutinin I (RCA-I) to VWF (suggestive of increased galactose, Gal, or N-acetylgalactosamine, GalNAc expression) in patients with VWD compared with controls.26,29 In addition, van Schooten et al reported significantly enhanced binding of the peanut agglutinin (suggesting increased O-linked T antigen expression) in a cohort of patients with type 1 VWD compared with healthy controls.30 Although these preliminary data are revealing, it is important to appreciate that the numbers of patients with VWD involved in these initial studies were relatively small (n = 19, 26, and 32, respectively), and that a variety of different VWD subtypes were studied. Nevertheless, these data support the hypothesis that changes in VWF carbohydrate structures may play a role in the pathogenesis underlying quantitative VWD, particularly in patients with low VWF, in the majority of whom the disease is not linked to the VWF gene locus. To specifically address this question, we have developed a novel lectin-panel methodology to systematically study VWF glycosylation profile in patients recently described in the Low VWF Ireland Cohort (LoVIC) study.31
Methods
Low-VWF patients and controls
As previously described, 126 adult (>18 years old) patients with low VWF levels were enrolled into the LoVIC.31 All patients had significant personal bleeding histories and lowest plasma VWF levels of at least 30 IU/dL, but less than 50 IU/dL, measured on 2 separate occasions at least 3 months apart. From this cohort, 110 patients with low VWF were selected for VWF glycan analysis. Blood samples were also collected from 68 healthy volunteer Irish adults. All patients and control subjects were blood group O and had an age median of 38 years for both groups. The study was approved by the Local Research Ethics Committee of St James’ Hospital, and written informed consent was obtained from all participants.
Plasma VWF:Ag and VWFpp levels
Plasma VWF:Ag levels were measured using a latex particle enhanced immunoturbidimetric assay on an automated coagulometer (ACL Top 700; Instrumentation Laboratories, Milan, Italy) at the same coagulation laboratory. Plasma VWF propeptide (VWF:pp) levels were determined by enzyme-linked immunosorbent assay (ELISA), using monoclonal antibodies CLB-Pro 35 and CLB-Pro 14.3-HRP (Sanquin, Amsterdam, The Netherlands), as previously described.32,33 For each low-VWF patient and control subject enrolled in the study, VWF:Ag half-life was calculated using their individual plasma VWF:Ag and VWFpp levels, as previously reported.32
Isolation of plasma-derived VWF
Plasma-derived VWF (pdVWF) was purified from the VWF-containing concentrate Haemate P (CSL Behring, Prussia, PA), as previously described.18,22 Briefly, Haemate P suspension was applied to a Sepharose CL-2B XK 16/70 gel filtration column (GE Healthcare, Amersham, Little Chalfont, United Kingdom). Elute fractions were then assessed for VWF:Ag, multimer distribution, and purity. After purification, pdVWF was treated with specific exoglycosidases, including α2-3 neuraminidase, α2-3,6,8,9 neuraminidase, β1-4 galactosidase, and N-glycosidase F (PNGase F), to generate a series of different VWF glycoforms with modified glycosylation profiles, as previously reported.22
Lectin plate-binding assessment of VWF glycans
VWF glycosylation profiles were assessed using a series of lectin plate-binding ELISAs. In brief, VWF:Ag concentration in purified VWF preparations and plasma samples were first determined by ELISA or immunoturbidimetric assay, respectively. Subsequently, all plasma samples were diluted in PBS-Tween 0.1% to achieve a final VWF concentration of 1 μg/mL. Before VWF incubation, the polyclonal anti-VWF capture antibody was digested with PNGase F (New England Biolabs, Hitchin, United Kingdom) overnight at 37°C. After blocking, VWF test samples were incubated for 2 hours at 37°C. Subsequently, biotinylated lectins, Sambucus nigra agglutinin (SNA; 0.1 μg/mL), Maackia amurensis lectin II (MAL-II; 2.5 μg/mL), wheat germ agglutinin (WGA; 2.5 μg/mL), Erythrina cristagalli agglutinin (ECA; 0.7 μg/mL), Ricinus communis agglutinin I (RCA-I; 0.5 μg/mL), and Ulex europaeus agglutinin I (UEA-I; 2 μg/mL; Vector Laboratories, Peterborough, United Kingdom), were incubated for 1 hour. Lectin binding was detected with high-sensitivity streptavidin-HRP (Pierce, Thermo Fisher Scientific, Hemel Hempstead, United Kingdom). Absorbance was read at 450 nm, and lectin binding was expressed as a percentage of pooled normal plasma. The intra-assay and inter-assay coefficients of variation were all less than 10% and less than 15%, respectively.
Neuraminidase activity in low-VWF plasma samples
Plasma neuraminidase activity was measured in selected low-VWF patients using a commercial Neuraminidase Assay Kit (Abcam, Cambridge, United Kingdom), according to the manufacturers’ instructions. As recommended, serial dilutions for neuraminidase standard and test samples was assessed.
Glycosylation gene sequencing
ST6GAL1 and ST6GAL2, ST3GAL1-6, FUT1 and FUT2, and NEU1-4 were sequenced using a custom genetic array, as previously described.31 In brief, pooled libraries were sequenced on MiSeq sequencer (Illumina) by the Genomics and Microarray Core of University of Colorado Denver, using the 300-bp paired-end sequencing. The average reads per bases quality for all samples is at least 79%. For bioinformatics analysis, reads that passed the quality filter step were mapped to the reference human genome sequence (hg19) with GSNAP (Genomic Short-read Nucleotide Alignment Program). The snpEff (variant annotation), dbNSFP (database for nonsynonymous SNPs’ functional predictions), ExAC, and 1000 genomes were used as before.31
Data presentation and statistical analysis
Statistical analyses were performed using GraphPad Prism software version 5.0 for Windows (GraphPad Software Inc., San Diego, CA). Normally distributed data were expressed as mean values ± standard error of the mean. To assess statistical differences, data were analyzed using the Mann-Whitney U test and the Spearman correlation coefficient where appropriate. For all statistical tests, P < .05 was considered significant.
