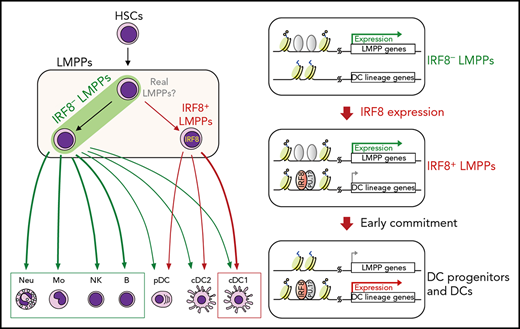
Key Points
IRF8+ LMPPs, derived from IRF8– LMPPs, preferentially generate DCs in vivo.
IRF8 epigenetically primes DC lineage genes in LMPPs.

Abstract
Dendritic cells (DCs), which are vital for immune responses, are derived from bone marrow hematopoietic stem cells via common DC progenitors (CDPs). DC lineage fate decisions occurring at stages much earlier than CDPs have recently been recognized, yet the mechanism remains elusive. By single-cell RNA-sequencing, in vivo cell transfer experiments, and an assay for transposase-accessible chromatin sequencing using wild-type, IRF8-GFP chimera knock-in or IRF8-knockout mice, we demonstrate that IRF8 regulates chromatin at the lymphoid-primed multipotent progenitor (LMPP) stage to induce early commitment toward DCs. A low but significant expression of IRF8, a transcription factor essential for DC and monocyte development, was initiated in a subpopulation within LMPPs. These IRF8+ LMPPs were derived from IRF8– LMPPs and predominantly produced DCs, especially classical DC1s, potentially via known progenitors, such as monocyte-DC progenitors, CDPs, and preclassical DCs. IRF8+ LMPPs did not generate significant numbers of monocytes, neutrophils, or lymphocytes. Although IRF8– and IRF8+ LMPPs displayed very similar global gene expression patterns, the chromatin of enhancers near DC lineage genes was more accessible in IRF8+ LMPPs than in IRF8– LMPPs, an epigenetic change dependent on IRF8. The majority of the genes epigenetically primed by IRF8 were still transcriptionally inactive at the LMPP stage, but were highly expressed in the downstream DC lineage populations such as CDPs. Therefore, early expression of the key transcription factor IRF8 changes chromatin states in otherwise multipotent progenitors, biasing their fate decision toward DCs.
Introduction
Hematopoietic stem cells (HSCs) generate various types of blood cells through intermediate progenitors.1,2 Pioneering studies have identified HSCs and multipotent progenitors based on the different patterns of cell surface markers. Recent advances in technologies such as single-cell RNA-sequencing (scRNA-seq) and in vivo lineage tracing have led to the realization that HSCs and early progenitors are highly heterogeneous and include subpopulations with distinct differentiation potentials,3-9 suggesting an early lineage specification during hematopoiesis. However, the mechanisms underlying the generation of their heterogeneity and early commitment are largely unknown.
Dendritic cells (DCs), indispensable for the elicitation of innate and acquired immune responses, are derived from HSCs.10 They are mainly composed of 3 subpopulations: classical DC1s (cDC1s; CD8+ XCR1+ in mice), cDC2s (CD8– XCR1– in mice), and plasmacytoid DCs (pDCs).11 Several progenitors with a DC differentiation potential have been identified. Lymphoid-primed multipotent progenitors (LMPPs) differentiate into lymphoid and myeloid cells including DCs but not erythrocytes and megakaryocytes.4,12 Monocyte-DC progenitors (MDPs) are bipotential for monocyte and DC differentiation, although a recent study challenges this view.13 Common DC progenitors (CDPs) are capable of generating cDCs and pDCs,14 whereas pre-cDCs produce cDCs only.15
Recently, DC lineage specification at earlier stages of hematopoiesis was suggested by several research groups.4,16-18 Naik et al analyzed the ability of individual LMPPs to generate different hematopoietic cell types using a lentivirus-based cell barcoding system and found that many single LMPPs produced only a few cell types, such as cDC1s.4 Lee et al performed comprehensive single-cell culture experiments of human HSCs and progenitors.16 The authors demonstrated that DC lineage specification starts near the HSC stage and suggested that the DC lineage-biased progenitors may be distinguished by the expression of the transcription factor IRF8. We and others have previously reported that IRF8 expression becomes uniformly high at the mononuclear phagocyte progenitor stages such as MDPs, CDPs, and common monocyte progenitors (cMoPs).15,19-22 In Irf8−/− mice, the numbers of CDPs, pre-cDCs, cDC1s, pDCs, and monocytes (especially the Ly6C+ subset) are severely reduced, whereas MDPs and cMoPs accumulate.19,22-24 Thus, IRF8 is clearly needed for the transitions from MDPs to CDPs and from cMoPs to monocytes. On the other hand, the in vivo differentiation potential of IRF8-expressing early progenitors near the HSC stage and the mechanism underlying IRF8’s effect on their lineage specification remain unknown.
In this study, we extensively analyzed the expression of IRF8 at early stages of hematopoiesis. scRNA-seq and flow cytometric analyses in wild-type (WT) and IRF8-GFP chimera knock-in mice25 revealed that low but significant IRF8 expression is initially induced within the LMPP population. IRF8+ LMPPs predominantly produce cDC1s but not monocytes, neutrophils, or lymphoid cells in vivo. Interestingly, chromatin regions that become newly accessible in IRF8+ LMPPs are associated with genes expressed in CDPs, pre-cDCs, and cDCs. Analysis of Irf8−/− mice showed that IRF8 deficiency causes a significant reduction in chromatin accessibility at the IRF8+ LMPP-specific open chromatin regions. These results suggest that IRF8 begins shaping the DC lineage chromatin landscape in LMPPs, leading to early DC lineage specification.
