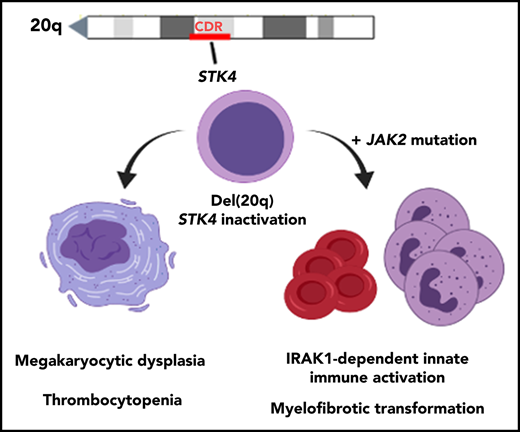
Key Points
Hippo kinase STK4 is downregulated in del(20q) MDS and MPN patients, and inactivation in mice recapitulates clinical disease features.
Heterozygous Hippo kinase loss cooperates with JAK2-V617F to promote myelofibrosis through IRAK1-mediated innate immune activation.

Abstract
Heterozygous deletions within chromosome 20q, or del(20q), are frequent cytogenetic abnormalities detected in hematologic malignancies. To date, identification of genes in the del(20q) common deleted region that contribute to disease development have remained elusive. Through assessment of patient gene expression, we have identified STK4 (encoding Hippo kinase MST1) as a 20q gene that is downregulated below haploinsufficient amounts in myelodysplastic syndrome (MDS) and myeloproliferative neoplasm (MPN). Hematopoietic-specific gene inactivation in mice revealed Hippo kinase loss to induce splenomegaly, thrombocytopenia, megakaryocytic dysplasia, and a propensity for chronic granulocytosis; phenotypes that closely resemble those observed in patients harboring del(20q). In a JAK2-V617F model, heterozygous Hippo kinase inactivation led to accelerated development of lethal myelofibrosis, recapitulating adverse MPN disease progression and revealing a novel genetic interaction between these 2 molecular events. Quantitative serum protein profiling showed that myelofibrotic transformation in mice was associated with cooperative effects of JAK2-V617F and Hippo kinase inactivation on innate immune-associated proinflammatory cytokine production, including IL-1β and IL-6. Mechanistically, MST1 interacted with IRAK1, and shRNA-mediated knockdown was sufficient to increase IRAK1-dependent innate immune activation of NF-κB in human myeloid cells. Consistent with this, treatment with a small molecule IRAK1/4 inhibitor rescued the aberrantly elevated IL-1β production in the JAK2-V617F MPN model. This study identified Hippo kinase MST1 (STK4) as having a central role in the biology of del(20q)-associated hematologic malignancies and revealed a novel molecular basis of adverse MPN progression that may be therapeutically exploitable via IRAK1 inhibition.
Introduction
Recurring chromosome abnormalities, such as large-scale deletion mutations, are frequent events in cancer and are especially prevalent in hematologic neoplasms.1 Modeling loss of function of critical genes affected by large deletions has significantly enhanced our understanding of the associated disease and revealed novel therapeutic approaches in myeloid malignancy.2-7 Somatic heterozygous deletions on chromosome 20q are detected in several hematopoietic malignancies, including myelodysplastic syndrome (MDS), classic myeloproliferative neoplasm (MPN), MDS/MPN overlap disorders, and acute leukemias.8-12 del(20q) is especially prevalent in MPN patients (∼10%-15%), where it is the most commonly detected cytogenetic abnormality associated with primary myelofibrosis (PMF) and post–polycythemia vera myelofibrosis (MF).13 These findings suggest that there are unique tumor-suppressor genes located in this region that, upon loss, contribute to diverse del(20q)-associated hematopoietic phenotypes.
Several studies have mapped a ∼3-Mb minimal common deleted region (CDR) spanning chromosome 20q12-20q13.12.14-16 There is no current evidence of homozygous gene inactivation in this region through mutation.17 To date, 2 transcriptional regulators, L3MBTL1 and MYBL2, are primarily implicated in the pathogenesis of del(20q)-associated malignancies. Knockdown of L3MBTL1 causes an erythroid differentiation bias in human CD34+ cells.18 MYBL2 is a MYB-related transcription factor that is downregulated in MDS patients, regardless of 20q status, and inactivation promotes MDS development in aged mice.19,20 Genetic inactivation of the 20q gene PTPN1, a nonreceptor tyrosine phosphatase that negatively regulates JAK/STAT signaling, was recently shown to contribute to MPN in mice.21 There remains a significant need to identify genes within this region that mediate the specific clinical features associated with del(20q) malignancy.
Located within the del(20q) CDR is STK4, encoding MST1, 1 of 2 mammalian orthologs of the evolutionarily conserved Hippo kinase. In the canonical signaling pathway, Hippo kinases MST1 and MST2 (STK4 and STK3) phosphorylate and activate a downstream kinase complex, which ultimately results in cytoplasmic sequestration and degradation of the oncoproteins YAP and TAZ.22 Involvement of the canonical Hippo pathway in hematopoietic cancers remains controversial,23 and enforced expression of a constitutively active YAP mutant does not affect normal hematopoietic stem cell (HSC) biology or promote malignancy.24 Instead, MST1 plays critical roles in the biology of hematopoietic cells via noncanonical mechanisms.25 In T lymphocytes, MST1 has an essential role in the regulation of thymic egress, adhesion, and apoptosis.26-29 MST1-deficient phagocytes show defects in migration and bactericidal activity,30,31 and MST1-deficient dendritic cells display altered cytokine secretion with detrimental effects on immune function.32,33 These hematopoietic-specific functions have significant implications in human disease, as rare individuals harboring germline homozygous inactivating mutations in STK4 have been identified with clinical presentation as a primary immunodeficiency characterized by susceptibility to chronic infection and frequent autoimmune manifestations.34-36
Here, we have identified STK4 as a subhaploinsufficient tumor-suppressor gene in MDS and MPN. Our data demonstrate that Hippo kinase loss in hematopoietic cells induces several phenotypes in mice that resemble specific clinical features associated with del(20q) malignancy. Importantly, using a JAK2-V617F model of MPN, we identified a novel genetic interaction with heterozygous Hippo kinase inactivation that led to accelerated disease progression to MF, which was caused by cooperative effects on chronic activation of innate immune signaling and NF-κB.
Materials and methods
Please see supplemental Methods (available on the Blood Web site) for full detailed descriptions of all methods.