Results
Application of novel multilectin binding panel to assess plasma VWF glycan expression
In previous studies that investigated VWF glycans in patients with VWD, only 3 different lectins were reported (RCA-I, ECA, and peanut agglutinin).26,29,30 We investigated lectin binding to VWF, using plate-binding ELISAs for a range of other lectins, but observed significant high background signals with SNA and RCA-I (supplemental Figure 1A-B, available on the Blood Web site). Because rabbit polyclonal anti-VWF antibody contains N-linked glycans, we assumed that these carbohydrate structures might be responsible for the background. To address this limitation, the anti-VWF capture antibody was treated with peptide N-glycosidase F (PNGase F). In plate-binding assays, we confirmed that PNGase F treatment had no significant effect on the binding of polyclonal anti-VWF to human VWF (supplemental Figure 1C). Importantly, however, PNGase F pretreatment of the capture antibody significantly reduced background signal in VWF plate-binding assays for SNA and RCA-I and surface plasmon resonance studies (supplemental Figure 1A-B,D).
Using this PNGase pretreatment approach, a panel of modified lectin ELISAs was developed to study VWF binding to SNA, RCA-I, ECA, MAL-II, WGA, and UEA-I, respectively. To validate these individual lectin ELISAs, we first tested a range of human plasma-derived VWF (pdVWF) glycoforms generated using a series of specific exoglycosidases (including α2-3,6,8,9 neuraminidase, α2-3 neuraminidase, β1-4 galactosidase, and PNGase F). SNA has specific affinity for terminal α(2-6)-linked sialic acid predominantly expressed on the N-glycans of pdVWF. Next, SNA binding was ablated after digestion with α2-3,6,8,9 neuraminidase and markedly reduced after PNGase F treatment (Figure 1A). In contrast, MAL-II has affinity for terminal α(2-3)-linked sialic acid linked to β1-3 galactose expressed on the O-glycans of pdVWF. Thus, MAL-II binding was undetectable after treatment with either α2-3 neuraminidase or α2-3,6,8,9 neuraminidase, but was unaffected by PNGase F digestion (Figure 1B). The lectin WGA recognizes sialic acid with a preference for α(2-3)-linked rather than α(2-6)-linked sialic acid residues, but also binds with higher affinity to exposed N-acetylglucosamine (GlcNAc) residues.34-37 As a consequence, its binding to VWF was significantly enhanced after digestion with neuraminidases and galactosidase (Figure 1C).
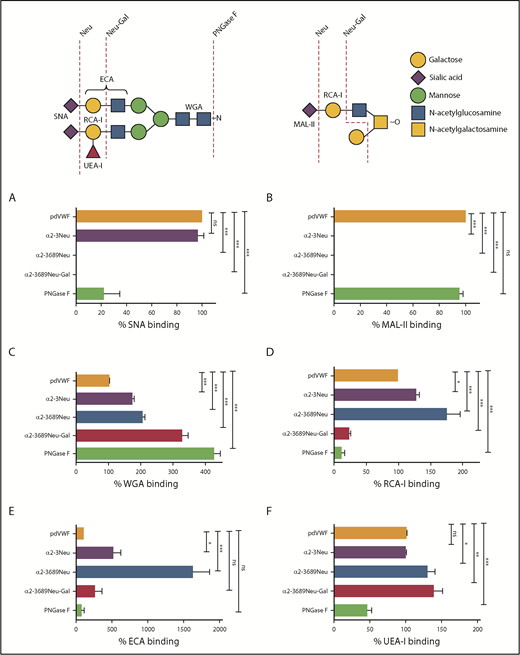
Generation of lectin plate-binding assays to assess plasma VWF glycan expression. (A-F) Lectin plate-binding assays were validated by assessing binding to a series of different VWF glycoforms generated by ex vivo treatment of purified pdVWF with specific exoglycosidases (summarized in schematic). After glycosidase digestions, residual glycan expression on VWF glycoforms were analyzed using SNA (A), MAL-II (B), WGA (C), RCA-I (D), ECA (E), and UEA-I (F). pdVWF was digested with α2-3 neuraminidase (purple bars), α2-3,6,8,9-neuraminidase (blue bars), α2-3,6,8,9-neuraminidase followed by β1-4 galactosidase (red bars), or PNGase F (green bars), respectively. All ELISAs were performed in triplicate, and results were expressed as a percentage of binding to untreated pdVWF. Results presented represent the mean values ± standard error of the mean. *P < .05; **P < .01; ***P < .001; Mann-Whitney U test. ns, not significant.
Generation of lectin plate-binding assays to assess plasma VWF glycan expression. (A-F) Lectin plate-binding assays were validated by assessing binding to a series of different VWF glycoforms generated by ex vivo treatment of purified pdVWF with specific exoglycosidases (summarized in schematic). After glycosidase digestions, residual glycan expression on VWF glycoforms were analyzed using SNA (A), MAL-II (B), WGA (C), RCA-I (D), ECA (E), and UEA-I (F). pdVWF was digested with α2-3 neuraminidase (purple bars), α2-3,6,8,9-neuraminidase (blue bars), α2-3,6,8,9-neuraminidase followed by β1-4 galactosidase (red bars), or PNGase F (green bars), respectively. All ELISAs were performed in triplicate, and results were expressed as a percentage of binding to untreated pdVWF. Results presented represent the mean values ± standard error of the mean. *P < .05; **P < .01; ***P < .001; Mann-Whitney U test. ns, not significant.
