Key Points
CALR mutants rescue cell surface localization of traffic-deficient TpoR, including R102P that causes congenital thrombocytopenia.
Oncogenic TpoR activation by CALR mutants requires interaction, stabilization, and cell surface localization of the TpoR-CALR complex.

Abstract
Calreticulin (CALR) +1 frameshift mutations in exon 9 are prevalent in myeloproliferative neoplasms. Mutant CALRs possess a new C-terminal sequence rich in positively charged amino acids, leading to activation of the thrombopoietin receptor (TpoR/MPL). We show that the new sequence endows the mutant CALR with rogue chaperone activity, stabilizing a dimeric state and transporting TpoR and mutants thereof to the cell surface in states that would not pass quality control; this function is absolutely required for oncogenic transformation. Mutant CALRs determine traffic via the secretory pathway of partially immature TpoR, as they protect N117-linked glycans from further processing in the Golgi apparatus. A number of engineered or disease-associated TpoRs such as TpoR/MPL R102P, which causes congenital thrombocytopenia, are rescued for traffic and function by mutant CALRs, which can also overcome endoplasmic reticulum retention signals on TpoR. In addition to requiring N-glycosylation of TpoR, mutant CALRs require a hydrophobic patch located in the extracellular domain of TpoR to induce TpoR thermal stability and initial intracellular activation, whereas full activation requires cell surface localization of TpoR. Thus, mutant CALRs are rogue chaperones for TpoR and traffic-defective TpoR mutants, a function required for the oncogenic effects.
Introduction
Calreticulin (CALR) mutants are associated with essential thrombocythemia and myelofibrosis, 2 major BCR-ABL–negative myeloproliferative neoplasms (MPNs).1,2 These mutants result from deletions and insertions in exon 9 of the CALR gene that generate a +1 frameshift. A new C terminus rich in positive and hydrophobic amino acids (supplemental Figure 1, available on the Blood Web site) is generated, leading to pathologic activation of the thrombopoietin receptor (TpoR), with persistent JAK2/STAT5 signaling and dysregulated megakaryopoiesis. In vitro experiments indicated that among cytokine receptors, TpoR is unique in its ability to be robustly and constitutively activated by CALR mutants and to induce transformation to cytokine independence of hematopoietic lines.3-6 The positively charged tail of the mutant CALR is absolutely required for this activation.3-9 In vivo, TpoR is essential for persistent JAK-STAT activation induced by mutant CALRs.3-5
The effect of the CALR C-terminal sequences on its chaperone activity, traffic, and secretion10 are still poorly understood. The present study queries whether: (1) entry of CALR mutants in the secretory pathway is required for TpoR activation; (2) CALR mutants are exposed at the surface; (3) interaction with TpoR is direct and which TpoR residues mediate activation; and (4) CALR mutants function as rogue chaperones, forcing exit from the endoplasmic reticulum (ER) of receptors that have not passed quality control, allowing premature progression through the secretory pathway. We assessed whether CALR mutants mediate transport to the cell surface of TpoR mutants that are known to be blocked in the ER and tested the role of cell surface localization in activation.
Methods
Cell lines and mouse primary bone marrow assays
Ba/F3 cells and γ2A JAK2-deficient fibrosarcoma cells were used for growth and transcriptional studies after transduction or transfection.3 Ba/F3 TpoR cells carrying a heterozygous Calr +1 frameshift mutation were engineered by using CRISPR/Cas9 editing.7 Transduced mouse bone marrow was assessed by using flow cytometry and megakaryocyte colony formation.3,4
Transcriptional and cell growth assays
Antibodies
Phospho-specific antibodies were from Cell Signaling Technologies,11 anti-CALR and anti-Mpl antibodies were from MilliporeSigma, and anti–β-actin antibodies were from MilliporeSigma. Antibodies detecting HA and FLAG were from Roche and MilliporeSigma, respectively. Rabbit polyclonal antibodies (SAT602) against CALR mutant tail were from MyeloPro.
Coimmunoprecipitation
NP40 extracts from HEK-293T cells transiently transfected with complementary DNAs (cDNAs) coding for HA-TpoR and Flag-CALR variants were incubated with anti-FLAG antibodies overnight at 4°C. Immunoprecipitation and western blotting were performed as previously described.3
Flow cytometry
Surface expression of the TpoR was measured by flow cytometry using an anti-human APC or PE-CD110 monoclonal antibody (Miltenyi Biotec). Surface localization of CALR wild type (WT) and mutants was measured by either anti-total CALR (N terminus; Abcam) or the antimutated CALR polyclonal antibodies SAT602 and Alexa647 anti-rabbit IgG secondary antibody (Thermo Fisher Scientific).
Confocal microscopy
Microscopy was performed on Calr−/− MEF paraformaldehyde-fixed and fluorescently labeled antibody-stained cells (mouse anti-GM130, CIS-Golgi matrix protein, and rabbit anti-CALR followed by anti-mouse IgG/Alexa568 and anti-rabbit IgG/Alexa488). A Zeiss Spinning Disc confocal microscope with a 3100 objective was used for analyses.
Nano–bioluminescence resonance energy transfer
cDNAs coding for TpoR and the erythropoietin receptor (EpoR) were cloned into a modified pNL-N vector (Promega) to generate an N-terminal fusion of the Nano-luciferase to the receptors. cDNAs coding for WT and the del52 mutant CALRs were cloned into the pHT-C vector to generate HaloTag fused constructs (Promega). HEK-EBNA cells transiently transfected with those constructs were analyzed for bioluminescence resonance energy transfer (BRET) on a GloMax Discover multiplate reader (Promega) at 37°C using the 450BP (donor) and 600LP (acceptor) built-in filters.
Analysis of oligomerization by size-exclusion chromatography
The extracellular domain of the human TpoR and human CALR mutant del52 produced in Drosophila S2 cells (Thermo Fisher Scientific) were fractionated by using size-exclusion chromatography by loading on a Superdex 200 Increase 10/300 column (GE Healthcare). Elution was performed at 0.5 mL/min with buffer TNG (Tris-NaCl-glycerol) pH 7.5, and 0.5 mL fractions were collected and analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and western blot. Fraction proteins were immunoprecipitated with anti-His5 antibody (Qiagen) and treated with Endoglycosidase-H (NEB) or N-glycosidase F (NEB).3
Statistical analysis
Statistical analyses were performed by using the unpaired nonparametric 2-tailed Student t test or the nonparametric multiple comparisons Steel test with a control group as indicated in the legend of figures. All the analyses with confidence intervals >95% are indicated as significant (*P < .05, **P < .01, and ***P < .001).
Results
The anterograde pathway is required for TpoR activation by CALR mutants
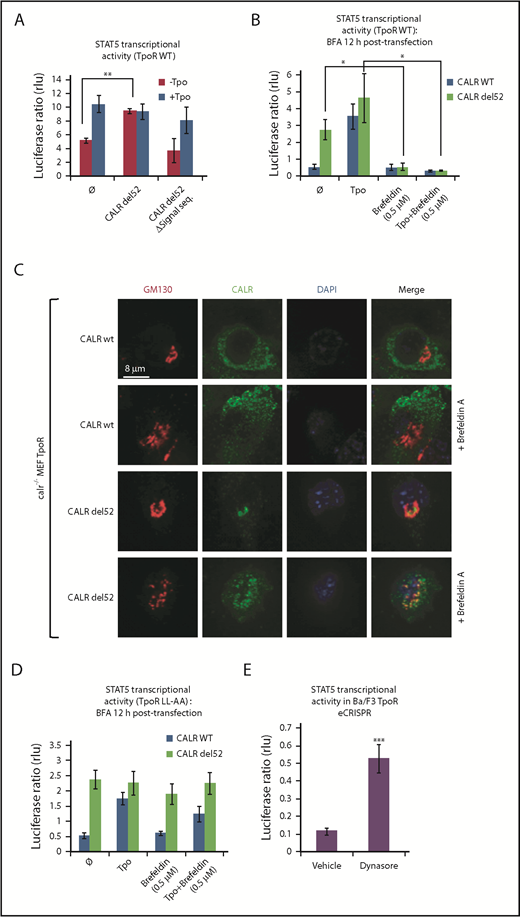
To show that entry into the secretory pathway is required for activation of TpoR by CALR del52, we found that a CALR del52 mutant lacking the signal peptide (CALR del52 Δ17) is unable, unlike CALR del52, to induce STAT5 transcriptional activity in transfected γ2A cells when coexpressed with TpoR (Figure 1A).