Methods
Mice
Male and female WT, Ly5.1, Irf8−/−, and Irf8Irf8Gfp/WT (IRF8-GFP)25 mice 8 to 10 weeks old in a C57BL/6 background were used. All animal experimentations were performed in accordance with the Guidelines for Proper Conduct of Animal Experiments (Science Council of Japan), and all protocols were approved by the institutional review boards of Yokohama City University (protocol F-A-17-018).
Cell isolation and flow cytometric analysis
Bone marrow and spleen cells were obtained by flushing the femur and tibia and with Liberase and DNase I (Roche) treatment, respectively.26 To isolate LMPPs from bone marrow, lineage markers (Lin; CD5, B220, CD11b, Ly6G/C, 7-4, Ter-119) negative cells were enriched using a Lineage Cell Depletion Kit (Miltenyi biotech). Lin− cells were further stained with fluorochrome labeled antibodies, followed by fluorescence-activated cell sorting (FACS). The purity of the sorted populations was >99%. FlowJo software (FlowJo, LLC) was used for data analysis.
scRNA-seq
Single-cell complementary DNA (cDNA) libraries were prepared using the Fluidigm C1 system. Sorted LMPPs were loaded onto a Fluidigm C1 Single-Cell Open App IFC (5-10 μm) at 4°C. cDNA was synthesized and amplified using SMARTer Ultra Low RNA Kit for the Fluidigm C1 System (Clontech). Each cDNA was subjected to tagmentation using a Nextera XT DNA Sample Preparation Kit (Illumina) and then polymerase chain reaction (PCR) amplification using Index Primers from a Nextera XT Index Kit (Illumina). The prepared libraries were sequenced on an Illumina HiSeq2500 instrument with 97-bp paired-end sequencing.
Transplantation experiments
LMPPs purified from WT, Irf8−/−, and IRF8-GFP mice (CD45.2+) were injected intravenously into lethally irradiated (9.5 Gy) or nonirradiated Ly5.1 mice (CD45.2−).
ATAC-seq
An assay for transposase-accessible chromatin with high throughput sequencing (ATAC-seq) was performed as described previously.27 Briefly, 7200 LMPPs are directly sorted into 1.5-mL tubes and pelleted by centrifugation for 10 minutes at 300g. The cells were then incubated with transposase solution (25 μL of 2× Tagment DNA buffer and 2.5 μL of Tagment DNA Enzyme [both Illumina Nextera DNA Library Prep Kit], 0.5 μL of 1% digitonin [Promega], and 22 μL of distilled water) at 37°C for 30 minutes with shaking at 300 rpm. Transposed DNA was then purified using a MinElute Reaction Cleanup kit (QIGEN). Purified transposed DNA was amplified using a NEBNext High Fidelity 2× PCR Master Mix (New England Biolabs) with indexed primers. Libraries were purified using SPRIselect beads (Beckman Coulter) to remove primer dimers. The prepared libraries were sequenced on a Miseq (Illumina) to generate paired-end 50-bp reads. ATAC-seq data from 2 independent experiments (biological duplicates) were obtained.
Markers and antibodies used for flow cytometric analysis, retroviral transduction, microarray, single-cell quantitative PCR with reverse transcription, scRNA-seq data analysis, ATAC-seq data analysis, reporter assay
Full details are provided in the supplemental Methods, available on the Blood Web site.
Results
An LMPP subpopulation expresses IRF8
A recent report showed that an IRF8-expressing subset of Lin– Sca-1+ CD117+ cells (LSKs) has an enhanced cDC differentiation potential in vitro.16 In addition, computational analysis of scRNA-seq data predicted that Irf8 messenger RNA-expressing early progenitors contain pDC lineage-biased progenitors in mice.18 To identify the exact differentiation stage at which the expression of IRF8 starts in mice, we analyzed bone marrow HSCs and early progenitors in IRF8-GFP chimera knock-in mice, which enable visualization of the IRF8 protein.25,28 IRF8-GFP was detected in ∼20% of LMPPs but not HSCs or Lin– Sca-1+ CD117+ CD150– CD34+ Flt3low multipotent progenitors (Figure 1A; supplemental Figure 1A). The expression levels of IRF8 in LMPPs were considerably lower than those in DC progenitors such as MDPs, CDPs, and pre-cDCs (supplemental Figure 1B).
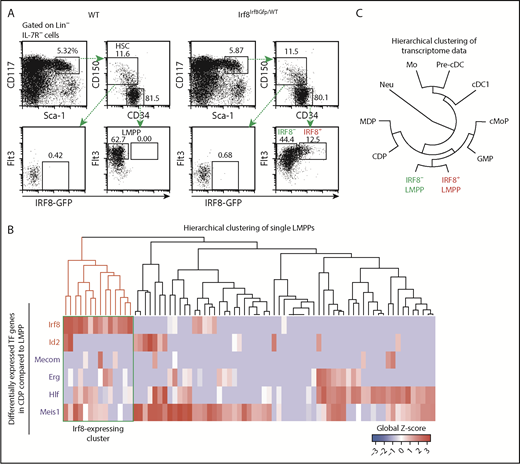
Identification of IRF8-expressing LMPPs. (A) Flow cytometry analysis of HSCs and early progenitors in IRF8-GFP mice. Data are representative of 5 independent experiments. (B) Hierarchical clustering of scRNA-seq data in WT LMPPs. Transcription factor genes upregulated or downregulated more than fivefold in LMPPs compared with CDPs were selected. Red and blue gene symbols denote upregulated and downregulated genes in CDPs, respectively. The Irf8 transcript-expressing cluster is enclosed by a green rectangle. (C) Hierarchical clustering of microarray transcriptome data in hematopoietic populations. Mo, monocyte; neu, neutrophil.
Identification of IRF8-expressing LMPPs. (A) Flow cytometry analysis of HSCs and early progenitors in IRF8-GFP mice. Data are representative of 5 independent experiments. (B) Hierarchical clustering of scRNA-seq data in WT LMPPs. Transcription factor genes upregulated or downregulated more than fivefold in LMPPs compared with CDPs were selected. Red and blue gene symbols denote upregulated and downregulated genes in CDPs, respectively. The Irf8 transcript-expressing cluster is enclosed by a green rectangle. (C) Hierarchical clustering of microarray transcriptome data in hematopoietic populations. Mo, monocyte; neu, neutrophil.