RCA-I binds to exposed terminal β-linked Gal residues. Thus, RCA-I binding was significantly enhanced after treatment with either α2-3 neuraminidase or α2-3,6,8,9 neuraminidase, and attenuated after pdVWF digestion with β1-4 galactosidase or PNGase F (Figure 1D). Importantly, using both modified ELISA assays and surface plasmon resonance (supplemental Figure 1E), we observed a clear inverse correlation between expression of terminal sialylation and the exposure of subterminal Gal residues, respectively. The lectin ECA also binds to Gal residues, particularly those present in polylactosamine repeat structures. Consequently, ECA binding was significantly increased after removal of terminal sialylation with either α2-3 neuraminidase or α2-3,6,8,9 neuraminidase (Figure 1E). The lectin UEA-I recognizes the ABO blood group, H-antigen, on VWF. In keeping with previous studies demonstrating that ABH carbohydrates are predominantly expressed on the N-glycans of VWF,11 UEA-I binding was significantly reduced after PNGase F digestion (Figure 1F). Taken together, the data from this series of ex vivo glycosidase digestions are in keeping with previous mass spectrometry studies performed on commercial VWF-containing concentrates and validate the utility of our modified lectin ELISA panel as a tool for screening glycan profile on pdVWF.
VWF glycan heterogeneity in normal population
The modified lectin ELISAs were first used to investigate variation in pdVWF glycan expression in a series of 68 healthy volunteers. For each of the lectins studied, VWF binding varied over a wide range, confirming that VWF glycosylation is heterogeneous in nature (Figure 2). Interestingly, despite the evidence that terminal sialylation plays a critical role in regulating VWF clearance, significant interindividual variation in VWF binding was observed for all 3 lectins (SNA, MAL-II, and WGA), recognizing terminal sialic acid expression. These findings suggest that quantitative N- and O-linked sialylation on VWF varies between normal individuals. In keeping with these data, exposure of subterminal β-Gal residues (RCA-I and ECA binding) also varied significantly between different individuals. Finally, despite the fact that all the control subjects were blood group O, there was also significant variation in H antigen expression (UEA-I binding). Interestingly, VWF multimer distribution had no significant effect on lectin binding, with similar binding observed for purified high- and low-molecular-weight multimer fractions, respectively (supplemental Figure 1F). Collectively, these data highlight that there is marked interindividual heterogeneity in VWF glycan expression, even among healthy controls.
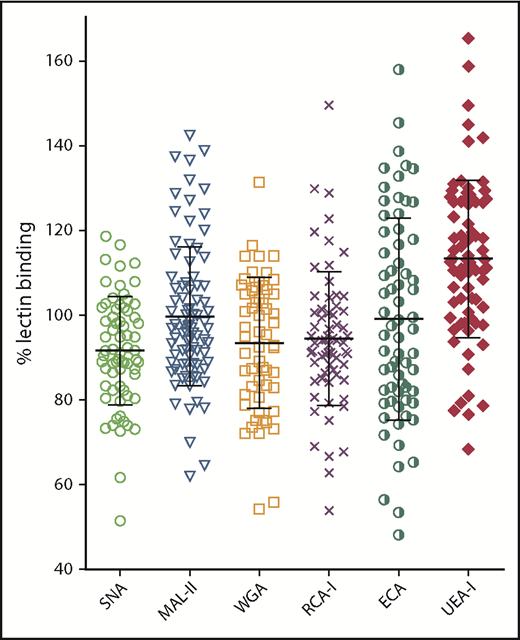
Glycan expression on plasma-derived VWF demonstrates marked heterogeneity in healthy controls. Using SNA-, MAL-II–, WGA-, RCA-I–, ECA-, and UEA-1–modified lectin plate-binding assays, pdVWF glycosylation profiles were examined in a cohort of 68 healthy volunteer blood donors. All ELISAs were performed in duplicate and results expressed as a percentage of binding to reference plasma.
Glycan expression on plasma-derived VWF demonstrates marked heterogeneity in healthy controls. Using SNA-, MAL-II–, WGA-, RCA-I–, ECA-, and UEA-1–modified lectin plate-binding assays, pdVWF glycosylation profiles were examined in a cohort of 68 healthy volunteer blood donors. All ELISAs were performed in duplicate and results expressed as a percentage of binding to reference plasma.
VWF glycans in patients with low VWF
Using our lectin panel, we next investigated VWF glycosylation profiles in a large cohort (n = 110) of patients with well-characterized low VWF levels (30-50 IU/dL) enrolled in the recently described LoVIC study.31 Importantly, all these patients were blood group O. Unsurprisingly, given the heterogeneity seen in the control population, significant interindividual variation in lectin-VWF binding was also observed among low-VWF patients (Figures 3 and 4). Interestingly, however, we observed that SNA binding was significantly reduced in the low-VWF cohort compared with controls (P < .01; Figure 3A). SNA preferentially recognizes α(2-6)-linked sialic acid residues, which are predominantly expressed as terminal sugars on the N-linked glycans of VWF. In contrast, MAL-II and WGA binding in the patient and control cohorts were similar (Figure 3B-C), suggesting that low-VWF patients do not have a quantitative difference in α(2-3)-linked sialylation.