Animals
All animal protocols were approved by the UCSD Institutional Animal Care and Use Committee. Mice were housed in standard conditions with up to 5 mice per cage, fed standard chow, and monitored in accordance with institutional guidelines. All relevant mouse strains were acquired from The Jackson Labs.
MDS gene expression analysis
For gene expression analysis in CD34+ bone marrow cells comparing del(20q) MDS patients with healthy control individuals, we used a previously published MDS patient gene expression data set (Gene Expression Omnibus accession number GSE58831). Individual Affymetrix CEL files from 6 del(20q) MDS patients (MDS097, MDS113, MDS129, MDS158, MDS187, and MDS202) and 10 normal controls (NBM01-NBM10) were downloaded from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) repository. Array normalization was performed using the robust multiarray average method. Differential gene expression analysis was performed using the limma R package from Bioconductor, with adjusted P-values calculated by the standard Benjamini-Hochberg (false discovery rate) method for multiple comparisons. Differentially expressed gene probes are ranked based on adjusted P-value (supplemental Table 1). For display purposes, a row-normalized gene expression heatmap of the 25 most significantly downregulated genes was generated using the HeatMapImage (version 6.0) module on the GenePattern server (https://genepattern.broadinstitute.org).
Peripheral blood analysis
Peripheral blood samples (∼100 μL per mouse) were collected in EDTA-coated microvettes (NC9299309; Fisher Scientific) by submandibular venipuncture with 5-mm animal lancets (GR5MM; Braintree Scientific). Analysis of differential blood cell counts and parameters was performed using a Scil Vet abc Plus+ instrument (Henry Schein Animal Health).
IRAK1/4 inhibitor treatments
IRAK1/4 inhibitor was purchased from Tocris Bioscience (5665-50). IRAK1/4 inhibitor was resuspended in dimethyl sulfoxide (DMSO) at a concentration of 10 mM and aliquoted prior to use. For injection into mice, working solutions were prepared by dilution of stock inhibitor solution 1:20 into 0.22-μm filtered phosphate-buffered saline. Working solutions were then heated to 55°C for ∼15 minutes and subjected to rocking at room temperature for 4 to 6 hours to aid in resuspension. Where applicable, an equal volume of 5% DMSO solutions were injected as controls (mock treatment). For in vivo IRAK1/4 inhibitor experiments, 4 mg/kg intraperitoneal injections were performed once every 3 days throughout the 4-week course of treatment. For cell culture experiments, working solutions were prepared via dilution in warmed (37°C) cell culture medium (supplemented RPMI 1640) before they were administered to the cells. THP-1 cells were cultured overnight in medium containing various concentrations of inhibitor or mock control medium (0.1% DMSO), before and during stimulation with lipopolysaccharide (LPS; 100 ng/mL).
Statistical analysis
Statistical analyses were conducted with GraphPad Prism (version 7.0) software. Individual statistical tests used for data analysis are indicated in the figure legends along with the resultant P-values. All data are displayed as means (bar graph, horizontal line, or point) with error bars always representing the standard error of the mean (SEM). All Student t tests were conducted as 2-tailed tests, with P < .05 indicating statistically significant results. In some cases, exact values are displayed in the figure panels. Where applicable, mouse sample sizes for measurement of various blood parameters were determined based on a minimal meaningful effect size of 1 standard deviation from a distribution of healthy wild-type control mice, assuming a power of 0.9 and α = 0.05.
Results
Hippo kinase STK4 is transcriptionally downregulated in del(20q) MDS and MPN patients
To identify the 20q genes that play a role in the biology of del(20q)-associated malignancies, we assessed gene expression data in bone-marrow–derived CD34+ cells from MDS patients compared with those from healthy controls. We used a published dataset that contained 6 patients harboring the 20q deletion.37 Given that not all 20q genes are expressed in hematopoietic cells and that genetic haploinsufficiency does not necessarily alter transcriptional output, we rationalized that genes involved in disease pathogenesis could be identified through consistent downregulation in del(20q) patients. We found that 9 (STK4, SERINC3, DDX27, ARFGEF2, IFT52, ADA, ADNP, RBL1, and RNF114) of the top 25 significantly downregulated genes in del(20q) MDS were located on chromosome 20q (Figure 1A; supplemental Table 1). The top hit located on 20q was STK4, which encodes Hippo kinase MST1. STK4 was downregulated to subhaploinsufficient amounts (mean, 3.6-fold) in the 6 del(20q)-harboring patients compared with the 10 healthy controls. We further assessed the expression of these 20q genes in independent cohorts of MF patients.38,39 This assessment revealed 2 of these genes to be significantly downregulated in patients with MF, regardless of 20q status, one of which was STK4, highlighting its potential importance in MPN progression regardless of 20q mutation (Figure 1B; supplemental Figure 1). As was observed in MDS, STK4 was downregulated to subhaploinsufficient amounts in 3 del(20q) MF patients (Figure 1C). Given the position within the defined 20q minimal CDR16 (Figure 1D), the stark downregulation in multiple patient cohorts, and the emerging roles in regulation of normal hematopoietic cell function and inflammatory signaling,25 STK4 was a high-priority candidate to functionally characterize in the context of hematological malignancy.