To understand the transcriptional profile of IRF8-expressing LMPPs at the single-cell level, we performed scRNA-seq in LMPPs derived from WT mice. As expected, Irf8 transcript-expressing LMPPs were detected at a percentage similar to that of IRF8-GFP+ LMPPs (Figure 1B; supplemental Table 1). Unsupervised clustering analysis using HOPACH,29 t-distributed stochastic neighbor embedding, and primary component analysis could not distinguish Irf8+ and Irf8– LMPPs (supplemental Figure 1C-D), suggesting that their global transcriptional profiles were not very different. Importantly, IRF8-expressing LMPPs were confirmed to indeed be LMPPs, not contamination by IRF8-expressing DC progenitors such as MDPs and CDPs, because hierarchical clustering of the microarray transcriptome data in hematopoietic populations also indicated that IRF8+ LMPPs and IRF8– LMPPs were grouped together (Figure 1C; supplemental Table 2).
We next used the expression of 6 transcription factor genes, upregulated or downregulated in CDPs compared with LMPPs, to perform hierarchical clustering of the single-cell data. This time, a distinct subpopulation expressing Irf8 was formed (Figure 1B). These results imply that the fate of LMPPs might be determined by a small number of key transcription factors such as IRF8.
IRF8+ LMPPs preferentially give rise to cDC1s in vivo
To understand the in vivo differentiation potential of the LMPP subpopulations demonstrated here, we transferred IRF8-GFP– LMPPs or IRF8-GFP+ LMPPs into lethally irradiated mice. Almost no progeny cells were detected on day 4 in the spleen, but large numbers of progenies were generated from IRF8– LMPPs, but not from IRF8+ LMPPs on days 7 and 10, indicating the differential proliferation capacity between the 2 LMPP subpopulations (CD45.2+ cells in Figure 2A-C).
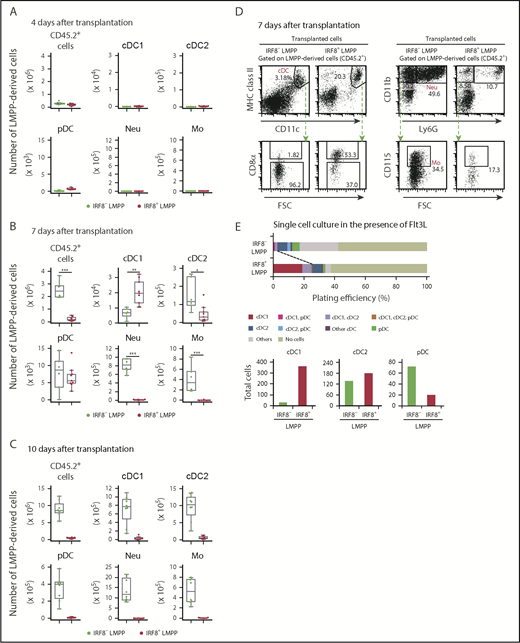
Differentiation potential of IRF8+ LMPPs in vivo. Flow cytometric analysis of splenic DC subpopulations, monocytes, and neutrophils 4 days (A), 7 days (B), or 10 days (C) after intravenous transplantation of LMPP subpopulations. A total of 1000 LMPPs (CD45.2+) were transplanted into irradiated Ly5.1 mice (CD45.2−), and donor-derived (CD45.2+) cells were analyzed. Absolute cell numbers (per spleen of a mouse) of the indicated progeny cells derived from transplanted LMPPs are shown in the boxplots. (A-C) Values from 3 independent experiments are shown. Representative FACS plots of cDCs (D, left) and monocytes and neutrophils (D, right) on day 7. (E) Single-cell differentiation analysis of LMPP subpopulations. LMPP subpopulations were single cell-sorted into 96-well plates and cultured with Flt3L for 7 days. Following staining, cells were analyzed by flow cytometry. A total of 192 single cells of each subpopulation were analyzed. cDC1, cDC2, and pDC differentiation potential in single LMPPs was determined from the staining patterns of cells (top). Total cell numbers of indicated DC subsets yielded in 192 wells are calculated (bottom). Data are representative of 2 independent experiments with similar results. *P < .05, **P < .01, ***P < .001 (Student t test). MHC, major histocompatibility complex.
Differentiation potential of IRF8+ LMPPs in vivo. Flow cytometric analysis of splenic DC subpopulations, monocytes, and neutrophils 4 days (A), 7 days (B), or 10 days (C) after intravenous transplantation of LMPP subpopulations. A total of 1000 LMPPs (CD45.2+) were transplanted into irradiated Ly5.1 mice (CD45.2−), and donor-derived (CD45.2+) cells were analyzed. Absolute cell numbers (per spleen of a mouse) of the indicated progeny cells derived from transplanted LMPPs are shown in the boxplots. (A-C) Values from 3 independent experiments are shown. Representative FACS plots of cDCs (D, left) and monocytes and neutrophils (D, right) on day 7. (E) Single-cell differentiation analysis of LMPP subpopulations. LMPP subpopulations were single cell-sorted into 96-well plates and cultured with Flt3L for 7 days. Following staining, cells were analyzed by flow cytometry. A total of 192 single cells of each subpopulation were analyzed. cDC1, cDC2, and pDC differentiation potential in single LMPPs was determined from the staining patterns of cells (top). Total cell numbers of indicated DC subsets yielded in 192 wells are calculated (bottom). Data are representative of 2 independent experiments with similar results. *P < .05, **P < .01, ***P < .001 (Student t test). MHC, major histocompatibility complex.