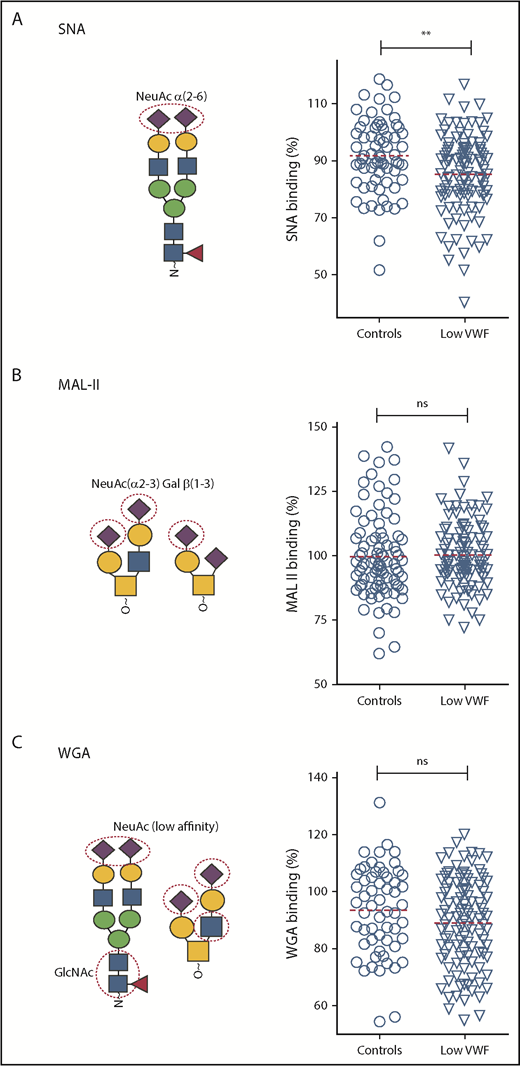
Terminal α(2-6)-linked sialylation on VWF is significantly reduced in patients with low VWF compared with controls. Lectin plate-binding ELISAs were performed in plasma samples collected from 110 patients with low VWF (▿) and compared with O blood group-matched control subjects (○). N- and O-linked sialylation on VWF were assessed using the lectins SNA, MAL-II, and WGA. As indicated in the illustrations, SNA (A) binds preferentially to terminal α(2-6)-linked sialic acid (NeuAc), which is predominantly expressed on the N-glycans of VWF; MAL-II (B) has affinity for terminal α(2-3)-linked sialic acid expressed on the O-glycans of VWF; and WGA (C) binds to sialic acid with a preference for α(2-3)-linked rather than α(2-6)-linked sialic acid residues, but also recognizes exposed N-acetylglucosamine (GlcNAc) residues. All ELISAs were performed in duplicate and results expressed as a percentage of binding to reference plasma (100%), using the slope of different dilutions. SNA binding was significantly reduced in patients with low VWF compared with controls. **P < .01, Mann-Whitney U test.
Terminal α(2-6)-linked sialylation on VWF is significantly reduced in patients with low VWF compared with controls. Lectin plate-binding ELISAs were performed in plasma samples collected from 110 patients with low VWF (▿) and compared with O blood group-matched control subjects (○). N- and O-linked sialylation on VWF were assessed using the lectins SNA, MAL-II, and WGA. As indicated in the illustrations, SNA (A) binds preferentially to terminal α(2-6)-linked sialic acid (NeuAc), which is predominantly expressed on the N-glycans of VWF; MAL-II (B) has affinity for terminal α(2-3)-linked sialic acid expressed on the O-glycans of VWF; and WGA (C) binds to sialic acid with a preference for α(2-3)-linked rather than α(2-6)-linked sialic acid residues, but also recognizes exposed N-acetylglucosamine (GlcNAc) residues. All ELISAs were performed in duplicate and results expressed as a percentage of binding to reference plasma (100%), using the slope of different dilutions. SNA binding was significantly reduced in patients with low VWF compared with controls. **P < .01, Mann-Whitney U test.
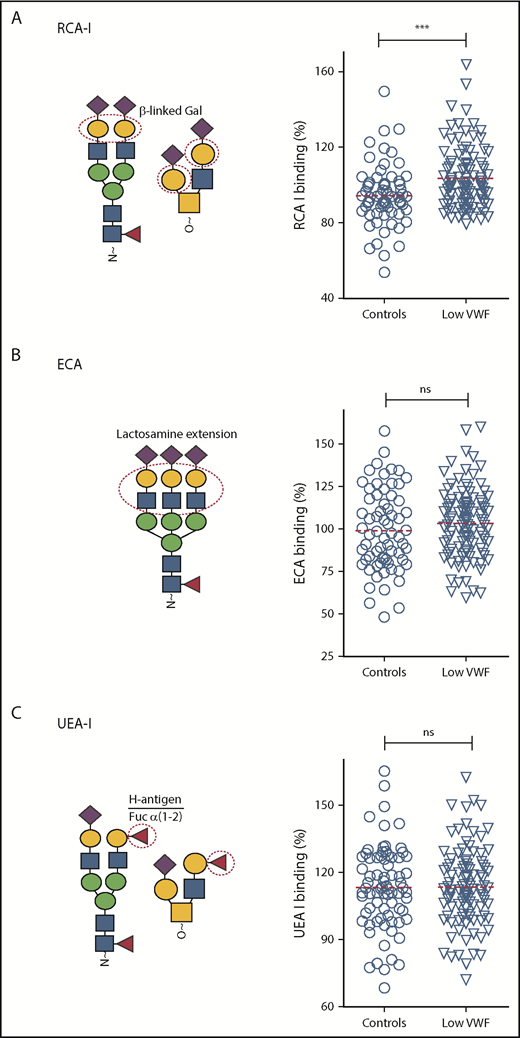
β-Gal expression is significantly increased in patients with low VWF compared with controls. Additional lectin plate-binding ELISAs were performed to further characterize VWF glycan expression in patients with low VWF (▿) compared with O blood group-matched control subjects (○). As indicated in the illustrations, RCA-I (A) binds to exposed terminal β-linked Gal residues; ECA (B) also binds to Gal residues, particularly those present in polylactosamine repeat structures; UEA-I (C) recognizes the ABO blood group, H-antigen, on VWF. All ELISAs were performed in duplicate and results expressed as a percentage of binding to reference plasma (100%) using the slope of different dilutions. RCA-I binding was significantly increased in patients with low VWF compared with controls. ***P < .001, Mann-Whitney U test.