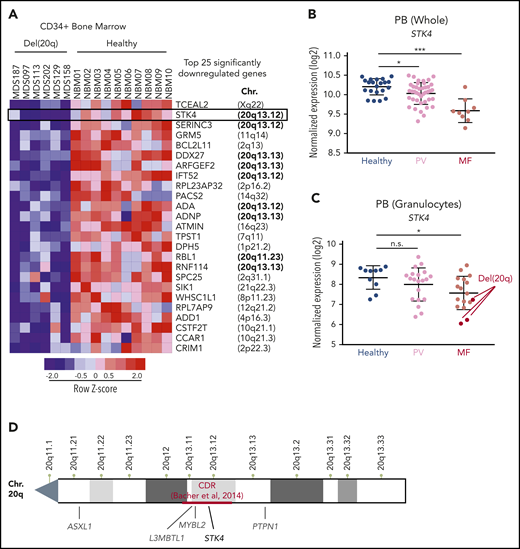
Hippo kinase STK4 is located in the 20q CDR and downregulated in MDS and MPN patients with del(20q). (A) Row-normalized heatmap of gene expression in CD34+ bone marrow cells from 6 del(20q) MDS patients (left) compared with CD34+ bone marrow cells from 10 healthy controls (right). The top 25 significantly downregulated genes in del(20q) patients, along with their chromosomal position in the genome are depicted. Data are from GEO accession number GSE58831. (B) STK4 gene expression measured by microarray in total peripheral blood from healthy controls (blue) and MPN patients with polycythemia vera (PV, pink) or primary MF (red). Data are from GEO accession number GSE26049. (C) STK4 gene expression measured by microarray in peripheral blood granulocytes from healthy controls (blue) and MPN patients with polycythemia vera (PV, pink) or MF (red). Samples identified as del(20q) are indicated. Data are from GEO accession number GSE54646. (D) An NCBI chromosome ideogram showing the q arm of chromosome 20. Minimal CDR measured by array comparative genomic hybridization in a cohort of 30 MDS patients16 is indicated in red. Positions of genes previously implicated in the pathogenesis of myeloid malignancies are indicated in gray. The position of STK4 is indicated in black. Statistical significance was determined by a 2-tailed Student t test. *P < .05, ***P < .001. n.s., not significant. Data in panel A are from Gerstung et al,37 data in panel B are from Skov et al38 and data in panel C are from Rampal et al.39
Hippo kinase STK4 is located in the 20q CDR and downregulated in MDS and MPN patients with del(20q). (A) Row-normalized heatmap of gene expression in CD34+ bone marrow cells from 6 del(20q) MDS patients (left) compared with CD34+ bone marrow cells from 10 healthy controls (right). The top 25 significantly downregulated genes in del(20q) patients, along with their chromosomal position in the genome are depicted. Data are from GEO accession number GSE58831. (B) STK4 gene expression measured by microarray in total peripheral blood from healthy controls (blue) and MPN patients with polycythemia vera (PV, pink) or primary MF (red). Data are from GEO accession number GSE26049. (C) STK4 gene expression measured by microarray in peripheral blood granulocytes from healthy controls (blue) and MPN patients with polycythemia vera (PV, pink) or MF (red). Samples identified as del(20q) are indicated. Data are from GEO accession number GSE54646. (D) An NCBI chromosome ideogram showing the q arm of chromosome 20. Minimal CDR measured by array comparative genomic hybridization in a cohort of 30 MDS patients16 is indicated in red. Positions of genes previously implicated in the pathogenesis of myeloid malignancies are indicated in gray. The position of STK4 is indicated in black. Statistical significance was determined by a 2-tailed Student t test. *P < .05, ***P < .001. n.s., not significant. Data in panel A are from Gerstung et al,37 data in panel B are from Skov et al38 and data in panel C are from Rampal et al.39
Hematopoietic-specific Hippo kinase inactivation results in lethal bone marrow failure with clinical features of del(20q) MDS
Two homologous mammalian Hippo kinases, MST1 and MST2, are encoded by STK4 and STK3, respectively. Although we did not identify evidence of STK3 downregulation in patient cohorts (supplemental Figure 2A), we found that, consistent with a previous report,25 STK4 is functionally dominant in the hematopoietic system, owing to its much more abundant expression in normal human and murine hematopoiesis (supplemental Figure 2B-C). Therefore, given the lack of available mice with floxed alleles for individual Hippo kinase genes, we modeled hematopoietic-specific loss of function by intercrossing Stk4/Stk3-floxed mice with Vav1-iCre transgenic mice, under the assumption that observed phenotypes would be primarily driven by Stk4 inactivation. Assessment of protein expression for MST1 (Stk4) and MST2 (Stk3), as well as their physiological phosphorylation target MOB1,40,41 confirmed Cre-mediated inactivation in hematopoietic cells (Figure 2A). Homozygous deletion within the hematopoietic system (Stk4−/−Stk3−/−) resulted in fully penetrant, postembryonic lethality caused by bone marrow failure (Figure 2B). This lethality was associated with progressive weight loss after weaning (Figure 2C-D), frequent splenomegaly (Figure 2E; supplemental Figure 3A), and a dramatic reduction in bone marrow cellularity (Figure 2F). Enlarged spleens showed evidence of extramedullary erythropoiesis and accumulation of CD71hi/Ter119+ (stage II) erythroblasts (supplemental Figure 3B). Pancytopenia was readily apparent in the peripheral blood of Stk4−/−Stk3−/− mice (Figure 2G-J). Based on our observations showing greater than twofold downregulation of STK4 expression (subhaploinsufficient) in del(20q) MDS and MPN patients, we also characterized Stk4+/−Stk3−/− littermates. These mice possess slightly less than 50% total Hippo kinase activity in hematopoietic cells and therefore most accurately reflect the physiological conditions observed in patients with subhaploinsufficient expression of STK4. A single copy of Stk4 was sufficient to rescue the weight loss, splenomegaly, and early lethality (Figure 2B-D; supplemental Figure 3A-B). Stk4+/−Stk3−/− mice typically showed thrombocytopenia with a trend toward mild anemia (Figure 2G-J). These findings are similar to the clinical presentation of MDS patients with isolated deletion of chromosome 20q.42,43 The peripheral leukopenia observed in Stk4−/−Stk3−/− mice was related to the reported absence of T lymphocytes from circulation and secondary lymphoid organs26,27 (supplemental Figure 3C-D). B lymphocytes were also drastically reduced in the bone marrow, and relative frequencies of granulocytes were increased in the bone marrow and spleen (supplemental Figure 3C-E). Assessment of immature cell populations revealed accumulation of cells at the Lin−Sca1+cKIT+ (LSK) progenitor stage in the bone marrow of Stk4−/−Stk3−/− mice, whereas phenotypic long-term hematopoietic stem cells (LSK+CD48−CD150+) were depleted (Figure 2K). Consistent with peripheral thrombocytopenia, the number of mature megakaryocytes in histological sections of Stk4−/−Stk3−/− bone marrow was significantly reduced compared with those in littermate controls (Figure 2L). We also frequently detected the presence of dysplastic megakaryocytes with highly irregular nuclear morphology in the bone marrow of Stk4−/−Stk3−/− mice (Figure 2M-N). To summarize, Hippo kinase loss results in megakaryocytic abnormalities that likely contribute to thrombocytopenia, which is a defining feature of del(20q) MDS.