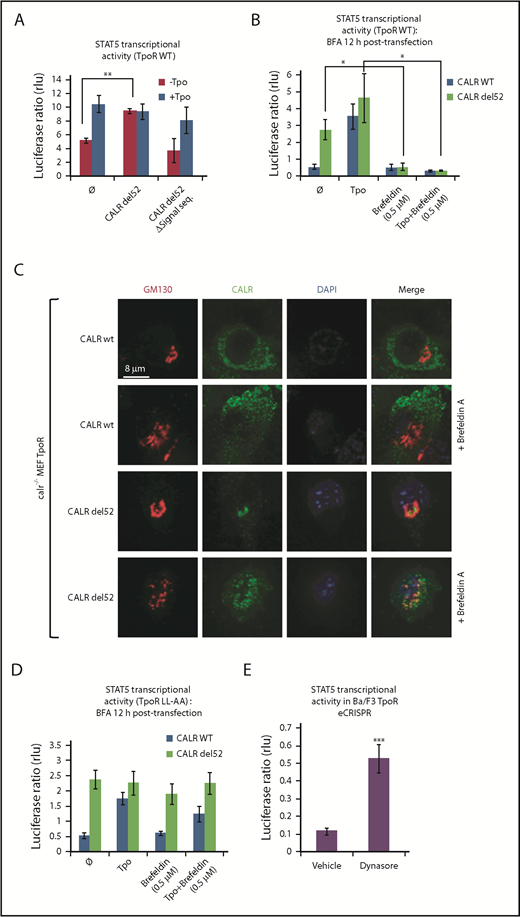
Traffic through the secretory pathway is required for TpoR activation by CALR del52. (A) CALR del52 mutant lacking the signal peptide does not activate STAT5 transcriptional activity in γ2A cells via TpoR. (B) Blocking the anterograde secretion pathway by BFA treatment of 16 hours (started 12 hours after transfection) leads to a complete inhibition of STAT5 transcriptional activity via WT TpoR in transfected γ2A cells. (C) Confocal analysis confirmed that 16 hours of BFA treatment of mouse embryo fibroblasts knock out for Calr and stably transduced with CALR variants and TpoR redistributes CALR del52 to the ER. (D) BFA treatment 12 hours posttransfection (for 16 hours) does not block activation of TpoR LL-AA by CALR del52 or Tpo, as assessed by STAT5 transcriptional activity in transfected γ2A cells. (E) Treatment of Ba/F3 TpoR eCRISPR/CALRdel52 with Dynasore hydrate (50 μM), which blocks endocytosis, increases STAT5 transcriptional activity. Shown are averages of 3 independent experiments each with 2 to 3 biological replicates. Statistical analysis (jmp pro12) was performed by using the nonparametric multiple comparisons Steel test with a control group (A-B) or by the unpaired nonparametric 2-tailed Student t test (E).
Traffic through the secretory pathway is required for TpoR activation by CALR del52. (A) CALR del52 mutant lacking the signal peptide does not activate STAT5 transcriptional activity in γ2A cells via TpoR. (B) Blocking the anterograde secretion pathway by BFA treatment of 16 hours (started 12 hours after transfection) leads to a complete inhibition of STAT5 transcriptional activity via WT TpoR in transfected γ2A cells. (C) Confocal analysis confirmed that 16 hours of BFA treatment of mouse embryo fibroblasts knock out for Calr and stably transduced with CALR variants and TpoR redistributes CALR del52 to the ER. (D) BFA treatment 12 hours posttransfection (for 16 hours) does not block activation of TpoR LL-AA by CALR del52 or Tpo, as assessed by STAT5 transcriptional activity in transfected γ2A cells. (E) Treatment of Ba/F3 TpoR eCRISPR/CALRdel52 with Dynasore hydrate (50 μM), which blocks endocytosis, increases STAT5 transcriptional activity. Shown are averages of 3 independent experiments each with 2 to 3 biological replicates. Statistical analysis (jmp pro12) was performed by using the nonparametric multiple comparisons Steel test with a control group (A-B) or by the unpaired nonparametric 2-tailed Student t test (E).
We next assessed whether accumulation of CALR mutants and TpoR in the ER is sufficient for activation of the TpoR. Treating γ2A cells also transfected with JAK2 with Brefeldin A (BFA), an inhibitor of ER-to-Golgi traffic,12-14 prevented activation of TpoR by both the ligand Tpo and by CALR mutants (Figure 1B), showing that activation of TpoR by CALR mutants occurs after the ER compartment. γ2A cells were used because they are JAK2-deficient and allowed us to show that JAK2 is absolutely required for activation of TpoR by CALR mutants.3 Using confocal microscopy, mutant CALR colocalized with the Golgi marker Giantin in mouse embryo fibroblasts knocked out for Calr, where WT and mutant CALRs were reconstituted together with TpoR3-5 (Figure 1C), consistent with the notion that CALR mutants are detectable in the Golgi apparatus.9 In hematopoietic cell lines, TpoR and CALR mutants colocalized in the cis-, medial-, and trans-Golgi network compartments (supplemental Figure 2A-B). BFA relocalized CALR del52 to the ER.12-15
Flow cytometry was used to detect cell surface levels of WT CALR and CALR del52 in stably transduced Ba/F3 cells expressing or not TpoR. Using a polyclonal antibody (SAT602) raised against the new tail of mutant CALR, we could detect the localization of mutant CALR in cells coexpressing TpoR, as well as in the absence of TpoR (supplemental Figure 3A). Complex formation between cell surface mutant CALR and TpoR was detected in UT7-Tpo cells by protein ligation assay (supplemental Figure 3B). Furthermore, colocalization of surface TpoR with mutant CALR del52 was detected in in vitro CD34+-derived megakaryocytes from 2 patients with MPN, as shown by the confocal microscopy (supplemental Figure 3C). Cell surface expression of CALR del52 was observed in CD34+ cells from 3 CALR del52 MPN patients by using flow cytometry (supplemental Figure 3D). Immunofluorescence was next used to identify subcellular localization of active TpoR complexes by studying the colocalization of TpoR and phospho-JAK2 Y1007/1008, as a marker for activated JAK2, in Ba/F3 cells. Strikingly, Ba/F3 cells coexpressing CALR del52 or CALR ins5 induced active TpoR complexes in early endosomes in the absence of Tpo. Moreover, TpoR and CALR del52/ins5 but not CALR WT colocalized in early endosomal vesicles (supplemental Figure 2D). Together, these data show that mutant CALRs promote cytokine-independent surface TpoR activation.
We questioned whether cell surface localization is sufficient to mediate activation of TpoR. The TpoR mutant in which the diLeu motif LL in the cytosolic domain has been mutated to AA is defective in internalization.16 Once arrived and stable at the surface, if activated by CALR mutant, this TpoR mutant would continue to signal when further traffic is prevented by BFA. Indeed, BFA failed to block TpoR LL-AA activation by mutant CALR but blocked TpoR WT activation (Figure 1B,D). Thus, signaling from the cell surface suffices for TpoR activation. Moreover, an inhibitor of internalization, Dynasore,17 enhanced TpoR activation by CALR mutants in Ba/F3 TpoR cells carrying a heterozygous Calr del52 mutation engineered by using CRISPR/Cas9 editing7 (Figure 1E).