Nevertheless, IRF8+ LMPPs generated a greater number of cDC1s than IRF8– LMPPs did on day 7 (Figure 2B,D; supplemental Figure 2A). In contrast, IRF8+ LMPPs gave rise to a fewer number of cDC2s than IRF8– LMPPs did. Comparable numbers of pDCs were produced by the 2 subsets at this time point (ie, day 7). Of note, CD11c− major histocompatibility complex class II− cells derived from IRF8+ LMPPs express CD117, Sca-1, and Flt3, but not CD150 and lineage-specific markers, indicating that they might be immature DC precursor cells (supplemental Figure 2B). Large numbers of neutrophils and monocytes were almost exclusively produced from IRF8– LMPPs. To test the DC differentiation potential of IRF8+ LMPPs at a single-cell level, we performed single-cell culture experiments in the presence of Flt3L. Consistent with the results of in vivo transplantation experiments, the percentage of single LMPPs that produced cDC1s, as well as the total cDC1 yield, was higher in the IRF8+ LMPP culture than in the IRF8– LMPP culture (Figure 2E). These results suggest that IRF8+ LMPPs have a cDC1-biased differentiation potential in vivo and in vitro.
On the other hand, when analyzed 3 days later (ie, on day 10 posttransplantation), IRF8– LMPPs produced large numbers of all mature myeloid populations including cDC1s, but few cells were detected in irradiated mice that received the IRF8+ LMPP transplant (Figure 2C). To clarify the relationship between IRF8– LMPPs and IRF8+ LMPPs, we transferred each LMPP subpopulation to see whether 1 subpopulation could generate the other. On day 3 posttransplantation, a fraction of IRF8– LMPPs gave rise to IRF8+ LMPPs, but not vice versa (Figure 3A). Besides, we noticed that IRF8+ LMPPs differentiated into MDPs, CDPs, and pre-cDCs at this early time point (Figure 3B-D). IRF8– LMPPs produced IRF8+ LMPPs, common myeloid progenitors, granulocyte-monocyte progenitors (GMPs), MDPs, and DC-committed progenitors such as CDPs on day 7 posttransplantation (supplemental Figure 3) and in the in vitro culture experiments (supplemental Figure 4). These results suggest that IRF8– LMPPs are upstream progenitors of IRF8+ LMPPs and that IRF8+ LMPPs rapidly give rise to DCs via known DC progenitor populations.

IRF8+ LMPPs are derived from IRF8− LMPPs. Flow cytometric analysis of bone marrow progenitors 3 days after intravenous transplantation of LMPP subpopulations. A total of 30 000 LMPPs were transplanted into a nonirradiated Ly5.1 mouse, and donor-derived cells were analyzed. Representative FACS plots of donor-derived LMPP subpopulations (A), MDPs (B), CDPs (B), and pre-cDCs (C) from 3 independent experiments. (D) Absolute cell numbers (per lower mouse limbs) of the indicated progeny cells derived from transplanted LMPPs are calculated. Values in the bar graph are the mean ± standard deviation from 3 independent experiments. *P < .05, **P < .01, ***P < .001 (Student t test).
IRF8+ LMPPs are derived from IRF8− LMPPs. Flow cytometric analysis of bone marrow progenitors 3 days after intravenous transplantation of LMPP subpopulations. A total of 30 000 LMPPs were transplanted into a nonirradiated Ly5.1 mouse, and donor-derived cells were analyzed. Representative FACS plots of donor-derived LMPP subpopulations (A), MDPs (B), CDPs (B), and pre-cDCs (C) from 3 independent experiments. (D) Absolute cell numbers (per lower mouse limbs) of the indicated progeny cells derived from transplanted LMPPs are calculated. Values in the bar graph are the mean ± standard deviation from 3 independent experiments. *P < .05, **P < .01, ***P < .001 (Student t test).
We also characterized the lymphoid differentiation potential of LMPP subpopulations. At 4 or 10 weeks posttransplantation, IRF8– LMPPs clearly gave rise to lymphocytes including T, B, and natural killer cells, whereas IRF8+ LMPPs did not (supplemental Figure 5A-B). At these time points, myeloid cells were barely produced from either LMPP subpopulation. We confirmed that IRF8– LMPPs produced a greater number of B cells than IRF8+ LMPPs in vitro (supplemental Figure 5C). Collectively, IRF8+ LMPPs predominantly produce DCs, especially cDC1s, but not monocytes, neutrophils, or lymphoid cells (supplemental Figure 6).
Because irradiation-induced inflammation might cause a differentiation bias of IRF8+ LMPPs toward DCs upon transplantation, we transferred the LMPP subpopulations into nonirradiated mice. IRF8+ LMPPs produced more cDC1s than IRF8– LMPPs even in nonirradiated recipients, whereas IRF8– LMPPs generated a greater number of monocytes than IRF8+ LMPPs on day 10 posttransplantation (supplemental Figure 7). In this experimental condition, neutrophils were barely detected in the spleen. These data confirm our finding that IRF8+ LMPPs have DC-biased differentiation potential.
We also examined the role of hepatic leukemia factor (HLF) and MEIS1 in DC development (Figure 1B). Unlike IRF8, the expression of these factors is lower in Irf8-expressing LMPPs and downstream DC lineage cells than in most other LMPPs. HLF, MEIS1, or IRF8 were retrovirally transduced into isolated LMPPs; DC differentiation was induced in vitro. As expected, IRF8 transduction promoted the generation of cDC1s (supplemental Figure 8). Interestingly, HLF introduction potently suppressed the generation of cDC1s but not cDC2s. MEIS1 transduction modestly suppressed the generation of both cDC1s and cDC2s. These results suggest that these factors, especially HLF, may also be involved in DC subpopulation fate decision at the LMPP stage.