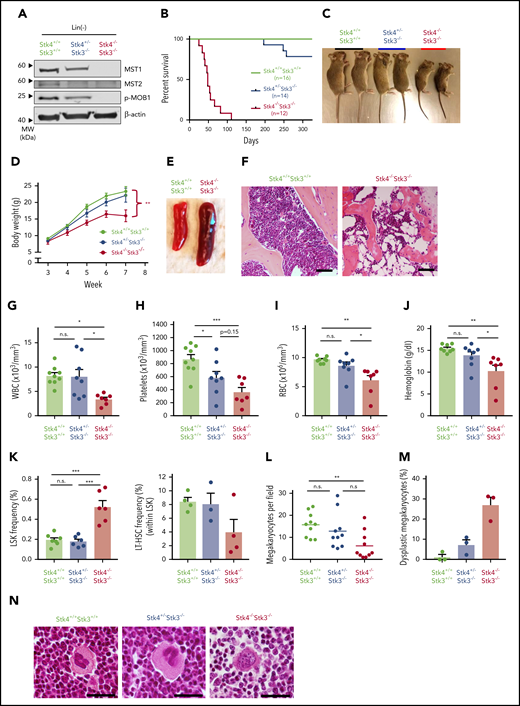
Hematopoietic-specific inactivation of Hippo kinases results in lethal bone marrow failure with features of del(20q) MDS. Genotypes analyzed for this figure include: Stk4f/fStk3f/f;Vav1-Cre+ (Stk4−/−Stk3−/−), Stk4f/+Stk3f/f; Vav1-Cre+ (Stk4+/−Stk3−/−), Stk4f/fStk3f/f;Vav1-Cre−, or Stk4f/+Stk3f/f;Vav1-Cre− (Stk4+/+Stk3+/+). All mice were analyzed in groups with littermates. Unless otherwise indicated, data are derived from mice of 6 to 9 weeks in age. (A) Western blot showing MST1 (Stk4), MST2 (Stk3), phosphorylated MOB1, and β-actin (loading control) proteins in Lin− hematopoietic cells. Bone-marrow– and spleen-derived mononuclear hematopoietic cells from 2 mice per genotype were pooled before Lin− isolation and protein lysate generation. (B) Kaplan-Meier survival plots for mice of indicated genotypes. (C) Representative image of 7-week-old, sex-matched littermates of mice of the indicated genotypes. (D) Weekly weight measurement (in grams) for multiple cohorts of mice between 3 and 7 weeks of age. n = 6 to 10 mice per genotype. Statistical significance was determined at week 7 by 1-way analysis of variance, followed by comparison against Stk4+/+Stk3+/+ values using the post hoc Tukey test. (E) Representative spleen images for 6- to 8-week-old mice of the indicated genetic backgrounds. (F) Representative hematoxylin and eosin (H&E)–stained bone marrow sections for 6- to 8-week-old mice. Scale bar, 200 μm. (G-J) Peripheral blood measurements for white blood cell (WBC) count (G), platelet count (H), red blood cell (RBC) count (I), and hemoglobin (J). Statistical significance was determined by 1-way ANOVA followed by the post hoc Tukey test for multiple comparisons. (K) Bone marrow frequencies of hematopoietic stem/progenitor populations as measured by flow cytometry in mice of the indicated genotypes. (L) The number of mature morphological megakaryocytes per ×20 magnification field in H&E-stained bone marrow sections. Data are derived from 5 representative sections each of 2 mice per genotype. Statistical significance was determined by 1-way ANOVA, followed by the post hoc Tukey test for multiple comparisons. (M) Frequency (%) of dysplastic megakaryocytes detected in representative H&E-stained bone marrow sections from 3 independent mice of the indicated genotypes. (N) Representative images of megakaryocytes observed in H&E-stained bone marrow sections in mice of the indicated genotypes. A representative dysplastic megakaryocyte with irregular nuclear morphology is indicated (right). Scale bar, 60 µm. Data are presented as mean values with error bars representing SEM and data points for individual mice. *P < .05, **P < .01, ***P < .001. LT-HSC, LSK+CD48−CD150+.
Hematopoietic-specific inactivation of Hippo kinases results in lethal bone marrow failure with features of del(20q) MDS. Genotypes analyzed for this figure include: Stk4f/fStk3f/f;Vav1-Cre+ (Stk4−/−Stk3−/−), Stk4f/+Stk3f/f; Vav1-Cre+ (Stk4+/−Stk3−/−), Stk4f/fStk3f/f;Vav1-Cre−, or Stk4f/+Stk3f/f;Vav1-Cre− (Stk4+/+Stk3+/+). All mice were analyzed in groups with littermates. Unless otherwise indicated, data are derived from mice of 6 to 9 weeks in age. (A) Western blot showing MST1 (Stk4), MST2 (Stk3), phosphorylated MOB1, and β-actin (loading control) proteins in Lin− hematopoietic cells. Bone-marrow– and spleen-derived mononuclear hematopoietic cells from 2 mice per genotype were pooled before Lin− isolation and protein lysate generation. (B) Kaplan-Meier survival plots for mice of indicated genotypes. (C) Representative image of 7-week-old, sex-matched littermates of mice of the indicated genotypes. (D) Weekly weight measurement (in grams) for multiple cohorts of mice between 3 and 7 weeks of age. n = 6 to 10 mice per genotype. Statistical significance was determined at week 7 by 1-way analysis of variance, followed by comparison against Stk4+/+Stk3+/+ values using the post hoc Tukey test. (E) Representative spleen images for 6- to 8-week-old mice of the indicated genetic backgrounds. (F) Representative hematoxylin and eosin (H&E)–stained bone marrow sections for 6- to 8-week-old mice. Scale bar, 200 μm. (G-J) Peripheral blood measurements for white blood cell (WBC) count (G), platelet count (H), red blood cell (RBC) count (I), and hemoglobin (J). Statistical significance was determined by 1-way ANOVA followed by the post hoc Tukey test for multiple comparisons. (K) Bone marrow frequencies of hematopoietic stem/progenitor populations as measured by flow cytometry in mice of the indicated genotypes. (L) The number of mature morphological megakaryocytes per ×20 magnification field in H&E-stained bone marrow sections. Data are derived from 5 representative sections each of 2 mice per genotype. Statistical significance was determined by 1-way ANOVA, followed by the post hoc Tukey test for multiple comparisons. (M) Frequency (%) of dysplastic megakaryocytes detected in representative H&E-stained bone marrow sections from 3 independent mice of the indicated genotypes. (N) Representative images of megakaryocytes observed in H&E-stained bone marrow sections in mice of the indicated genotypes. A representative dysplastic megakaryocyte with irregular nuclear morphology is indicated (right). Scale bar, 60 µm. Data are presented as mean values with error bars representing SEM and data points for individual mice. *P < .05, **P < .01, ***P < .001. LT-HSC, LSK+CD48−CD150+.