CALR mutants directly interact with TpoR, which remains partially immature after Golgi processing, and induce TpoR dimerization
As reported,5,6,18 HA-TpoR coimmunoprecipitated with mutant CALR del52 tagged with a Flag tag. This interaction was dependent on the N-glycosylation on TpoR and on the lectin domain and chaperone effects of CALR (Figure 2A),19 as also shown for the interaction of nontagged proteins.3,4 In addition, we found that W319 located in the C terminus of CALR mutants but situated in close proximity to the globular domain, being part of the lectin-binding domain,20 is required for binding to TpoR.
![Figure 2. TpoR and CALR del52 interact directly to form stable complexes. (A) Coimmunoprecipitation analysis showed that TpoR and CALR del52 interaction is dependent on N-linker sugars of TpoR, especially N117-sugars and on the lectin binding domain of CALR. (B) Oligomerization analysis of CALR and TpoR by size-exclusion chromatography. Expression vectors coding for CALR del52 or extracellular domain (ECD) of TpoR and containing a metallothionein promoter were stably transfected (calcium phosphate) in S2 Drosophila cells with G418-containing medium. Expression of the proteins of interest was induced by copper treatment, and secreted forms of TpoR and CALR del52 were further collected in the supernatant. His-tagged ECD-TpoR was enriched by using a Nickel column. Finally, molecular-size distribution of CALR del52 and TpoR was measured by size-exclusion chromatography and fractions analyzed on MOPS 4% to 12% gel in the presence of DTT. (C) Glycosylation analysis with Endoglycosidase H and Peptide N-glycosidase F (N-glyc F or PNGase F) of the indicated fractions obtained from size-exclusion chromatography. (D) Cartoon representation of our Nano-BRET set-up. Because CALR del52 (del52) mutant interacts longer and in close proximity (<10 nm) to TpoR compared with the WT CALR, it enables BRET from the Nano-luciferase fused to the N-terminus of TpoR (NL) to the 618-ligand fluorophore (depicted by a red star) bound to the HaloTag (H) fused to the C-terminal tail of CALR. The BRET is symbolized by a shift in wavelength (460 nm [blue] to 618 nm [red]) of the bioluminescence emanating from the Nano-luciferase. (E) BRET between Nano-luciferase fused cytokine receptors and CALR fused to HaloTag using the Nano-BRET system. There is a strong energy transfer between TpoR and CALR del52 but not between EpoR and CALR del52. Black bars show standard deviation. ****P < .0001, unpaired nonparametric 2-tailed Student t test, 4 independent experiments each with 3 biological replicates.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/25/10.1182_blood-2018-09-874578/3/m_blood874578f2.png?Expires=1769111038&Signature=xxTjJiYCXJmX4Uro~J6snP-T3I2V93r6aOLYiCiSoZnldqN~l3BedQjNycPbcJsFGq48HBJWBYHDQjLKhnjCr6hNsTrrf6anI~uBjk6NgzDjxtHah8Hn~jcjhyEH9wrwgG93Qj8~GoPEZAMFWcVKrtVXVgGvoeIMcOmSt5s2NVOi183DvSdqxWBeozp-y423WrLjbbNZ4eJWh0iL7jRSgn944mhWniZglc4cgzB8uvhPHWkm1UQx4boFAJsHSx3usG-47K4MI-a1ohnVZqWKYWlAZBMqRAJNn85X1tzH5iEervJhdSrbvQAU-5eKKZ-NkT5rbbnhqdxRkQiaKD4bFg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TpoR and CALR del52 interact directly to form stable complexes. (A) Coimmunoprecipitation analysis showed that TpoR and CALR del52 interaction is dependent on N-linker sugars of TpoR, especially N117-sugars and on the lectin binding domain of CALR. (B) Oligomerization analysis of CALR and TpoR by size-exclusion chromatography. Expression vectors coding for CALR del52 or extracellular domain (ECD) of TpoR and containing a metallothionein promoter were stably transfected (calcium phosphate) in S2 Drosophila cells with G418-containing medium. Expression of the proteins of interest was induced by copper treatment, and secreted forms of TpoR and CALR del52 were further collected in the supernatant. His-tagged ECD-TpoR was enriched by using a Nickel column. Finally, molecular-size distribution of CALR del52 and TpoR was measured by size-exclusion chromatography and fractions analyzed on MOPS 4% to 12% gel in the presence of DTT. (C) Glycosylation analysis with Endoglycosidase H and Peptide N-glycosidase F (N-glyc F or PNGase F) of the indicated fractions obtained from size-exclusion chromatography. (D) Cartoon representation of our Nano-BRET set-up. Because CALR del52 (del52) mutant interacts longer and in close proximity (<10 nm) to TpoR compared with the WT CALR, it enables BRET from the Nano-luciferase fused to the N-terminus of TpoR (NL) to the 618-ligand fluorophore (depicted by a red star) bound to the HaloTag (H) fused to the C-terminal tail of CALR. The BRET is symbolized by a shift in wavelength (460 nm [blue] to 618 nm [red]) of the bioluminescence emanating from the Nano-luciferase. (E) BRET between Nano-luciferase fused cytokine receptors and CALR fused to HaloTag using the Nano-BRET system. There is a strong energy transfer between TpoR and CALR del52 but not between EpoR and CALR del52. Black bars show standard deviation. ****P < .0001, unpaired nonparametric 2-tailed Student t test, 4 independent experiments each with 3 biological replicates.
TpoR and CALR del52 interact directly to form stable complexes. (A) Coimmunoprecipitation analysis showed that TpoR and CALR del52 interaction is dependent on N-linker sugars of TpoR, especially N117-sugars and on the lectin binding domain of CALR. (B) Oligomerization analysis of CALR and TpoR by size-exclusion chromatography. Expression vectors coding for CALR del52 or extracellular domain (ECD) of TpoR and containing a metallothionein promoter were stably transfected (calcium phosphate) in S2 Drosophila cells with G418-containing medium. Expression of the proteins of interest was induced by copper treatment, and secreted forms of TpoR and CALR del52 were further collected in the supernatant. His-tagged ECD-TpoR was enriched by using a Nickel column. Finally, molecular-size distribution of CALR del52 and TpoR was measured by size-exclusion chromatography and fractions analyzed on MOPS 4% to 12% gel in the presence of DTT. (C) Glycosylation analysis with Endoglycosidase H and Peptide N-glycosidase F (N-glyc F or PNGase F) of the indicated fractions obtained from size-exclusion chromatography. (D) Cartoon representation of our Nano-BRET set-up. Because CALR del52 (del52) mutant interacts longer and in close proximity (<10 nm) to TpoR compared with the WT CALR, it enables BRET from the Nano-luciferase fused to the N-terminus of TpoR (NL) to the 618-ligand fluorophore (depicted by a red star) bound to the HaloTag (H) fused to the C-terminal tail of CALR. The BRET is symbolized by a shift in wavelength (460 nm [blue] to 618 nm [red]) of the bioluminescence emanating from the Nano-luciferase. (E) BRET between Nano-luciferase fused cytokine receptors and CALR fused to HaloTag using the Nano-BRET system. There is a strong energy transfer between TpoR and CALR del52 but not between EpoR and CALR del52. Black bars show standard deviation. ****P < .0001, unpaired nonparametric 2-tailed Student t test, 4 independent experiments each with 3 biological replicates.