β-Gal expression is significantly increased in patients with low VWF compared with controls. Additional lectin plate-binding ELISAs were performed to further characterize VWF glycan expression in patients with low VWF (▿) compared with O blood group-matched control subjects (○). As indicated in the illustrations, RCA-I (A) binds to exposed terminal β-linked Gal residues; ECA (B) also binds to Gal residues, particularly those present in polylactosamine repeat structures; UEA-I (C) recognizes the ABO blood group, H-antigen, on VWF. All ELISAs were performed in duplicate and results expressed as a percentage of binding to reference plasma (100%) using the slope of different dilutions. RCA-I binding was significantly increased in patients with low VWF compared with controls. ***P < .001, Mann-Whitney U test.
In preliminary experiments, we showed that removal of terminal sialylation from the N-glycan of human VWF resulted in enhanced expression of subterminal Gal residues, and thus increased RCA-I binding (Figure 1C,F; supplemental Figure 1C). In support of the observation that terminal α(2-6)-linked sialylation is reduced in a subset of patients with low VWF, we further observed that RCA-I binding was significantly increased in these patients compared with in healthy controls (P < .001; Figure 4A). No significant difference in ECA binding was seen between the low-VWF cohort and the healthy control population (Figure 4B). Furthermore, in spite of the major effect of ABO blood group in regulating VWF clearance,38 similar levels of H antigen expression (UAE-I binding) were seen in both groups (Figure 4C).
Of the 110 low-VWF patients investigated, previous studies had identified potential pathological VWF sequence variations in 35 subjects.31 Consequently, we further investigated whether lectin binding in the presence or absence of VWF gene mutations in our low-VWF cohort. Interestingly, differences in RCA-I binding were observed between VWF mutation-negative low-VWF patients compared with VWF mutation-positive low-VWF and healthy control subjects, respectively (supplemental Figure 2). Although it did not quite achieve statistical significance, we observed higher RCA-I binding in VWF mutation-negative patients vs VWF mutation-positive low-VWF subjects (P = .056). In addition, RCA-I binding was significantly higher in VWF mutation-negative low-VWF subjects compared with controls (P < .005). In contrast, no significant difference in RCA-I binding was observed for VWF mutation-positive low-VWF patients compared with healthy controls (P = .52). For all the other lectins studied, no differences in binding were observed for VWF mutation-negative compared with VWF mutation-positive low-VWF patients (supplemental Figure 3). Cumulatively, these findings demonstrate that VWF glycan expression is different in patients with low VWF compared with healthy control subjects. In particular, there is a significant reduction in α(2-6)-linked sialic acid that results in a secondary increase in subterminal Gal exposure.
Etiology underlying reduced α2-6 sialylation in low VWF
We hypothesized that the reduction in α(2-6)-linked sialic acid in patients with low VWF might be a result of enhanced plasma neuraminidase activity. To address this question, plasma neuraminidase activity levels were determined in a subset of 14 LoVIC patients with the highest levels of SNA binding to VWF compared with the 13 LoVIC patients with the lowest levels of SNA binding (Figure 5A). Similar levels of neuraminidase activity were observed in both groups (Figure 5B). To investigate endothelial cell sialylation and the glycan trimming in circulation in patients with low VWF, plasma was collected from a subgroup (n = 23) of LoVIC patients immediately before and 1 hour after 1-deamino-8-D-arginine vasopressin (DDAVP) administration. All patients demonstrated significantly increased plasma VWF:Ag levels post-DDAVP (median, 61.2 vs 174.2 IU/dL; P < .001; Figure 5C). Interestingly, we found that SNA binding to VWF secreted after DDAVP was significantly elevated compared with circulating steady-state plasma VWF (median, 87.3% vs 103.3%; P < .05; Figure 5D). This observation suggests that high-molecular-weight VWF multimers stored within Weibel Palade bodies are highly sialylated compared with VWF in circulation, and is in keeping with recent evidence suggesting that plasma protein glycans undergo progressive glycan trimming after secretion into the plasma.39 In contrast to the increased SNA binding observed after DDAVP administration, no significant differences in UEA-I binding to blood group H expression on VWF were seen (supplemental Figure 4). To further investigate the biology underlying the reduction in terminal α(2-6)-linked sialic acid expression in patients with low VWF, next-generation sequence analysis studies were performed for a series of genes including sialyltransferases (ST3GAL and ST6GAL), fucosyltransferases (FUT), and neuraminidases (NEU). No enrichment of variants in the coding regions of ST6GAL1 and ST6GAL2 were identified in low-VWF patients (supplemental Table 1). Both these genes encode for α2-6 sialyltransferases. Although a single nucleotide variant in the 3′ untranslated region of ST6GAL2 (rs776819723) was observed with higher frequency in our cohort compared with existing databases (supplemental Table 1), the potential consequences of this 3′ untranslated region alteration remain unknown.