A limited effect of IRF8 on immediate gene expression in IRF8+ LMPPs
We next sought to clarify the role of IRF8 in IRF8+ LMPPs. In Irf8−/− mice, LSKs and LMPPs were slightly diminished (supplemental Figure 9A). However, the ratio of LMPPs to LSKs was not significantly different between WT and Irf8−/− mice, indicating that IRF8 deficiency does not specifically affect LMPP development. Consistent with the phenotype of Irf8−/− mice,22 Irf8−/− LMPPs failed to produce cDC1s, and instead generated a large number of neutrophils in vivo (supplemental Figure 9B).
Single-cell quantitative PCR with reverse transcription (RT-qPCR) revealed that WT and Irf8−/− LMPPs contained similar percentages of Irf8-expressing cells (Figure 4A; supplemental Table 3). Of note, the mutant Irf8 gene in Irf8−/− mice retain the enhancers and promoters and expresses a short Irf8 transcript that does not produce IRF8 protein.30 These data suggest that the IRF8 protein is not required for the expression of Irf8 in the LMPP subpopulation. To further test whether IRF8 affects gene expression in Irf8+ LMPPs, we performed scRNA-seq on Irf8−/− LMPPs (supplemental Table 1). t-Distributed stochastic neighbor embedding analysis of all the detected genes in scRNA-seq data could not distinguish Irf8+ and Irf8– LMPPs regardless of the genotype (ie, WT or Irf8−/−; supplemental Figure 9C), suggesting that global gene expression profiles of LMPPs are barely affected by the absence of IRF8.
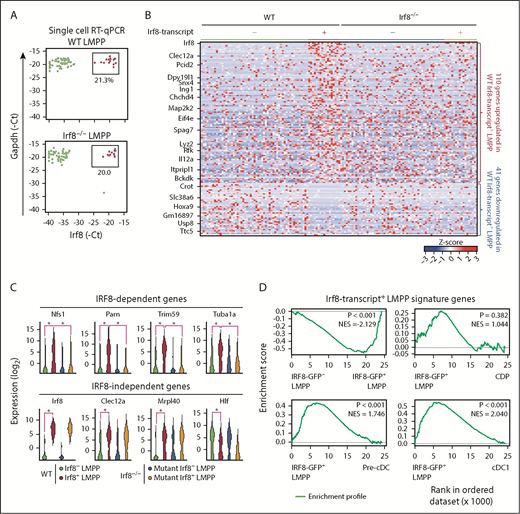
IRF8+ LMPP signature genes are barely expressed in DC lineage cells. (A) Single-cell RT-qPCR analysis of Irf8 and Gapdh in WT and Irf8−/− LMPPs. Expression levels of Irf8 and Gapdh are expressed in −1 × Ct units. (B) A heat map showing z scores for the expression of LMPP subpopulation-specific genes. A total of 151 genes, significantly upregulated or downregulated in WT Irf8-transcript+ LMPPs compared with WT Irf8-transcript− LMPPs, were selected by using the scRNA-seq data (1-way analysis of variance, P < .05). Irf8-transcript+ cells were defined as cells with Irf8 fragments per kilobase million >10. (C) Representative LMPP subpopulation-specific genes. IRF8-dependent or IRF8-independent genes are shown as violin plots. *P < .05 (1-way analysis of variance). (D) GSEA comparing hematopoietic populations for 110 Irf8-transcript+ LMPP signature genes.
IRF8+ LMPP signature genes are barely expressed in DC lineage cells. (A) Single-cell RT-qPCR analysis of Irf8 and Gapdh in WT and Irf8−/− LMPPs. Expression levels of Irf8 and Gapdh are expressed in −1 × Ct units. (B) A heat map showing z scores for the expression of LMPP subpopulation-specific genes. A total of 151 genes, significantly upregulated or downregulated in WT Irf8-transcript+ LMPPs compared with WT Irf8-transcript− LMPPs, were selected by using the scRNA-seq data (1-way analysis of variance, P < .05). Irf8-transcript+ cells were defined as cells with Irf8 fragments per kilobase million >10. (C) Representative LMPP subpopulation-specific genes. IRF8-dependent or IRF8-independent genes are shown as violin plots. *P < .05 (1-way analysis of variance). (D) GSEA comparing hematopoietic populations for 110 Irf8-transcript+ LMPP signature genes.
There were 151 genes differentially expressed between Irf8– and Irf8+ LMPPs (Figure 4B-C; supplemental Table 4); 110 genes were upregulated and 41 genes were downregulated in Irf8+ LMPPs. We confirmed the expression of the differentially expressed genes in isolated IRF8-GFP– LMPPs and IRF8-GFP+ LMPPs by RT-qPCR, and found similar results (supplemental Figure 9D; supplemental Table 3). Gene set enrichment analysis (GSEA) of these 110 genes, however, revealed that the expression of these genes was only transiently induced in IRF8+ LMPPs and then decreased in DC lineage populations such as CDPs, pre-cDCs, and cDC1s (Figure 4D). Although the expression of the previously mentioned 41 genes was further decreased in the downstream DC lineage populations, such a decrease was also observed in cells of other lineages including monocytes and neutrophils (supplemental Figure 10A). Of the 151 genes differentially expressed between the LMPP subpopulations, the expression of 23 genes was regulated in an IRF8-dependent manner (Figure 4B-C). Again, GSEA yielded no evidence that these IRF8-dependent genes are related to DC development (supplemental Figure 10B). Overall, these data imply that IRF8 plays only a minimal role in the cDC1-biased LMPP subpopulation at the immediate gene expression level.