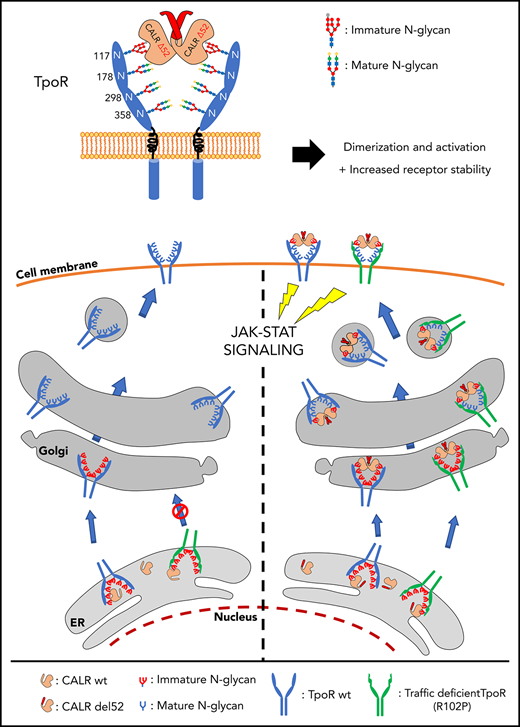
Careful analysis of the size of the coprecipitated TpoR band indicated that it was slightly heavier than the TpoR in the total cell lysate, suggesting a difference in N-glycosylation, in agreement with immature TpoR detected in CALR mutant cells.3,4 We then coexpressed the extracellular domain of TpoR with mutant CALR del52, or just the extracellular domain of TpoR alone in Schneider Drosophila S2 cells. By size-exclusion chromatography, we observed complex formation at ≈200 kDa in a fraction that contained both proteins (Figure 2B). Mass spectrometry detected a 5-times higher proportion of high mannose sugars in TpoR in this fraction than in the case for TpoR-soluble domain purified in the same cells from a ≈60 kDa molecular fraction. The latter contained mature types of sugars, which in insect cells are fucose-containing paucimannosidic N-glycans.21 Such mature structure was completely absent from the TpoR Asn117 when in complex with mutant CALR del52 (supplemental Figure 4; supplemental Table 1). Gel migration of the mature band of TpoR from the 200 kDa fraction was also slower with partial sensitivity to Endo H, unlike the TpoR from the 60 kDa fraction or the TpoR from the equivalent fraction when expressed alone in S2 cells (Figure 2C). Mass spectrometry showed mature glycosylation for the other Asn residues of TpoR (178, 298, 358) in complexes with CALR mutants, suggesting that Asn117 is the key masked asparagine by CALR mutants. Thus, a stable complex is formed between mutant CALR and partially immature TpoR, which traffics to Golgi. These findings, as well as previous results,3 provide evidence that TpoR in complex with mutant CALR carries immature high-mannose sugars.
To further test for direct interaction of TpoR and CALR in living cells, we used the Nano-BRET system.22 In this approach, a short distance of 10 nm maximum is required for an efficient BRET between 2 proteins.22 We fused the C terminus of WT or mutant CALR to the HaloTag, and the N-terminus of TpoR and EpoR to the Nano-luciferase (Figure 2D). A strong BRET was detected from the TpoR to CALR del52 but not to the CALR WT (Figure 2E). Although a very small but significant BRET was detected for CALR mutant and EpoR, this finding was minor in comparison with TpoR.
Because activation of the TpoR requires receptor dimerization/oligomerization, we tested the capacity of CALR mutants to induce dimerization of cytosolic tails of TpoR by using the Nano-BiT protein complementation approach.23 TpoR and EpoR monomers were C-terminally fused to either the LgBiT or the SmBiT split-luciferase units. Coexpression of TpoR with CALR del52, but not CALR WT, led to the dimerization of receptor cytosolic tails in a JAK2-dependent manner, which was not the case for the EpoR (supplemental Figure 12). Thus, CALR mutants specifically interact with TpoR, leading to close apposition of cytosolic domains.
MPN-associated CALR mutants rescue traffic of TpoR R102P known to be completely absent from the cell surface
One hypothesis is that CALR mutants are rogue chaperones that promote pathologic traffic/activation of TpoR carrying immature N-glycosylation to the surface. We questioned whether receptor mutants that are blocked in the ER can be rescued for their traffic to the cell surface by CALR mutants.
Homozygous mutation in the extracellular domain of TpoR at position R102 to proline (R102P; Figure 3A) leads to a receptor blocked in the ER.24 Children born with this homozygous mutation experience congenital amegakaryocytic thrombocytopenia (CAMT) that progresses to marrow failure.25,26 No other endogenous ligand can replace Tpo, and no agent can rescue TpoR R102P for traffic to the cell surface.24
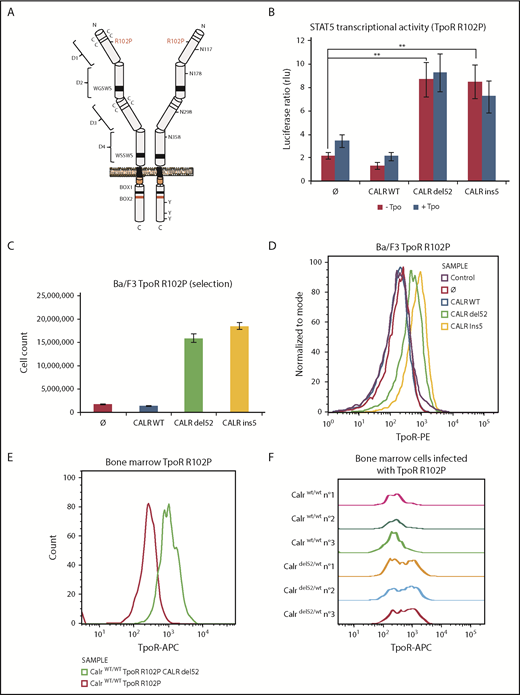
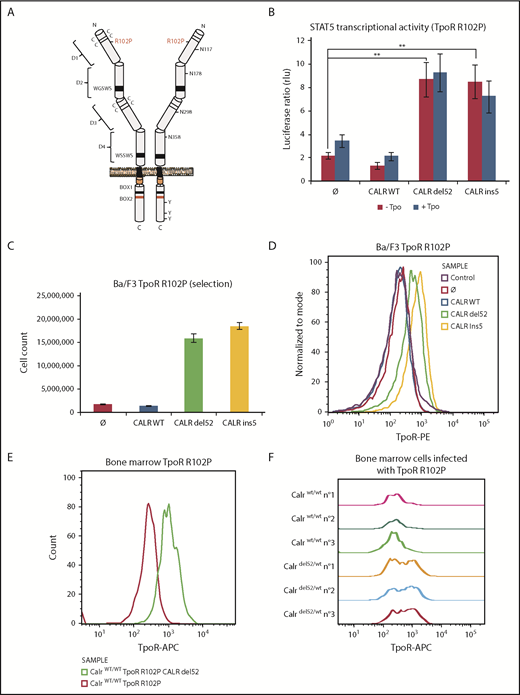
CALR mutants activate TpoR mutant R102P, which is traffic-deficient and is not localized at the cell surface. (A) Schematic representation of TpoR R102P with the position of the mutation and N-glycosylations in the extracellular domain. (B) Transcriptional activity of CALR mutants in the presence of TpoR R102P. Values shown represent the average of 3 independent experiments, each performed with 3 biological replicates ± standard error of the mean. **P < .01. Statistical analysis (jmp pro12) was performed by the nonparametric multiple comparisons Steel test with a control group. (C) Long-term (selection) proliferation assays for TpoR R102P in the presence of CALR mutants. (D) Cell surface localization of TpoR R102P in the presence of CALR mutants measured by flow cytometry. Flow cytometry analysis using an anti-human TpoR (CD110) of retrovirally transduced bone marrow cells from C57Bl6 mice expressing either TpoR R102P alone or together with CALR del52 (E), or bone marrow cell from KI-Calrdel52/WT mice transduced or not with TpoR R102P (F).
CALR mutants activate TpoR mutant R102P, which is traffic-deficient and is not localized at the cell surface. (A) Schematic representation of TpoR R102P with the position of the mutation and N-glycosylations in the extracellular domain. (B) Transcriptional activity of CALR mutants in the presence of TpoR R102P. Values shown represent the average of 3 independent experiments, each performed with 3 biological replicates ± standard error of the mean. **P < .01. Statistical analysis (jmp pro12) was performed by the nonparametric multiple comparisons Steel test with a control group. (C) Long-term (selection) proliferation assays for TpoR R102P in the presence of CALR mutants. (D) Cell surface localization of TpoR R102P in the presence of CALR mutants measured by flow cytometry. Flow cytometry analysis using an anti-human TpoR (CD110) of retrovirally transduced bone marrow cells from C57Bl6 mice expressing either TpoR R102P alone or together with CALR del52 (E), or bone marrow cell from KI-Calrdel52/WT mice transduced or not with TpoR R102P (F).