Inducible Hippo kinase inactivation recapitulates features of MDS and MPN in adult mice
We also assessed the consequences of inducible Hippo kinase inactivation in adult mice by using the interferon-inducible Mx1-Cre system. To ensure the hematopoietic-intrinsic specificity of the observed phenotypes, we generated bone marrow chimeric mice prior to treatment with polyinosinic:polycytidylic acid (pIpC), to induce gene deletion (Figure 3A). Inducible gene inactivation was confirmed in both homozygous (Stk4Δ/ΔStk3Δ/Δ) and heterozygous (Stk4Δ/+Stk3Δ/+) genotypes relative to Mx1-Cre− littermate controls (Stk4+/+Stk3+/+) in bone marrow Lin−cKIT+ cells (Figure 3B). Peripheral leukopenia, peripheral thrombocytopenia, and splenomegaly were readily apparent after inducible homozygous Hippo kinase deletion (Figure 3C-E). Surprisingly, heterozygous inactivation in the adult hematopoietic system led to a chronic increase in peripheral leukocyte counts (Figure 3C), which was related to elevated numbers of granulocytes and granulocytic skewing in the peripheral blood (Figure 3F-G). Thrombocytopenia in mice was accompanied by a pronounced increase in mean platelet volume (MPV), further highlighting defects in the megakaryocytic lineage (Figure 3H-I). In contrast to Vav1-Cre, only 2 of 14 (∼15%) Stk4Δ/ΔStk3Δ/Δ mice experienced lethal bone marrow failure during the course of long-term observations (up to 1 year after pIpC), and there were no indications of anemia (Figure 3J). This result was most likely related to selection pressure toward nonexcised alleles in the Mx1-Cre model, as Hippo kinase protein and mRNA expression were more abundant in Lin− bone marrow cells 1 year after pIpC-induced gene inactivation (supplemental Figure 4). Regardless, we found consistent evidence supporting both heterozygous and homozygous Hippo kinase inactivation contributing to specific clinical features associated with del(20q) MDS and MPN.
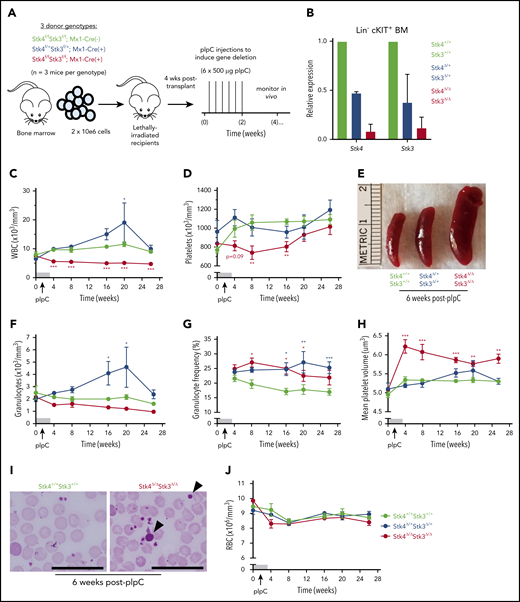
Inducible Hippo kinase deletion in the adult mouse hematopoietic system recapitulates MDS and MPN phenotypes. Recipient mice analyzed in this figure were derived from cells of the following genotypes: Stk4f/fStk3f/f;Mx1-Cre+ (Stk4Δ/ΔStk3Δ/Δ, red), Stk4f/+Stk3f/+;Mx1-Cre+ (Stk4Δ/+Stk3Δ/+, blue), and Stk4f/fStk3f/f;Mx1-Cre− (Stk4+/+Stk3+/+, green); n = 9 mice per genotype. (A) Experimental schematic depicting generation of bone marrow chimeras and pIpC treatment to induce hematopoietic-specific gene deletion for the indicated genotypes. (B) Gene expression for Stk4 and Stk3 measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in flow-sorted Lin− cKIT+ hematopoietic progenitor cells derived from 2 representative mice of the indicated genotypes, 2 weeks after completion of pIpC treatment. Peripheral blood white blood cell (WBC) counts (C); peripheral blood platelet counts (D); representative spleens from mice of the indicated genotypes, 6 weeks after pIpC treatment (E); peripheral blood granulocyte counts (F); peripheral blood granulocyte frequencies (G); and peripheral blood MPV (H). (I) Representative Wright-Giemsa–stained peripheral blood smears from mice of the indicated genotypes, 6 weeks after pIpC treatment. Arrowheads indicate abnormally large platelets. Scale bar, 50 μm. (J) Peripheral blood red blood cell (RBC) counts. Data are representative of 2 independent experiments. Homozygous and heterozygous groups were independently tested for statistical significance against controls by multiple t tests with the Holm-Sidak correction. *P < .05, **P < .01, ***P < .001.
Inducible Hippo kinase deletion in the adult mouse hematopoietic system recapitulates MDS and MPN phenotypes. Recipient mice analyzed in this figure were derived from cells of the following genotypes: Stk4f/fStk3f/f;Mx1-Cre+ (Stk4Δ/ΔStk3Δ/Δ, red), Stk4f/+Stk3f/+;Mx1-Cre+ (Stk4Δ/+Stk3Δ/+, blue), and Stk4f/fStk3f/f;Mx1-Cre− (Stk4+/+Stk3+/+, green); n = 9 mice per genotype. (A) Experimental schematic depicting generation of bone marrow chimeras and pIpC treatment to induce hematopoietic-specific gene deletion for the indicated genotypes. (B) Gene expression for Stk4 and Stk3 measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in flow-sorted Lin− cKIT+ hematopoietic progenitor cells derived from 2 representative mice of the indicated genotypes, 2 weeks after completion of pIpC treatment. Peripheral blood white blood cell (WBC) counts (C); peripheral blood platelet counts (D); representative spleens from mice of the indicated genotypes, 6 weeks after pIpC treatment (E); peripheral blood granulocyte counts (F); peripheral blood granulocyte frequencies (G); and peripheral blood MPV (H). (I) Representative Wright-Giemsa–stained peripheral blood smears from mice of the indicated genotypes, 6 weeks after pIpC treatment. Arrowheads indicate abnormally large platelets. Scale bar, 50 μm. (J) Peripheral blood red blood cell (RBC) counts. Data are representative of 2 independent experiments. Homozygous and heterozygous groups were independently tested for statistical significance against controls by multiple t tests with the Holm-Sidak correction. *P < .05, **P < .01, ***P < .001.