IRF8 establishes a DC lineage chromatin landscape in IRF8+ LMPPs
What then is the major action of IRF8 in IRF8+ LMPPs? Promoter-distal enhancers are essential for cell type-specific gene expression and are established before the expression of associated genes during hematopoiesis.31,32 We have recently shown that IRF8 promotes the enhancer establishment of monocyte- and DC-related genes in MDPs and cMoPs.33 To analyze the in vivo role of IRF8 in the establishment of chromatin landscape in IRF8+ LMPPs, we performed ATAC-seq, a sensitive technique to probe genome-wide chromatin accessibility using hyperactive Tn5 transposase.27,34 Distribution analysis of ATAC-seq peaks in enhancer regions revealed that large parts of ATAC-seq peaks overlapped between IRF8-GFP– LMPPs and IRF8-GFP+ LMPPs, whereas relatively small numbers of subpopulation-specific peaks were observed (Figure 5A; supplemental Table 5). Representative genome browser images of IRF8– LMPP-specific or IRF8+ LMPP-specific ATAC-seq peaks, associated with genes Jak2 and Irf8, respectively, are depicted in Figure 5B. We validated our ATAC-seq data by qPCR of ATAC-seq libraries and reporter assays for ATAC-seq peak regions (supplemental Figures 11 and 12; supplemental Table 3).
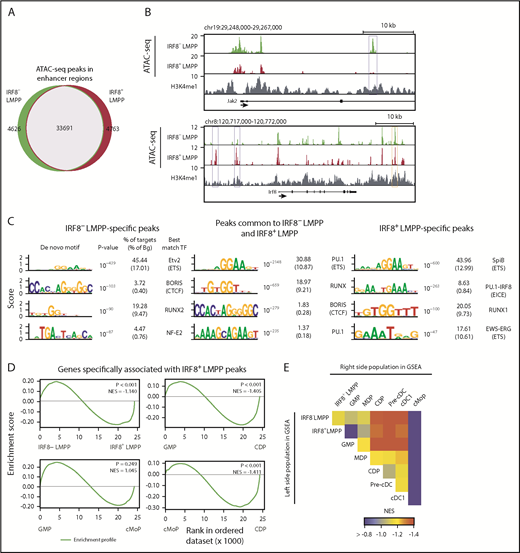
ATAC-seq analysis of IRF8+ LMPPs. ATAC-seq analyses were performed in IRF8− and IRF8+ LMPPs isolated from IRF8-GFP mice. Distal ATAC-seq peaks within H3K4me1-marked regions were selected. (A) A Venn diagram showing the overlap of ATAC-seq peaks between LMPP subpopulations. (B) Genome browser images of ATAC-seq data at the Jak2 and Irf8 gene loci. Blue rectangles indicate IRF8− LMPP (top) and IRF8+ LMPP (bottom) specific peaks. The orange rectangle indicates a previously reported Irf8 enhancer region.24 (C) De novo motif analysis of ATAC-seq peak regions specific for each subpopulation and those common to 2 subpopulations. (D) GSEA comparing hematopoietic populations for genes associated with IRF8+ LMPP-specific ATAC-seq peaks. (E) Heat map showing NES of GSEA. NES, normalized enrichment score.
ATAC-seq analysis of IRF8+ LMPPs. ATAC-seq analyses were performed in IRF8− and IRF8+ LMPPs isolated from IRF8-GFP mice. Distal ATAC-seq peaks within H3K4me1-marked regions were selected. (A) A Venn diagram showing the overlap of ATAC-seq peaks between LMPP subpopulations. (B) Genome browser images of ATAC-seq data at the Jak2 and Irf8 gene loci. Blue rectangles indicate IRF8− LMPP (top) and IRF8+ LMPP (bottom) specific peaks. The orange rectangle indicates a previously reported Irf8 enhancer region.24 (C) De novo motif analysis of ATAC-seq peak regions specific for each subpopulation and those common to 2 subpopulations. (D) GSEA comparing hematopoietic populations for genes associated with IRF8+ LMPP-specific ATAC-seq peaks. (E) Heat map showing NES of GSEA. NES, normalized enrichment score.
To predict the transcription factors that regulate chromatin landscapes at LMPP stages, we performed computational DNA motif analysis in the identified ATAC-seq peak regions. RUNX and PU.1 (ETS) DNA-binding motifs were overrepresented in ATAC-seq peaks both in those common and specific to the subpopulations (Figure 5C), as expected from their critical roles in HSCs and early hematopoietic progenitors.8,35 Interestingly, a PU.1-IRF8 composite-binding motif was significantly enriched in only IRF8+ LMPP-specific ATAC-seq peaks (Figure 5C, right), suggesting that IRF8, known to heterodimerize with PU.1, is involved in the regulation of open chromatin regions newly established at the IRF8+ LMPP stage.
We next performed gene ontology analysis of the genes associated with subpopulation-specific ATAC-seq peaks (supplemental Figure 13). Genes associated with IRF8– LMPP-specific ATAC-seq peaks were enriched for proliferation and HSC-related annotations, whereas genes associated with IRF8+ LMPP-specific ATAC-seq peaks were enriched for immune response-related annotations such as “positive regulation of phagocytosis,” “inflammatory response,” and “response to lipopolysaccharide.” GSEA revealed that the expression levels of genes associated with IRF8+ LMPP-specific ATAC-seq peaks were only slightly higher in IRF8+ LMPPs than in IRF8– LMPPs (Figure 5D-E). However, these genes became highly expressed in CDPs, pre-cDCs, and cDC1s but not in GMPs or cMoPs. These results suggest that the open chromatin regions of enhancers near DC lineage genes emerge at the IRF8+ LMPP stage before the high expression.