Expressing CALR del52 or ins5 led to significant activation of STAT5 transcriptional activity in γ2A cells via TpoR R102P (Figure 3B). This required similar N-glycosylation on TpoR and intact lectin binding and chaperone activity for the CALR mutants (supplemental Figure 5D-E). In parallel, we stably expressed TpoR R102P in Ba/F3 cells and then coexpressed WT CALR or CALR del52 or ins5. We observed that CALR del52 and ins5 were able to induce constitutive signaling of TpoR R102P in Ba/F3 cells, leading to short- and long-term cytokine-independent growth (Figure 3C; supplemental Figure 5F). Using flow cytometry, we could detect cell surface TpoR R102P only in cells expressing CALR del52 or ins5 but not in cells expressing CALR WT or empty vector (Figure 3D).
An Endo H–resistant TpoR R102P was only detected in CALR del52 and ins5 cells (supplemental Figure 5A). We interpret this result as the consequence of the processing of 3 of 4 Asn residues in the Golgi apparatus, as also shown for TpoR WT extracellular domain in insect cells (supplemental Table 1). The presence of TpoR R102P was confirmed in the Golgi apparatus in the presence of CALR mutants (supplemental Figure 5B). Importantly, on the cell surface, TpoR R102P could be further activated by the small molecule TpoR agonist eltrombopag but not by the ligand, Tpo (supplemental Figure 5F-G). Since Tpo requires residues around R102 for binding, we used eltrombopag which binds His499 at the outset of the transmembrane domain.27
We next examined cell surface localization of TpoR R102P in primary bone marrow cells. Figure 3E shows a significant cell surface localization of the human TpoR R102P in cells also transduced with CALR del52. Furthermore, endogenous expression of CALR del52 in bone marrow from a heterozygous knock-in mouse model28 is sufficient to restore cell surface localization of exogenous human TpoR R102P, detected by a human TpoR-specific antibody (Figure 3F).
MPN-associated CALR mutants rescue traffic of TpoR mutants that are ER-retained via cytosolic KKXX retention signals
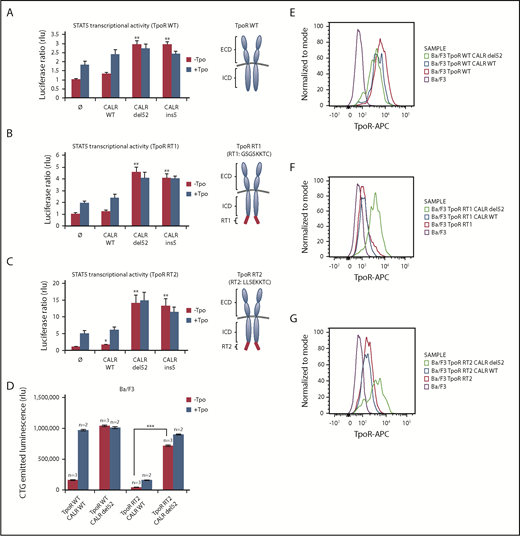
Given the rescue to the cell surface of TpoR R102P from ER by CALR mutants, we questioned whether we could retain TpoR in the ER by a cytosolic ER retention signal (KKXX)29 and then tested the effects of CALR mutants. We created TpoR RT1 (GSGSSKKTC), which contains the C-terminal KKXX signal after a flexible GSGS linker, and TpoR RT2 (LLSEKKTC), which contains a more rigid LLSE linker before the ER retention KKXX motif (Figure 4A-C, right panels) previously used to retain truncated CD4 mutants.29 Both mutants were largely retained intracellularly compared with WT TpoR (Figure 4E-G), with only a minor response to ligand (Figure 4B-D). Upon CALR del52 expression, these TpoR mutants were rescued for traffic, as detected by flow cytometry, for STAT5 activation and autonomous growth in Ba/F3 cells. Thus, CALR mutants overcome retention signals on TpoR and rescue ER-retained receptor chains to cell surface localization.
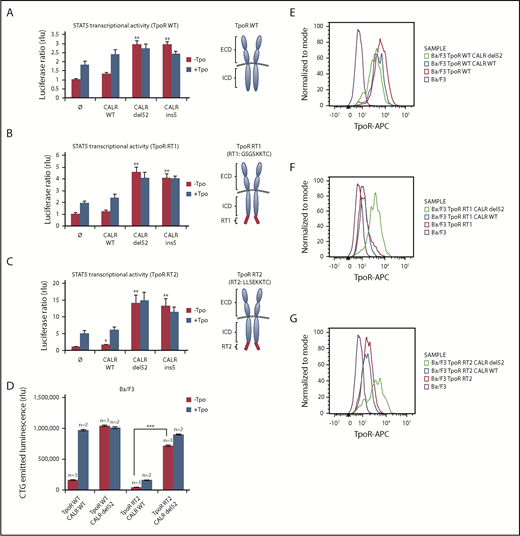
CALR mutants activate and rescue cell surface expression of mutated TpoR harboring a retention signal in its C-terminal domain. STAT5 transcriptional activity in γ2A cells of CALR mutants when coexpressed with TpoR WT (A), TpoR RT1 (B), and TpoR RT2 (C). Values shown represent the average of 3 independent experiments each done with 3 biological repeats ± standard error of the mean. (D) Autonomous growth at 72 hours, as determined by CTG assays of Ba/F3 cells expressing the indicated CALR and TpoR WT or RT2. Cells were infected with retroviruses coding for WT or mutant CALR and WT TpoR or TpoR RT2. Similar expression was determined by flow cytometry for markers downstream of the IRES of the retroviral vectors (mCherry for CALR and GFP for TpoR variants). Shown are means ± standard error of the mean. Kruskal-Wallis test with Steel posttest at 5% significance level. ***P < .001. Flow cytometry analysis of cell surface expression of the TpoR WT (E), TpoR RT1 (F), and TpoR RT2 (G) coexpressed or not with CALR WT or CALR del52. ICD, intracellular domain; RT, retention signal.
CALR mutants activate and rescue cell surface expression of mutated TpoR harboring a retention signal in its C-terminal domain. STAT5 transcriptional activity in γ2A cells of CALR mutants when coexpressed with TpoR WT (A), TpoR RT1 (B), and TpoR RT2 (C). Values shown represent the average of 3 independent experiments each done with 3 biological repeats ± standard error of the mean. (D) Autonomous growth at 72 hours, as determined by CTG assays of Ba/F3 cells expressing the indicated CALR and TpoR WT or RT2. Cells were infected with retroviruses coding for WT or mutant CALR and WT TpoR or TpoR RT2. Similar expression was determined by flow cytometry for markers downstream of the IRES of the retroviral vectors (mCherry for CALR and GFP for TpoR variants). Shown are means ± standard error of the mean. Kruskal-Wallis test with Steel posttest at 5% significance level. ***P < .001. Flow cytometry analysis of cell surface expression of the TpoR WT (E), TpoR RT1 (F), and TpoR RT2 (G) coexpressed or not with CALR WT or CALR del52. ICD, intracellular domain; RT, retention signal.