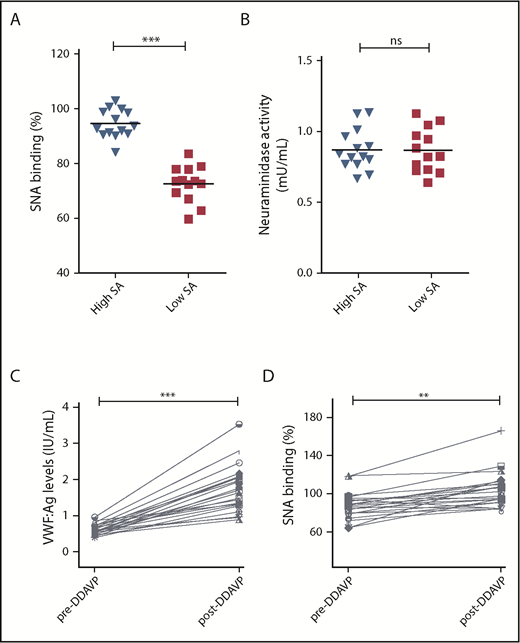
Etiology underlying reduced α(2-6)-linked sialylation in patients with low VWF. (A-B) To investigate the pathophysiology involved in the reduction in α(2-6)-linked sialylation on VWF in low-VWF patients. (A) Two subsets of low-VWF patients were defined with highest (blue) and lowest (red) levels of SNA binding (sialic acids) to VWF (n = 13-14 in each group). (B) Plasma neuraminidase activity was not significantly different between the 2 subgroups (ns = not significant). (C-D) To study endothelial cell sialylation of VWF in patients with low VWF, samples were collected from a subgroup (n = 23) of LoVIC patients immediately before and 1 hour after DDAVP administration. (C) All patients demonstrated significantly increased plasma VWF:Ag levels post-DDAVP (median, 61.2 vs 174.2 IU/dL; ***P < .001). (D) SNA binding to VWF secreted after DDAVP was significantly elevated compared with circulating steady-state plasma VWF (median, 87.3% vs 103.3%; **P < .01).
Etiology underlying reduced α(2-6)-linked sialylation in patients with low VWF. (A-B) To investigate the pathophysiology involved in the reduction in α(2-6)-linked sialylation on VWF in low-VWF patients. (A) Two subsets of low-VWF patients were defined with highest (blue) and lowest (red) levels of SNA binding (sialic acids) to VWF (n = 13-14 in each group). (B) Plasma neuraminidase activity was not significantly different between the 2 subgroups (ns = not significant). (C-D) To study endothelial cell sialylation of VWF in patients with low VWF, samples were collected from a subgroup (n = 23) of LoVIC patients immediately before and 1 hour after DDAVP administration. (C) All patients demonstrated significantly increased plasma VWF:Ag levels post-DDAVP (median, 61.2 vs 174.2 IU/dL; ***P < .001). (D) SNA binding to VWF secreted after DDAVP was significantly elevated compared with circulating steady-state plasma VWF (median, 87.3% vs 103.3%; **P < .01).
Increased Gal exposure is associated with enhanced clearance in low VWF
On the basis of previous murine data studies,13,26 we hypothesized that reduced α2-6 sialylation may be contributing to the pathophysiology underlying low VWF by promoting enhanced VWF clearance. Elevated VWFpp/VWF:Ag ratios have been used to define patients with VWD with enhanced clearance. In particular, previous studies demonstrated that an increased VWFpp/VWF:Ag ratio higher than 3 could be used to identify patients with type 1C VWD.40 Despite the reduction in terminal sialylation, we observed increased VWFpp/VWF:Ag ratios higher than 3 in only 6% of the total LoVIC cohort (Figure 6A). However, in our control population, we found that the upper limit for the VWFpp/VWF:Ag ratio normal range was 1.48. Interestingly, we observed that although only a minority of low-VWF patients had a VWFpp/VWF:Ag ratio higher than 3, there was nonetheless clear evidence of subtly enhanced VWF clearance in a significant subset of LoVIC patients (Figure 6A; P < .01). Thus, 28 (25%) of the low-VWF cohort had estimated VWF half-lives less than 6 hours (Figure 6B). Furthermore, an inverse correlation between enhanced RCA-I binding (exposure Gal) and estimated VWF half-life was also observed in this subset of low-VWF patients with shorter half-life and higher VWFpp/VWF:Ag ratios (Figure 6C). Together, these findings support that loss of terminal sialylation may contribute to the pathophysiology underpinning low VWF in at least a subgroup of patients by promoting enhanced clearance. Importantly, this enhanced clearance is more subtle compared with that previously reported in patients with archetypal type 1C VWD mutations.40,41
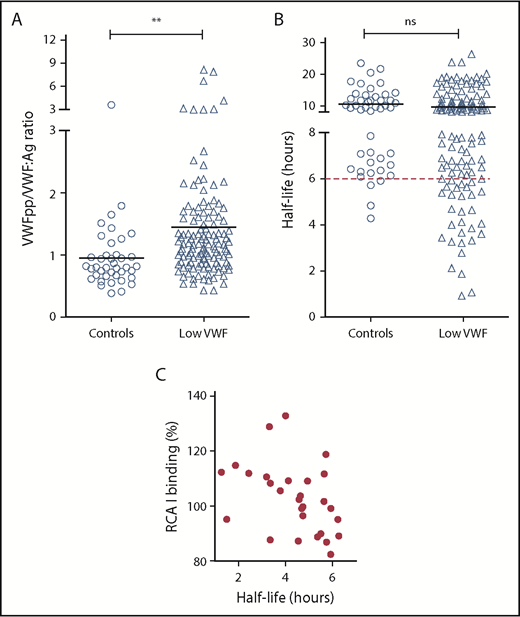
Increased Gal exposure is associated with enhanced clearance in a subset of patients with Low VWF. (A) Steady-state plasma VWF:Ag and VWFpp levels were determined by ELISA for control subjects and patients with low VWF, respectively. All ELISAs were performed in triplicate. Plasma VWFpp/VWF:Ag ratios were significantly elevated in patients with low VWF (△) compared with healthy controls (○). Mean values for each group are indicated by a black line; **P < .01. (B) For each control (○) and patient with low VWF (△), plasma VWF half-life was calculated using the VWFpp and VWF:Ag levels, as has been described.49 The red dotted line designates a VWF:Ag half-life of 6 hours. (C) For the 28 low-VWF patients with plasma VWF:Ag half-life of less than 6 hours, a significant inverse correlation was observed between half-life and RCA-I binding (Gal exposure) to VWF (Spearman ranking = −0.47; P < .05).