To examine whether IRF8 protein is required for the establishment of the IRF8+ LMPP-specific chromatin landscape, we performed ATAC-seq in LMPPs isolated from WT and Irf8−/− mice (supplemental Table 6). Because IRF8+ subpopulation was ∼20% of total LMPPs (Figure 1), only part of IRF8+ LMPP-specific ATAC-seq peaks were detected in WT LMPPs (1434 of 4763 regions). We found that the chromatin accessibility in the 1434 regions was significantly reduced in Irf8−/− LMPPs, and 846 (59%) regions were no longer judged as peaks (Figure 6A). De novo motif analysis of these 846 ATAC-seq peaks lost in Irf8−/− LMPPs revealed significant enrichment of the PU.1-IRF8 composite element (Figure 6B). Representative genome browser images of IRF8-dependent IRF8+ LMPP-specific ATAC-seq peaks at the Ifi44 gene locus are shown (Figure 6C). To test whether IRF8 directly acts on the IRF8+ LMPP-specific open chromatin regions, we analyzed previously published IRF8 chromatin immunoprecipitation sequencing (ChIP-seq) data of MDPs,33 and found that IRF8 binding is enriched at the IRF8+ LMPP-specific open chromatin regions (Figure 6D). These data indicate that IRF8 is important for the establishment of many open chromatin regions newly formed in IRF8+ LMPPs (supplemental Figure 14). Of note, genes associated with IRF8-dependent, IRF8+ LMPP-specific open chromatin regions included not only those encoding regulators of DC functions such as Btla36 and Tyk2,37 but also those encoding transcriptional regulators implicated in DC differentiation, such as Irf8 itself, Hdac9,38,39 and Smad7.40
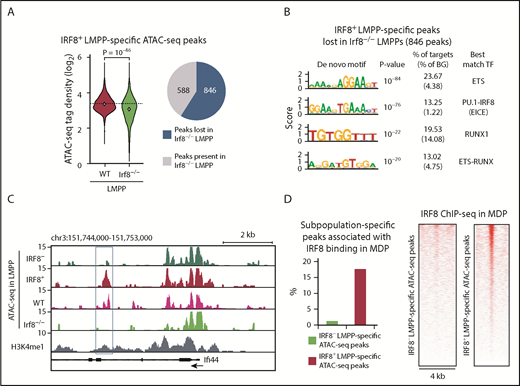
IRF8 governs the chromatin accessibility of DC lineage enhancers in IRF8+ LMPPs. ATAC-seq analyses were performed in WT and Irf8−/− LMPPs. IRF8+ LMPP-specific 1434 ATAC-seq peak regions detected in WT LMPPs were analyzed. (A, left) ATAC-seq tag-densities of the peak regions in WT and Irf8−/− LMPPs are shown in violin plots. Student t test was performed for statistical significance. (A, right) The presence or absence of the 1434 ATAC-seq peaks were judged in Irf8−/− LMPPs. (B) De novo motif analysis of IRF8+ LMPP-specific peaks lost in Irf8−/− LMPPs (ie, 846 peaks). (C) Genome browser image of ATAC-seq data at the Ifi44 gene locus. Blue rectangle indicates an IRF8-dependent IRF8+ LMPP-specific peak. (D, left) Overlap between LMPP subpopulation-specific ATAC-seq peaks and IRF8 ChIP-seq peaks in MDPs. (D, right) Heat maps of IRF8 binding in MDPs at the LMPP subpopulation-specific ATAC-seq peak regions. Each horizontal line represents the density of IRF8 ChIP-seq tags in the 4-kb region centered on the ATAC-seq peak summit.
IRF8 governs the chromatin accessibility of DC lineage enhancers in IRF8+ LMPPs. ATAC-seq analyses were performed in WT and Irf8−/− LMPPs. IRF8+ LMPP-specific 1434 ATAC-seq peak regions detected in WT LMPPs were analyzed. (A, left) ATAC-seq tag-densities of the peak regions in WT and Irf8−/− LMPPs are shown in violin plots. Student t test was performed for statistical significance. (A, right) The presence or absence of the 1434 ATAC-seq peaks were judged in Irf8−/− LMPPs. (B) De novo motif analysis of IRF8+ LMPP-specific peaks lost in Irf8−/− LMPPs (ie, 846 peaks). (C) Genome browser image of ATAC-seq data at the Ifi44 gene locus. Blue rectangle indicates an IRF8-dependent IRF8+ LMPP-specific peak. (D, left) Overlap between LMPP subpopulation-specific ATAC-seq peaks and IRF8 ChIP-seq peaks in MDPs. (D, right) Heat maps of IRF8 binding in MDPs at the LMPP subpopulation-specific ATAC-seq peak regions. Each horizontal line represents the density of IRF8 ChIP-seq tags in the 4-kb region centered on the ATAC-seq peak summit.
Discussion
In this study, we investigated the role of IRF8 in early DC lineage priming by using scRNA-seq, in vivo transplantation, and ATAC-seq. Our data showed that IRF8 expression is initially induced at the LMPP stage in mice. The IRF8+ LMPPs differentiate into DC progenitors and eventually produce DCs, particularly cDC1s. Moreover, the chromatin of DC lineage enhancers becomes accessible in IRF8+ LMPPs in an IRF8-dependent manner. These results imply that hematopoietic cell fate decisions at very early stages are epigenetically dictated by lineage-determining transcription factors such as IRF8. Our data are consistent with the recent elegant report by Lee et al showing that IRF8 expression in human HSCs and multipotent progenitors mark lineage specification toward DCs,16 and further provide insights into the DC differentiation pathway in vivo. Moreover, we clarified the mechanism by which IRF8 guides cell fate.
Based on the expression patterns of transcription factor genes, LMPPs appear to be classified into several subpopulations, including Irf8+HlflowMeis1low, Irf8–Hlf+, Irf8–Meis1+, and Irf8–Hlf+Meis1+ cells (Figure 1B). We demonstrated that IRF8– LMPPs give rise to IRF8+ LMPPs; thus, the former contain those upstream of the latter. In addition, HLF and MEIS1 have been reported to be required for HSC maintenance and self-renewing capabilities.41 Therefore, we envisage that HLF and MEIS1-expressing IRF8– LMPPs may be the “real” LMPPs with multilineage differentiation potential (supplemental Figure 6). The exact nature of these IRF8– LMPP subpopulations will need to be fully characterized in the future, possibly by using reporter mice in which the expression of key transcription factors can be simultaneously monitored. Interestingly, forced HLF expression potently suppressed the differentiation of LMPPs to cDC1s, suggesting that HLF downregulation in IRF8+ LMPPs may be important for DC subpopulation fate decision. Regarding the route of differentiation between LMPPs and DCs, we showed that not only IRF8– LMPPs but IRF8+ LMPPs can differentiate into MDPs (Figure 3B-D). It has been reported that ∼10% of MDPs express cDC signature genes.42 Indeed, human and mouse MDPs include cells that produce cDCs only, but not monocytes.13,16 Our observations, together with previously published results, indicate that a fraction of MDPs is already committed to the cDC fate and is likely to include IRF8+ LMPP-derived, cDC1-biased MDPs.