Identification of a hydrophobic patch in TpoR extracellular domain that is required for activation by CALR mutants but not for binding
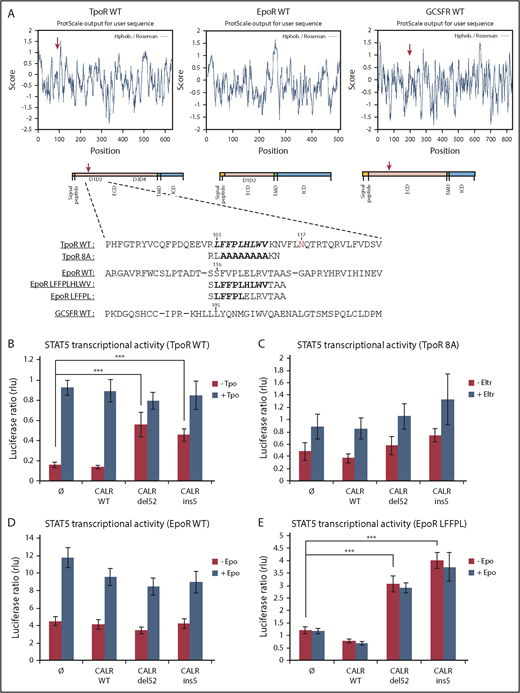
We searched for conserved motifs in the extracellular domains of cytokine receptors shared by TpoR and G-CSFR, which can also be weakly activated by CALR mutants,3 and not by EpoR and other receptors that are not activated by CALR mutants. A hydrophobic patch was identified only in the extracellular domains of TpoR and G-CSFR. This patch is conserved in TpoR from different species and overlaps with the sequence where R102 is located (Figure 5A; supplemental Figure 6A-B).
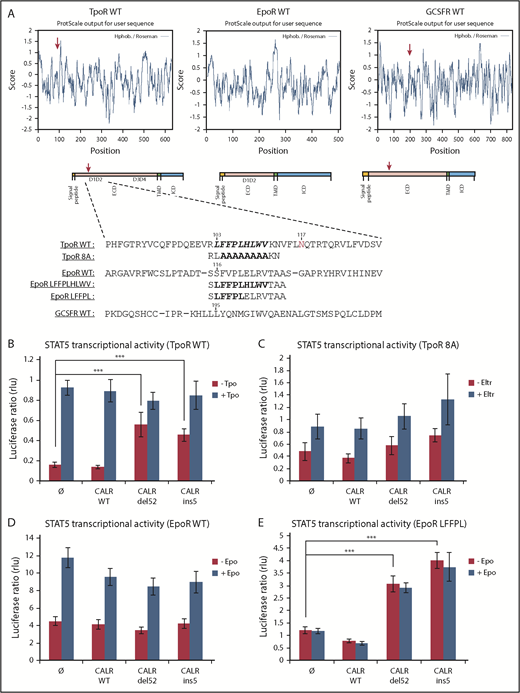
The importance of the hydrophobic sequence in D1D2 domain for TpoR activation. (A) In silico analysis showing that the hydrophobic sequence in TpoR, EpoR, and GCSFR was performed with ExPASy ProtScale by using the scale Hphob. / Roseman. The presence of a hydrophobic patch in the extracellular domain is observed in TpoR and GCSFR but not EpoR, as shown in the graphs. Below the graphs are schematic representations of receptor domains comprising a hydrophobic signal peptide, ECD, hydrophobic transmembrane domain (TMD), and ICD. The red arrows indicate the hydrophobic cluster in ECD corresponding to the maximal hydrophobic peak in the TpoR and GCSFR sequences. Eventually, sequence alignment of the region surrounding hydrophobic cluster (bold residues) was performed with CLUSTAL Ω (1.2.1) multiple sequence alignment tool. Sequence of mutants TpoR 8A and EpoR LFFPL were also indicated. STAT5 transcriptional activity of CALR mutants when coexpressed with TpoR WT (B), TpoR 8A (C), EpoR WT (D), and the minimally changed EpoR LFFPL (E). Values shown represent the average of 3 independent experiments each done with 3 biological repeats ± standard error of the mean. ***P < .001. Statistical analysis (jmp pro12) was performed by using the nonparametric multiple comparisons Steel test with a control group (±1 µg/ml Eltrombopag; ±10 U/ml Epo; ±10 ng/ml Tpo).
The importance of the hydrophobic sequence in D1D2 domain for TpoR activation. (A) In silico analysis showing that the hydrophobic sequence in TpoR, EpoR, and GCSFR was performed with ExPASy ProtScale by using the scale Hphob. / Roseman. The presence of a hydrophobic patch in the extracellular domain is observed in TpoR and GCSFR but not EpoR, as shown in the graphs. Below the graphs are schematic representations of receptor domains comprising a hydrophobic signal peptide, ECD, hydrophobic transmembrane domain (TMD), and ICD. The red arrows indicate the hydrophobic cluster in ECD corresponding to the maximal hydrophobic peak in the TpoR and GCSFR sequences. Eventually, sequence alignment of the region surrounding hydrophobic cluster (bold residues) was performed with CLUSTAL Ω (1.2.1) multiple sequence alignment tool. Sequence of mutants TpoR 8A and EpoR LFFPL were also indicated. STAT5 transcriptional activity of CALR mutants when coexpressed with TpoR WT (B), TpoR 8A (C), EpoR WT (D), and the minimally changed EpoR LFFPL (E). Values shown represent the average of 3 independent experiments each done with 3 biological repeats ± standard error of the mean. ***P < .001. Statistical analysis (jmp pro12) was performed by using the nonparametric multiple comparisons Steel test with a control group (±1 µg/ml Eltrombopag; ±10 U/ml Epo; ±10 ng/ml Tpo).
To explore the relevance of this region for mutant CALR effects, we replaced the 8 hydrophobic residues (FFPLHLWV) in TpoR (from 104 to 111) with alanines, resulting in construct TpoR 8A and tested activation by CALR mutants. We also transplanted this hydrophobic patch into the EpoR at the homologous position. In contrast to the TpoR WT, we found that CALR mutants cannot activate STAT5 via TpoR 8A, as assessed by the STAT transcriptional assay (Figure 5A,C).
Surprisingly, TpoR 8A still interacted with mutant CALR (Figure 2A). Thus, this hydrophobic region might stabilize an active dimeric interface for TpoR, rather than mediating the interaction with the mutant CALR.
Transplanting the full TpoR hydrophobic motif (LFFPLHLWV) to EpoR restored activation by CALR mutants in STAT5 transcriptional assays in transiently transfected γ2A cells (supplemental Figure 6C). We also created a more minimally mutated EpoR mutant (EpoR LFFPL), which as EpoR LFFPLHLWV was activated by CALR mutants (Figure 5E), unlike EpoR WT3 (Figure 5D). Surprisingly, the EpoR LFFPL mutant was more active than the EpoR LFFPLHLWV when coexpressed with CALR mutants (del52 and ins5). Thus, a minimal hydrophobic threshold needs to be passed for CALR mutants to activate EpoR, at least in overexpression transient assays.
We next assessed the ability of CALR mutants to induce autonomous growth of Ba/F3 cells by CTG assay. As expected, TpoR 8A was not able to mediate autonomous growth (supplemental Figure 7B). Interestingly, EpoR LFFPL failed to induce autonomous growth of Ba/F3 cells in the presence of CALR mutants (supplemental Figure 7D). Thus, EpoR LFFPL exhibits only partial activation, reminiscent of G-CSFR.3
To confirm this partial activation, we next transduced CALR mutants along with TpoR and EpoR variants in primary murine Mpl−/− (TpoR knockout) bone marrow cells. Only TpoR, and not TpoR 8A, could be activated by CALR mutants to yield megakaryocytic colonies in the absence of Tpo (supplemental Figure 7E-F). Conversely, the EpoR LFFPL mutant, but not EpoR WT, could support cytokine-independent megakaryocytic colonies in the presence of CALR mutants. Thus, in this very sensitive system, the EpoR mutant can be activated by CALR mutants.
CALR mutants promote enhanced stability of TpoR and of folding-deficient mutants but not of EpoR
Given the direct interaction, we assessed whether CALR mutants exerted a stabilization effect on TpoR folding by using the thermal shift assay,30 in which protein misfolding/aggregation induced by increments in temperature is measured according to western blotting of cell lysates. In Table 1 and supplemental Figure 8, we show that: (1) CALR del52 did not change the melting temperature of EpoR in stably transduced Ba/F3 EpoR cells; (2) CALR del52 increased the stability of TpoR WT and TpoR R102P, unlike WT CALR; (3) the stability of TpoR NtQ and of TpoR 8A was not affected by CALR mutant; and (4) the EpoR containing the transplanted hydrophobic sequence was weakly stabilized. We validated our results in primary mouse platelets derived from CRISPR/Cas9-engineered Calr del52 heterozygous knockin mice. Again, TpoR exhibited increased thermal stability in the presence of CALR del52. Thus, the presence of N-glycosylation and the hydrophobic patch confers to TpoR the ability to be stabilized by the CALR mutants, which correlates with CALR mutant–induced activation in sensitive assays.