Increased Gal exposure is associated with enhanced clearance in a subset of patients with Low VWF. (A) Steady-state plasma VWF:Ag and VWFpp levels were determined by ELISA for control subjects and patients with low VWF, respectively. All ELISAs were performed in triplicate. Plasma VWFpp/VWF:Ag ratios were significantly elevated in patients with low VWF (△) compared with healthy controls (○). Mean values for each group are indicated by a black line; **P < .01. (B) For each control (○) and patient with low VWF (△), plasma VWF half-life was calculated using the VWFpp and VWF:Ag levels, as has been described.49 The red dotted line designates a VWF:Ag half-life of 6 hours. (C) For the 28 low-VWF patients with plasma VWF:Ag half-life of less than 6 hours, a significant inverse correlation was observed between half-life and RCA-I binding (Gal exposure) to VWF (Spearman ranking = −0.47; P < .05).
Discussion
Carbohydrate structures play critical roles in regulating many aspects of VWF biology, including its biosynthesis, susceptibility to proteolysis, and clearance.13,17,22,26,42 Given this functional importance, it is perhaps unsurprising that abnormalities of VWF glycosylation have been shown to cause significantly reduced plasma VWF levels in a number of murine models.13,27 On the basis of these cumulative data, glycan abnormalities may also play a role in the pathogenesis of human VWD. To address this hypothesis, we developed a novel panel of lectin-binding ELISAs to facilitate high-throughput screening of glycan expression on human plasma VWF. Using this lectin panel, we observed significant interindividual heterogeneity in VWF glycan expression among healthy controls. This variability was consistent across each of the different lectins studied. Interestingly, although terminal sialylation and ABO(H) determinants have previously been shown to play key roles in regulating VWF proteolysis and clearance, significant variation in SNA, MAL-II, RCA-I, and UEA-I lectin binding were all observed among individual normal individuals. Further larger studies and multivariate analysis will be needed to define the molecular mechanisms responsible for this marked interindividual heterogeneity in VWF glycosylation. Nonetheless, our findings suggest that alterations in VWF carbohydrate expression are likely to contribute to quantitative and qualitative variations in VWF levels in the normal population. Given the effect of plasma VWF levels on both thrombotic and bleeding risks, elucidation of the molecular mechanisms involved in regulating interindividual variation in VWF glycan expression has clear translational relevance.
Previous studies have consistently shown that VWF gene mutations are less common in patients with low VWF (30-50 IU/dL) compared with patients with type 1 VWD (<30 IU/dL).31,43 For example, potential detrimental VWF sequence variants were identified in less than 40% of the subjects enrolled in the LoVIC study.31 These data clearly support that novel pathological mechanisms contribute to reduced plasma VWF levels in subjects with low VWF. Using our lectin panel, we observed that VWF glycans were abnormal in a significant subset of LoVIC patients. In particular, we found significantly increased RCA-I binding (Gal exposure) to VWF in a cohort of 110 patients with low VWF compared with O-blood group-matched healthy controls. To investigate the mechanism responsible for this increased RCA-I binding in some patients with low VWF, we studied quantitative sialylation on the N- and O-linked glycans of VWF. MAL-II and WGA lectin binding were similar in low-VWF patients compared with controls, suggesting that the increased Gal exposure cannot be explained by a significant reduction in terminal α(2-3)-linked sialylation on the O-glycans of VWF. Conversely, however, a significant reduction in SNA binding was detected. These data demonstrate that expression of terminal α(2-6)-linked sialylation (predominantly expressed on the N-glycans of human VWF) is reduced in a significant subgroup of patients with low VWF, with an associated secondary increase in Gal exposure (Figure 7). Of note, we observed no significant difference in ECA binding between low-VWF patients and controls, suggesting that increased N-linked Gal expression is independent of any changes in polylactosamine repeat structures. Interestingly, the increase in RCA-I binding appeared more marked than the reduction in SNA binding, which may reflect the fact that not all the N- and O-glycan chains are capped by terminal sialic acid. Rather, some VWF glycan antennae terminate with Gal or GalNAc residues. In addition, SNA binds only to α(2-6)-linked sialic acid residues, whereas some α(2-3)-, α(2-8)-, and α(2-9)-linked sialylation has also been reported on plasma-derived VWF.

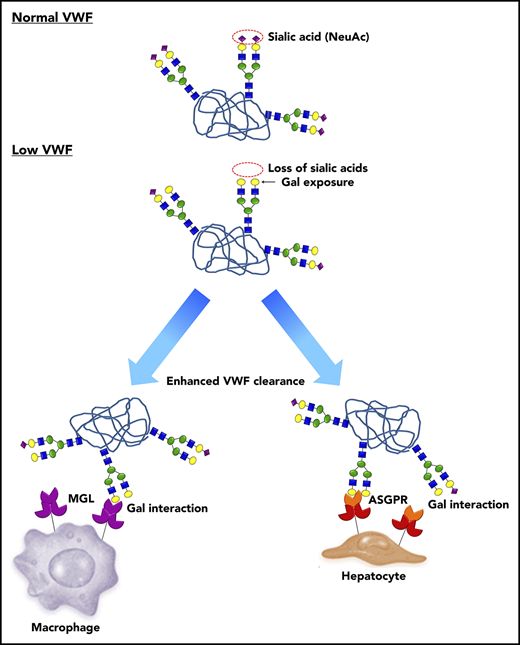
A novel pathophysiology underlying low VWF levels: reduced N-linked sialylation leads to increased galactose (Gal) exposure, which in turn triggers enhanced VWF clearance. In normal human plasma, the N-linked glycans of VWF exist predominantly as bi-antennary complex-type chains that are capped by terminal sialic acid residues (purple diamonds), which play an important role in protecting against clearance. In a subgroup of patients with low VWF, a quantitative reduction in N-linked sialylation results in enhanced exposure of subterminal Gal residues (yellow circles). These exposed Gal residues trigger enhanced VWF clearance through a number of different pathways including the asialoglycoprotein receptor (ASGPR; also known as Ashwell-Morell receptor) on hepatocytes and macrophage galactose lectin receptor (MGL) on macrophages.