Hippo kinase inactivation promotes macrothrombocytopenia upon aging, even in the absence of malignant clonal HSC expansion
In addition to malignancy, somatic deletions of chromosome 20q are detected at low frequency in otherwise healthy individuals who experience clonal hematopoiesis during aging,44,45 suggesting that loss of 20q genes promotes clonal/premalignant expansion of HSCs. We therefore performed serial competitive bone marrow transplantation assays to determine whether Hippo kinase inactivation contributes to HSC clonal expansion. Hippo kinase deletion resulted in a gene dosage-dependent defect in initial engraftment in the bone marrow of recipient mice. This was indicated by a near complete lack of Stk4−/−Stk3−/− CD45.2 cells in the peripheral blood at 4 weeks after transplantation (Figure 4A-D). We attempted to transplant cells in a noncompetitive manner and confirmed a defect in engraftment potential (supplemental Figure 5). After the initial engraftment, heterozygous inactivation resulted in no detectable competitive advantage over 48 weeks in primary and secondary transplantations (Figure 4A-D). Stk4+/−Stk3+/− HSCs maintained relatively normal output to all mature lineages except for the T-cell (CD3+) lineage, which was consistently reduced (Figure 4D). At 48 weeks, Stk4+/−Stk3+/− CD45.2 donor cell frequencies showed a comparable distribution to control donor cells within the total bone marrow and LSK populations (Figure 4E). No mice developed any overt malignancy; however, upon aging (40-48 weeks), assessment of peripheral blood revealed that several mice that received Stk4+/−Stk3+/− donor cells developed macrothrombocytopenia (Figure 4F). This observation demonstrates that the presence of Stk4+/−Stk3+/− hematopoietic cells, even as a minor clone within the bone marrow, exerts a dominant negative effect on platelet production upon aging.
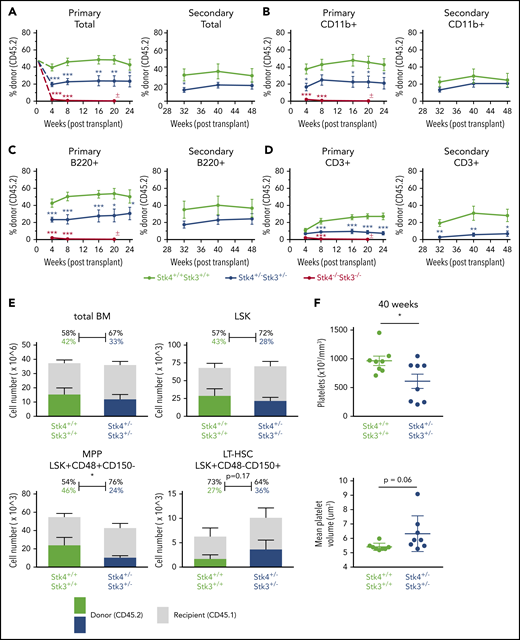
Hippo kinase inactivation promotes macrothrombocytopenia upon aging, even in the absence of malignant clonal HSC expansion. Genotypes analyzed for this figure include Stk4f/fStk3f/f;Vav1-Cre+ (Stk4−/−Stk3−/−, red), Stk4f/+Stk3f/+;Vav1-Cre+ (Stk4+/−Stk3+/−, blue), and Stk4f/fStk3f/f;Vav1-Cre− (Stk4+/+Stk3+/+, green). Primary transplantations: n = 10 (Stk4+/+Stk3+/+), n = 10 (Stk4+/−Stk3+/−), and n = 9 (Stk4−/−Stk3−/−). Secondary transplantations: n = 8 (Stk4+/+Stk3+/+) and n = 8 (Stk4+/−Stk3+/−). (A-D) Donor-derived frequencies (CD45.2%) in the peripheral blood over the course of primary and secondary bone marrow transplantations are shown for total mononuclear cells (A), myeloid-lineage cells (CD11b+) (B), B-lineage cells (B220+) (C), and T-lineage cells (CD3+) (D). (E) Analysis of hematopoietic stem/progenitor cell donor–derived frequencies (CD45.2%) in total bone marrow at the experimental end point (48 weeks). Total cell counts for the indicated populations are shown (left axis). Mean donor- and recipient-derived frequencies are indicated as a percentage. Statistical significance was determined by 2-tailed Student t test. (F) Platelet count and MPV peripheral blood of mice receiving the indicated donor genotypes at 40 weeks after transplant. Data are presented as the mean ± SEM. Statistical significance was determined by 2-tailed Student t test. *P < .05, **P < .01, ***P < .001.
Hippo kinase inactivation promotes macrothrombocytopenia upon aging, even in the absence of malignant clonal HSC expansion. Genotypes analyzed for this figure include Stk4f/fStk3f/f;Vav1-Cre+ (Stk4−/−Stk3−/−, red), Stk4f/+Stk3f/+;Vav1-Cre+ (Stk4+/−Stk3+/−, blue), and Stk4f/fStk3f/f;Vav1-Cre− (Stk4+/+Stk3+/+, green). Primary transplantations: n = 10 (Stk4+/+Stk3+/+), n = 10 (Stk4+/−Stk3+/−), and n = 9 (Stk4−/−Stk3−/−). Secondary transplantations: n = 8 (Stk4+/+Stk3+/+) and n = 8 (Stk4+/−Stk3+/−). (A-D) Donor-derived frequencies (CD45.2%) in the peripheral blood over the course of primary and secondary bone marrow transplantations are shown for total mononuclear cells (A), myeloid-lineage cells (CD11b+) (B), B-lineage cells (B220+) (C), and T-lineage cells (CD3+) (D). (E) Analysis of hematopoietic stem/progenitor cell donor–derived frequencies (CD45.2%) in total bone marrow at the experimental end point (48 weeks). Total cell counts for the indicated populations are shown (left axis). Mean donor- and recipient-derived frequencies are indicated as a percentage. Statistical significance was determined by 2-tailed Student t test. (F) Platelet count and MPV peripheral blood of mice receiving the indicated donor genotypes at 40 weeks after transplant. Data are presented as the mean ± SEM. Statistical significance was determined by 2-tailed Student t test. *P < .05, **P < .01, ***P < .001.