Full TpoR activation by CALR mutants requires TpoR cell surface localization
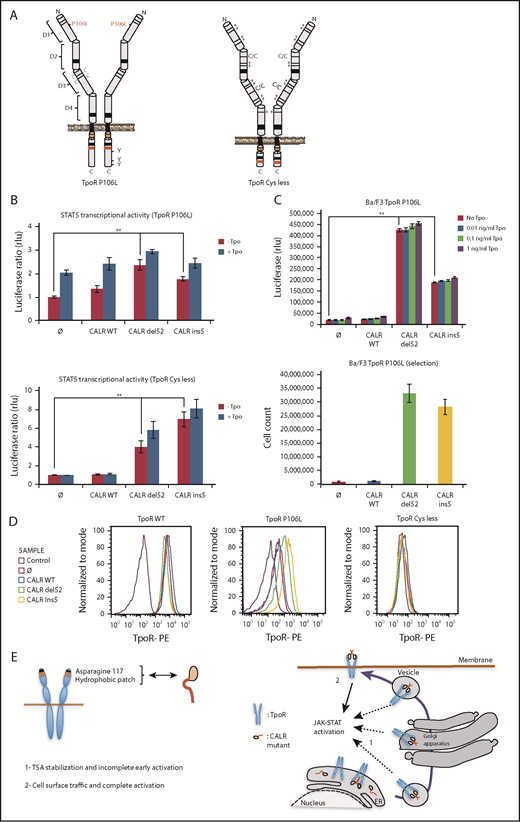
To test this model, we examined a series of traffic-defective TpoRs (Figure 6; supplemental Table 2) and assessed whether cell surface localization was induced by CALR mutants and correlated with full activation. TpoR Cys less was engineered to have 11 Cys residues of 15 in the extracellular domain mutated to Ser, resulting in misfolding and complete retention in the ER (supplemental Figures 2E and 9); TpoR Cys less does not respond to Tpo due to lack of transport to the cell surface. Another traffic-defective pathogenic TpoR mutant, TpoR P106L,31 induces paradoxical thrombocytosis due to insufficient clearing of Tpo by platelets.32 TpoR D1D2 lacks extracellular domains D3D4 and is absent from the cell surface (supplemental Figure 10C). TpoR G509N33 has been engineered to contain a substitution at the end of the transmembrane domain, which activates the murine TpoR; however, in the human TpoR, it is not active and is not localized at the cell surface,33 whereas TpoR K39N (Baltimore mutant) is known to be traffic-defective.34
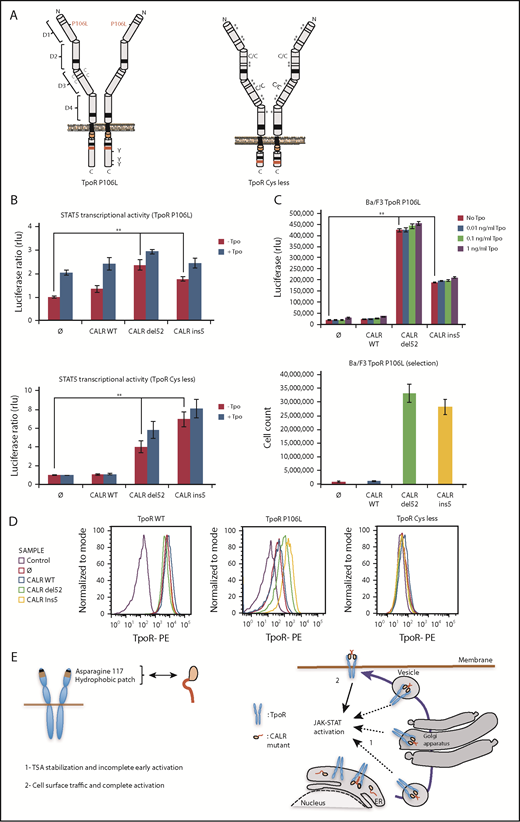
Receptor cell surface localization is required for complete transforming activity of CALR mutants in Ba/F3 cells. (A) Schematic representation of TpoR P106L and TpoR Cys less showing the mutated residues. The activating effect of CALR mutants on a series of mutated receptors measured by luciferase assay (B), short-term proliferation assay (C, upper graph) and long-term proliferation assay (C, under graph), and the cell surface localization of the receptors measured by flow cytometry (D). Values shown in the luciferase assay and short-term proliferation assay represent the average of 3 independent experiments each done with 3 biological repeats ± standard error of the mean, and values shown in the long-term proliferation assay correspond to 3 replicates ± standard error of the mean. Statistical analysis (jmp pro12) was performed by using the nonparametric multiple comparisons Steel test with a control group. (E) Cartoon showing mechanisms of action for CALR mutants requiring binding to TpoR through Asn117 and a hydrophobic patch near the Asn117 site (left cartoon). The oncogenic effect of CALR mutants requires binding and stabilization of the receptor, exit from the ER, and traffic through the secretory pathway. Intracellular TpoR/CALR mutant complexes can induce an incomplete early signal (right cartoon, 1). Only cell surface localization of this complex leads to complete activation and cell transformation (right cartoon, 2).
Receptor cell surface localization is required for complete transforming activity of CALR mutants in Ba/F3 cells. (A) Schematic representation of TpoR P106L and TpoR Cys less showing the mutated residues. The activating effect of CALR mutants on a series of mutated receptors measured by luciferase assay (B), short-term proliferation assay (C, upper graph) and long-term proliferation assay (C, under graph), and the cell surface localization of the receptors measured by flow cytometry (D). Values shown in the luciferase assay and short-term proliferation assay represent the average of 3 independent experiments each done with 3 biological repeats ± standard error of the mean, and values shown in the long-term proliferation assay correspond to 3 replicates ± standard error of the mean. Statistical analysis (jmp pro12) was performed by using the nonparametric multiple comparisons Steel test with a control group. (E) Cartoon showing mechanisms of action for CALR mutants requiring binding to TpoR through Asn117 and a hydrophobic patch near the Asn117 site (left cartoon). The oncogenic effect of CALR mutants requires binding and stabilization of the receptor, exit from the ER, and traffic through the secretory pathway. Intracellular TpoR/CALR mutant complexes can induce an incomplete early signal (right cartoon, 1). Only cell surface localization of this complex leads to complete activation and cell transformation (right cartoon, 2).
All traffic-defective mutants, such as TpoR R102P, could be activated by mutant CALR del52 in γ2A cells (Figure 6). The cell surface localization of TpoR P106L and G509N was significantly enhanced by CALR mutants (supplemental Figure 9E), whereas TpoR K39N was localized at the surface without and slightly enhanced with CALR mutants (supplemental Figure 10F). In contrast, mutant CALRs could not rescue traffic of TpoR Cys less and D1D2 (supplemental Figures 2E and 10C). TpoR P106L, G509N, and K39N were able to support autonomous growth in Ba/F3 cells induced by CALR mutants, with long-term growth after selection, as with TpoR R102P. TpoR Cys less and D1D2 failed to support short- and long-term autonomous growth (supplemental Figures 9A and 10B). Thus, full activation by CALR mutants requires cell surface localization of TpoR.