A novel pathophysiology underlying low VWF levels: reduced N-linked sialylation leads to increased galactose (Gal) exposure, which in turn triggers enhanced VWF clearance. In normal human plasma, the N-linked glycans of VWF exist predominantly as bi-antennary complex-type chains that are capped by terminal sialic acid residues (purple diamonds), which play an important role in protecting against clearance. In a subgroup of patients with low VWF, a quantitative reduction in N-linked sialylation results in enhanced exposure of subterminal Gal residues (yellow circles). These exposed Gal residues trigger enhanced VWF clearance through a number of different pathways including the asialoglycoprotein receptor (ASGPR; also known as Ashwell-Morell receptor) on hepatocytes and macrophage galactose lectin receptor (MGL) on macrophages.
The etiology underlying the abnormal VWF glycosylation in some patients with low VWF remains unclear. The reduction in α(2-6)-linked sialylation may reflect aberrant glycosylation during VWF biosynthesis as a result of constitutive glycosylation pathway abnormalities within endothelial cells. Because glycosylation occurs co-translationally, it is also possible that mutations affecting VWF intracellular trafficking and/or folding could also affect the carbohydrate structures expressed on the final VWF monomer. Alternatively, the reduced VWF sialylation observed in patients with low VWF may reflect enhanced loss of terminal sialylation in the circulation after VWF secretion, but no changes in neuraminidase activity in plasma between patients with low and high sialic acids expression were found. Although previous studies reported alterations in VWF glycan expression in patients with underlying liver cirrhosis, mildly abnormal liver functions tests (alkaline phosphatase, γ glutamyl transferase, aspartate aminotransferase and alanine aminotransferase) were identified in only 4 of 100 low-VWF subjects tested (data not shown).
Interestingly, SNA binding was significantly increased in VWF secreted post-DDAVP vs pre-DDAVP administration. This finding is in keeping with recent studies demonstrating that progressive trimming of glycans occurs with glycoprotein aging in normal plasma by glycosidases as neuraminidases.39 Although we found no significant increase in plasma neuraminidase activity in patients with low VWF, this does not exclude the possibility that secreted VWF in these patients could demonstrate enhanced susceptibility to neuraminidase digestion.
Notwithstanding the mechanisms responsible for the reduction in capping α(2-6)-linked sialylation in some patients with low VWF, our results showed that this aberrant glycosylation is likely to play an important role in the etiology of their reduced plasma VWF levels. In particular, the secondary increase in Gal residue exposure is associated with significantly enhanced VWF clearance (Figure 7). This reduced half-life is likely to be modulated through a number of different lectin-mediated pathways, including the ASGPR on hepatocytes and the MGL receptor on macrophages.44-46 In addition, reduction in terminal sialylation may also enhance VWF clearance through macrophage scavenger receptor-mediated pathways (including low-density lipoprotein receptor–related protein 1 and scavenger receptor class A1).47,48 From a clinical perspective, it is interesting that although enhanced clearance was observed in patients with low VWF and abnormal glycans compared with healthy controls, the clearance rates were significantly lower than those previously reported in patients with type 1C VWD.40 As a consequence, the net increase in steady state plasma VWFpp/VWF:Ag ratios in low-VWF patients was less marked. Nevertheless, our data clearly demonstrate that subtle increased VWF clearance contributes to the pathogenesis underlying reduced VWF levels in a significant proportion of patients with low VWF.
Presented as an oral communication at the International Society of Thrombosis and Haemostasis Congress, Berlin, Germany, 11 July 2017.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Katrina Diener and Colin Larson from Genomic and Microarray Core, University of Colorado, Denver.
This work was supported by an Irish Health Research Board Health Research Award (HRA-POR-2014-529) (J.S.O.) and a Science Foundation Ireland Principal Investigator Award (11/PI/1066) (J.S.O.).
Authorship
Contribution: S.A., M.L., N.D., S.P., G.D.T., and K.L.J. performed experiments; S.A., M.L., A.C., J.D.P., and J.S.O. designed the research and analyzed data; N.M.O., C.K., K.R., M.B., M.N., and A.P. recruited patients and collected and organized DNA and plasma samples; and S.A., M.L., A.C, T.M.B., R.J.S.P., P.J., J.D.P., J.M.O., and J.S.O. were involved in writing and reviewing the paper.
Conflict-of-interest disclosure: M.L. has received research funding from Baxalta and has served on advisory boards for Baxalta. J.S.O. has served on the speaker’s bureau for Baxter, Bayer, Novo Nordisk, Boehringer Ingelheim, Leo Pharma, and Octapharma. He has also served on the advisory boards of Baxter, Bayer, Octapharma CSL Behring, Daiichi Sankyo, Boehringer Ingelheim, and Pfizer. J.S.O. has also received research grant funding awards from Baxter, Bayer, Pfizer, and Novo Nordisk. P.J. receives research funding from CSL Behring, Octapharma, and Shire and has served on advisory boards for CSL Behring and Shire. The remaining authors declare no competing financial interests.
Correspondence: Sonia Aguila, Department of Molecular and Cellular Therapeutics, Irish Centre for Vascular Biology, Royal College of Surgeons in Ireland, 123 St Stephen’s Green, Dublin 2, Ireland; e-mail soniamartinez@rcsi.ie.