Heterozygous Hippo kinase inactivation cooperates with JAK2-V617F to promote lethal MF
Given the high frequency of del(20q) in MPN, especially in JAK2-mutant polycythemia vera and PMF,46,47 we hypothesized that heterozygous Hippo kinase inactivation cooperates with this driver mutation to modify disease phenotypes and progression. We used a human JAK2-V617F retroviral transduction/transplantation model of MPN that presents as polycythemia vera with a low rate of progression to MF in C57BL/6-recipient mice.48 HSC-enriched bone marrow cells from Stk4+/−Stk3+/− and littermate control (Vav1-Cre−) mice were transduced at equal efficiencies (supplemental Figure 6) with JAK2-V617F (V617F) or empty vector control (Vector), and transplants were monitored by peripheral blood sampling for 36 weeks (Figure 5A). Engraftment efficiencies of V617F-expressing (GFP+) cells were similar between genotypes at 4 weeks after transplantation, and GFP+ cell chimerism in the peripheral blood gradually increased over time, with mean frequencies of approximately 15% at 20 weeks (Figure 5B). Both V617F-expressing genotypes showed a similar degree of polycythemia (Figure 5C). Platelet levels were variable, with no significant differences between groups (Figure 5D). The most pronounced difference detected was elevated granulocytes in the V617F-Stk4+/−Stk3+/− group compared with V617F-Stk4+/+Stk3+/+ (Figure 5E). Importantly, mice receiving V617F-Stk4+/−Stk3+/− cells were significantly more likely to experience adverse MPN progression and to succumb to lethal MF within the course of the 36-week analysis (Figure 5F). Upon necessary euthanasia of moribund mice, massive splenomegaly was apparent (supplemental Figure 7A) and mice were both thrombocytopenic and anemic (supplemental Figure 7B). High-grade reticulin fibrosis was readily apparent in bone marrow sections from moribund mice (supplemental Figure 7C). Enlarged spleens showed increased vascularity with greater reticulin fiber deposition outside of vasculature (supplemental Figure 7D). None of the moribund mice showed detectable myeloid blasts or further evidence of transformation to acute myelogenous leukemia (not shown). These phenotypes were consistent with a diagnosis of post–polycythemia vera MF. We terminated the experiment at an end point of 36 weeks and assessed the relative degree of MPN progression in remaining mice. Higher grade bone marrow reticulin fibrosis and relative bone marrow hypocellularity were apparent within the total population of V617F-Stk4+/−Stk3+/− compared with V617F-Stk4+/+Stk3+/+, whereas both groups showed splenomegaly relative to vector controls (Figure 5G-I; supplemental Figure 7E). These results demonstrate that heterozygous Hippo kinase inactivation contributed to accelerated MPN progression toward myelofibrotic transformation.
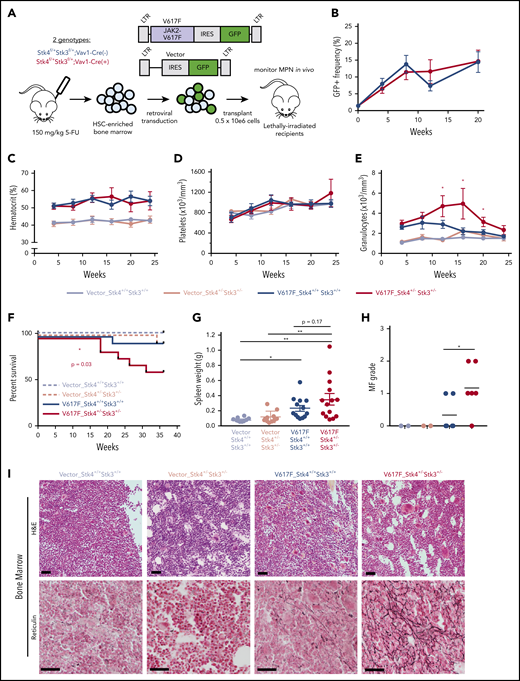
Heterozygous Hippo kinase inactivation cooperates with JAK2-V617F to promote lethal MF in mice. Genotypes used in this model are Stk4f/+Stk3f/+;Vav1-Cre+ (Stk4+/−Stk3+/−) and Stk4f/+Stk3f/+; Vav1-Cre− (Stk4+/+Stk3+/+). (A) Experimental schematic demonstrating the strategy for JAK2-V617F retroviral transduction/transplantation murine model of MPN. Mice were monitored up 36 weeks after transplant. Fluorouracil (5-FU)-treated bone marrow was pooled from 9 mice per genotype per transduction/transplantation experiment. Total mice analyzed in this model were as follows: n = 11 (Vector-Stk4+/+Stk3+/+), n = 10 (Vector-Stk4+/−Stk3+/−), n = 14 (V617F-Stk4+/+Stk3+/+), and n = 15 (V617F-Stk4+/−Stk3+/−). (B) Peripheral blood GFP+ (JAK2-V617F expressing) cell frequencies at the indicated time points measured via flow cytometry: n = 14 (V617F-Stk4+/+Stk3+/+) and n = 15 (V617F-Stk4+/−Stk3+/−). Data are means ± SEM. (C) Peripheral blood hematocrit (%) measurements at the indicated time points: n = 11 (Vector-Stk4+/+Stk3+/+), n = 10 (Vector-Stk4+/−Stk3+/−), n = 7 (V617F-Stk4+/+Stk3+/+), and n = 8 (V617F-Stk4+/−Stk3+/−). (D) As in panel C, but for platelet counts. (E) As in panel C, but for granulocyte counts. (F) Kaplan-Meier survival plot indicating overall survival for the indicated groups over the course of 36 weeks. Statistical significance is determined by log rank (Mantel-Cox) test: n = 11 (vector-Stk4+/+Stk3+/+), n = 10 (vector-Stk4+/−Stk3+/−), n = 14 (V617F-Stk4+/+Stk3+/+), and n = 15 (V617F-Stk4+/−Stk3+/−). (G) Spleen weights for mice of the indicated groups analyzed at the 36-week end point or upon euthanasia for disease progression: n = 11 (vector-Stk4+/+Stk3+/+), n = 10 (vector-Stk4+/−Stk3+/−), n = 13 (V617F-Stk4+/+Stk3+/+), and n = 14 (V617F-Stk4+/−Stk3+/−). Statistical significance was determined by one-way ANOVA followed by the post hoc Tukey test for multiple comparisons. (H) Quantification of MF grades 0-3 in murine bone marrow sections 36 weeks after transplant, based on an established fibrosis grading scale (see methods). Statistical significance between V617F groups was determined by 2-tailed Student t test. (I) Images of representative H&E-stained (top) and reticulin-stained (bottom) sections from the 3 groups indicated. Vector are representative of grade 0 MF, V617F-Stk4+/+Stk3+/+ is representative of grades 0 to 1 MF, and V617F-Stk4+/−Stk3+/− is representative of grades 1 to 2 MF. Scale bar, 100 µm. Data from 2 independent transduction/transplantation experiments with similar group size that demonstrated comparable results are combined. Data are means ± SEM. *P < .05, **P < .01.