Discussion
The present study clarifies how a mutant chaperone (CALR) specifically activates a cytokine receptor, the TpoR, and also reports that TpoR mutants which are defective in traffic to the cell surface are rescued by such CALR mutants. The switch from a chaperone to a TpoR activating protein requires acquisition by CALR of a new sequence at the C terminus via the highly prevalent +1 frameshift in MPNs, which leads to a loss of the ER-retention KDEL sequence and significant transport via the secretory pathway to the surface. This scenario also endows the mutant CALRs with rogue chaperone activity, taking with it TpoR proteins that would fail quality control, such as TpoR R102P, or immature TpoR chains to the cell surface. In contrast to WT CALR that needs to detach from immature N-glycosylated proteins for those client proteins to progress through the secretory pathway,35 CALR mutants form stable complexes with TpoR that maintain immature N-glycosylation at Asn117. These complexes are then transported through the secretory pathway. Of interest, CALR mutants were shown to interact with MHC-I carrying immature N-glycosylation containing 9 mannoses and 1-glucose.15 Our mass spectrometry data clearly validate the presence of 9 mannoses at TpoR Asn117 (supplemental Table 1). It remains to be determined whether the CALR mutants also require the 1-glucose to maintain interaction with TpoR, the terminal 1- glucose being required for the interactions of the CALR WT with N-glycosylated proteins.36-38 Further studies are required to assess whether the positively charged C terminus of the CALR mutants may alter lectin domain binding to N-linked sugars. The interaction with TpoR requires W319 of the CALR C-terminal domain, which is close to residues D135 and Y109.20,39
Of interest, CALR mutants were recently shown to be dimers/homomultimers.40 The interaction with TpoR is stabilizing an active dimer of TpoR cytosolic domains, as shown by the Nano-BiT experiment (supplemental Figure 12). As a consequence, the signaling of TpoR is activated, reflected by phosphorylation of cytosolic Y626, which is the main docking phosphorylation site for downstream signaling. Interestingly, several dimeric orientations of TpoR are compatible with signaling,11,41 unlike EpoR,42 possibly explaining the sensitivity of TpoR to activation.
We uncovered a rogue chaperone activity for CALR mutants on TpoR and on several of its pathogenic mutants (R102P, P106L, G509N, and K39N) in which stimulation of traffic to the cell surface correlated with full-transforming activity. We identified receptors in which CALR mutant fails to promote cell surface localization; for those receptors, only partial activation was detected. One explanation could be a lower capacity of those extracellular mutated receptors to bind dimers/oligomers of CALR mutant proteins.40 Another would be that signaling is prolonged by endosome localization post-internalization, and that cell surface localization is a prerequisite for endosomal localization.
Our data showing cell surface TpoR-CALR mutant complexes and their key role in full activation might pave the way for the development of new therapeutic strategies to target exposed CALR mutants in ET and PMF, by producing antibodies that specifically target the CALR mutant; cell surface TpoR activation would thereby be prevented. However, CAMT is frequently severe and lethal in the absence of bone marrow transplantation. The fact that CALR mutants rescued the traffic to the cell surface and function of TpoR R102P might open new therapeutic perspectives in CAMT with testing modified versions of CALR mutants that would be retro-translocated to the ER or by transient gene therapy approaches.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Lidvine Genet for expert technical support, Nicolas Dauguet for flow cytometry assistance, and Marito Araki for sharing Flag-tagged CALR retroviral constructs. A.R. was supported by de Duve Institute Morange Funds postdoctoral fellowship and by a FRS-FNRS fellowship. T.B. was supported by a Télévie fellowship. S.N.C. is Honorary Research Director at FRS-FNRS Belgium.
Funding to S.N.C. is acknowledged from Ludwig Institute for Cancer Research, Fondation contre le cancer, Salus Sanguinis and Fondation “Les avions de Sébastien,” projects Action de recherché concertée 16/21-073 and Walloon Excellence in Life Sciences and Biotechnology F 44/8/5-MCF/UIG–10955. Funding of I.P., C.M., and W.V. is acknowledged from Ligue Nationale Contre le Cancer (“Equipe labellisée 2016”) (H.R.); Institut National du Cancer (INCA-PLBIO-2015) (I.P.), and Institut National de la Santé et de la Recherche Médicale (Inserm).
Authorship
Contribution: C.P., I.C., A.R., and T.B. planned paper strategy, performed experiments, interpreted data, and wrote the paper; G.V., E.L., J.-P.D., Y.O.-A., C. Mouton, and M.M.S. planned specific experiments and interpreted data; D.C. expressed and purified proteins in insect cells and interpreted data concerning complex formation and N-glycosylation; D.V. performed mass spectrometry analysis; C. Marty, R.-I.A., I.P., and W.V. provided experimental data on traffic of receptors in hematopoietic cells; A.R. planned, performed, and interpreted experiments related to thermal stability and confocal immunofluorescence; H.N., E.H., E.X., and R.K. produced specific antibodies against the mutated segment of CALR and provided data related to cell surface complex formation between CALR mutants and TpoR; and S.N.C. planned research, interpreted data, and wrote the paper.
Conflict-of-interest disclosure: R.K. and S.N.C. are cofounders of MyeloPro Research and Diagnostics GmbH, Vienna, Austria. The remaining authors declare no competing financial interests.
Correspondence: Stefan N. Constantinescu, Ludwig Institute for Cancer Research, Université catholique de Louvain and de Duve Institute, Ave Hippocrate 74 UCL 74-4, 1200 Brussels, Belgium; e-mail: stefan.constantinescu@bru.licr.org.

![Figure 2. TpoR and CALR del52 interact directly to form stable complexes. (A) Coimmunoprecipitation analysis showed that TpoR and CALR del52 interaction is dependent on N-linker sugars of TpoR, especially N117-sugars and on the lectin binding domain of CALR. (B) Oligomerization analysis of CALR and TpoR by size-exclusion chromatography. Expression vectors coding for CALR del52 or extracellular domain (ECD) of TpoR and containing a metallothionein promoter were stably transfected (calcium phosphate) in S2 Drosophila cells with G418-containing medium. Expression of the proteins of interest was induced by copper treatment, and secreted forms of TpoR and CALR del52 were further collected in the supernatant. His-tagged ECD-TpoR was enriched by using a Nickel column. Finally, molecular-size distribution of CALR del52 and TpoR was measured by size-exclusion chromatography and fractions analyzed on MOPS 4% to 12% gel in the presence of DTT. (C) Glycosylation analysis with Endoglycosidase H and Peptide N-glycosidase F (N-glyc F or PNGase F) of the indicated fractions obtained from size-exclusion chromatography. (D) Cartoon representation of our Nano-BRET set-up. Because CALR del52 (del52) mutant interacts longer and in close proximity (<10 nm) to TpoR compared with the WT CALR, it enables BRET from the Nano-luciferase fused to the N-terminus of TpoR (NL) to the 618-ligand fluorophore (depicted by a red star) bound to the HaloTag (H) fused to the C-terminal tail of CALR. The BRET is symbolized by a shift in wavelength (460 nm [blue] to 618 nm [red]) of the bioluminescence emanating from the Nano-luciferase. (E) BRET between Nano-luciferase fused cytokine receptors and CALR fused to HaloTag using the Nano-BRET system. There is a strong energy transfer between TpoR and CALR del52 but not between EpoR and CALR del52. Black bars show standard deviation. ****P < .0001, unpaired nonparametric 2-tailed Student t test, 4 independent experiments each with 3 biological replicates.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/25/10.1182_blood-2018-09-874578/3/m_blood874578f2.png?Expires=1769151581&Signature=OUqNvHarlOh1d0IqrnQCO-xkjPUDEqDhYHnF6yvLQ~-ISC4qB-A0jM2FI~P2k0mzxBfNzvUdFkiHgqcpH59bIDro4n7UA~wYIU0APUcIsxYHu8lOFfb9ZR1Paru4tOcW8mZbs04F7YhfH31dnV5pfvOxNxpM8Izk5RQl~sHKOc~D~yv05nSDAcfaIKEWZeaNx79uTos~2tr4BqQxQN5O7pEoV2Gn-UFcv5JjaHWAaZ5P0TRcuT3B2bbDACF2QPDDm8RArfObO5U4WdlILt-TSZI9niWE2ZwXn4-y4Jw89pMtyYj2BBPnQtIftANFVG5Lwo4KG6xUqys-yhsh6Btdkg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)