We elucidated the mechanism by which IRF8 regulates early DC lineage specification by genome-wide approaches such as scRNA-seq and ATAC-seq. Initial extensive attempts at finding global changes in gene expression between IRF8– and IRF8+ LMPP subpopulations was unsuccessful. Consistent with our results, scRNA-seq analysis in human HSCs and early progenitors demonstrated the transcriptional state of single cells is highly interconnected without hierarchical relationships.43 Nevertheless, we identified IRF8+ LMPP-specific open chromatin regions whose emergence was dependent on IRF8. Genes associated with these open chromatin regions were not yet actively transcribed in LMPPs, but highly expressed in the downstream DC lineage populations such as CDPs, pre-cDCs, and cDCs. Similar time lags between epigenetic and transcriptional changes has been reported for mouse monocyte and DC differentiation from MDPs in vivo,33 for in vitro differentiation of several hematopoietic cell types in mice,44 and very recently for human in vivo hematopoiesis.45 Yu et al also showed that the differentiation bias of mouse HSCs is associated with distinct epigenetic states such as DNA methylation and chromatin accessibility.5 Collectively, these data illustrate that key transcription factors such as IRF8 induce epigenetic changes in lineage-specific enhancers in a subset of otherwise multipotent progenitors to prepare for future expression of the associated genes, thereby controlling biased differentiation behaviors.
An important question is the biological meaning of the presence of IRF8+, cDC1-primed LMPP subpopulation. On the basis of the cell yields in LMPP transfer experiments using irradiated mice, we can speculate that the percentage of cDC1s derived from IRF8+ LMPPs in the steady state is relatively small. We noticed, however, that IRF8+ LMPPs require only 7 days to generate DCs, whereas IRF8– LMPPs require 10 days. This indicates that IRF8+ LMPPs give rise to cDC1s more quickly than IRF8– LMPPs do in vivo. It is conceivable that IRF8+ LMPPs serve as a rapid source of cDC1s, for example in response to infection. During Plasmodium chabaudi infection, Flt3L is secreted from mast cells to induce cDC1 expansion.46 In addition, Tussiwand et al demonstrated that cDC1s are accumulated in response to infection by intracellular pathogens such as Listeria monocytogenes and Mycobacterium tuberculosis.47 Interestingly, interleukin-12 secreted during these microbial infections boosts cDC1 production through the action of interferon-γ (IFN-γ). It has also been reported that IFN-γ upregulates IRF8 expression in macrophages and myeloid progenitors48 ; thus, Flt3L and the interleukin-12–IFN-γ axis stimulated by microbes might activate the DC differentiation pathway through IRF8+ LMPPs to eradicate infections. Further investigation is needed to test this hypothesis in future studies.
Several transcription factors, such as PU.1, CBFβ/RUNX, and BATF3/IRF8 are reported to be required for Irf8 expression in the MDP, GMP/MDP, and pre-cDC stages, respectively.20,24,35 However, the mechanism underlying the expression of Irf8 in LMPPs remains unknown, except for our finding that IRF8 itself is not required. It is tempting to speculate that HLF may repress Irf8 transcription. In this regard, our ATAC-seq data, showing distinct open chromatin regions between IRF8– and IRF8+ LMPPs, are also worthy of further investigation. Clarifying the mechanism how Irf8 is induced in LMPPs will deepen our understanding of DC lineage fate decisions.
The single-cell RNA-sequencing and assay for transposase-accessible chromatin with high throughput sequencing data have been deposited in the DNA Data Bank of Japan (accession numbers DRA005985, DRA005986, and DRA006834). The microarray data for lymphoid-primed multipotent progenitors have been deposited in the Gene Expression Omnibus database (accession GSE113748). The microarray data for granulocyte-monocyte progenitors, monocyte-dendritic cell (DC) progenitors, common DC progenitors, preclassical DCs, common monocyte progenitors, classical DC1s, monocytes, and neutrophils were previously published in the Gene Expression Omnibus database (accession codes GSE50052 and GSE84509).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Kyoko Ochiai at Tohoku University for her valuable advice on B-cell differentiation experiments in vitro, and Hideaki Nakajima, Koichi Murakami, Tatsuma Ban, Go R. Sato, and Ayano Shinozuka at Yokohama City University for their help with experiments.
This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science/Ministry of Education, Culture, Sports, Science and Technology (MEXT; JP15H04860 and JP18K19345 [T.T.], JP221S0002 and JP16H06279 [Y.S.], JP16K21271 and JP17KK0171 [D.K.]); the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases (H.C.M.) and National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development (K.O.); Yokohama Foundation for Advancement of Medical Science (D.K.); and the MEXT Joint Usage/Research Center Program at the Advanced Medical Research Center, Yokohama City University (T.T.).
Authorship
Contribution: D.K. and T.T. designed the study. D.K., W.K., H.S., A.N., and Y.S. performed the experiments; D.K., W.K., H.S., J.N., Y.S., and T.T. analyzed the data; D.K. and T.T. wrote the manuscript. H.C.M., K.O., and Y.S. provided key resources; and T.T. supervised the project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daisuke Kurotaki, Department of Immunology, Yokohama City University Graduate School of Medicine, 3-9 Fukuura, Kanazawa-ku, Yokohama 236-0004, Japan; e-mail: kurotaki@yokohama-cu.ac.jp; and Tomohiko Tamura, Department of Immunology, Yokohama City University Graduate School of Medicine, 3-9 Fukuura, Kanazawa-ku, Yokohama 236-0004, Japan; e-mail: tamurat@yokohama-cu.ac.jp.