Heterozygous Hippo kinase inactivation cooperates with JAK2-V617F to promote lethal MF in mice. Genotypes used in this model are Stk4f/+Stk3f/+;Vav1-Cre+ (Stk4+/−Stk3+/−) and Stk4f/+Stk3f/+; Vav1-Cre− (Stk4+/+Stk3+/+). (A) Experimental schematic demonstrating the strategy for JAK2-V617F retroviral transduction/transplantation murine model of MPN. Mice were monitored up 36 weeks after transplant. Fluorouracil (5-FU)-treated bone marrow was pooled from 9 mice per genotype per transduction/transplantation experiment. Total mice analyzed in this model were as follows: n = 11 (Vector-Stk4+/+Stk3+/+), n = 10 (Vector-Stk4+/−Stk3+/−), n = 14 (V617F-Stk4+/+Stk3+/+), and n = 15 (V617F-Stk4+/−Stk3+/−). (B) Peripheral blood GFP+ (JAK2-V617F expressing) cell frequencies at the indicated time points measured via flow cytometry: n = 14 (V617F-Stk4+/+Stk3+/+) and n = 15 (V617F-Stk4+/−Stk3+/−). Data are means ± SEM. (C) Peripheral blood hematocrit (%) measurements at the indicated time points: n = 11 (Vector-Stk4+/+Stk3+/+), n = 10 (Vector-Stk4+/−Stk3+/−), n = 7 (V617F-Stk4+/+Stk3+/+), and n = 8 (V617F-Stk4+/−Stk3+/−). (D) As in panel C, but for platelet counts. (E) As in panel C, but for granulocyte counts. (F) Kaplan-Meier survival plot indicating overall survival for the indicated groups over the course of 36 weeks. Statistical significance is determined by log rank (Mantel-Cox) test: n = 11 (vector-Stk4+/+Stk3+/+), n = 10 (vector-Stk4+/−Stk3+/−), n = 14 (V617F-Stk4+/+Stk3+/+), and n = 15 (V617F-Stk4+/−Stk3+/−). (G) Spleen weights for mice of the indicated groups analyzed at the 36-week end point or upon euthanasia for disease progression: n = 11 (vector-Stk4+/+Stk3+/+), n = 10 (vector-Stk4+/−Stk3+/−), n = 13 (V617F-Stk4+/+Stk3+/+), and n = 14 (V617F-Stk4+/−Stk3+/−). Statistical significance was determined by one-way ANOVA followed by the post hoc Tukey test for multiple comparisons. (H) Quantification of MF grades 0-3 in murine bone marrow sections 36 weeks after transplant, based on an established fibrosis grading scale (see methods). Statistical significance between V617F groups was determined by 2-tailed Student t test. (I) Images of representative H&E-stained (top) and reticulin-stained (bottom) sections from the 3 groups indicated. Vector are representative of grade 0 MF, V617F-Stk4+/+Stk3+/+ is representative of grades 0 to 1 MF, and V617F-Stk4+/−Stk3+/− is representative of grades 1 to 2 MF. Scale bar, 100 µm. Data from 2 independent transduction/transplantation experiments with similar group size that demonstrated comparable results are combined. Data are means ± SEM. *P < .05, **P < .01.
Stk4 loss cooperates with JAK2-V617F to activate innate immune responses and proinflammatory cytokine production
Altered cytokine production is important for MPN pathogenesis in both human patients and mouse models.49-51 To identify the biological basis through which loss of Hippo kinase signaling contributes to MPN progression, we performed in vivo quantitative cytokine profiling using a multiplex Quantibody array52 (Figure 6A). We reliably measured the abundance of 153 unique proteins in 3 mice each in the 4 experimental groups in our MPN model (Figure 6A; supplemental Table 2). Principal component analysis and hierarchical clustering indicated that JAK2-V617F expression had a greater overall effect on variance in cytokine abundance than did genotype (supplemental Figure 8). The analysis identified a core subset of proteins that were uniquely more or less abundant in serum from the V617F-Stk4+/−Stk3+/− group (Figure 6B). To further explore the unique differences in our MPN model, we compared significantly elevated proteins in each of the V617F-expressing groups against vector-transduced control mice from both genotypes (Figure 6C). A total of 18 proteins were significantly elevated in V617F-expressing mice relative to vector controls. Among these, 7 were common between genotypes, including CXCL1, CXCL9 (MIG), and TGF-β1, whereas another 7 were unique to V617F-Stk4+/−Stk3+/−, including IL-6, IL-1β, IL-15, and MMP-3. We also performed Ingenuity Pathway Analysis, using a ranked list based on z score against a background reference of the entire set of measured proteins, to predict upstream regulators in hematopoietic cells defining the unique cytokine profile associated with the V617F-Stk4+/−Stk3+/− group (Figure 6D). This analysis predicted several activated upstream regulators associated with innate immune signaling, including TLR4/2, MYD88, and IKKα, consistent with significantly elevated cytokines in the double-mutant context being canonical proinflammatory mediators associated with NF-κB activation.
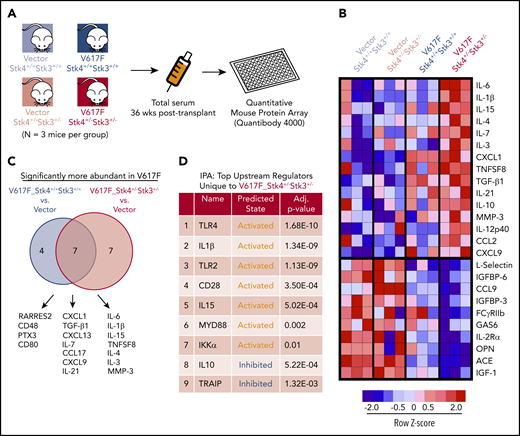
Hippo kinase deletion cooperates with JAK2-V617F to activate innate immune response and proinflammatory cytokine production. (A) Experimental schematic for array-based serum cytokine profiling in JAK2-V617F MPN model in vivo. (B) Heatmap displaying the relative abundance of the most uniquely enriched (top) or depleted (bottom) cytokines in V617F-Stk4+/−Stk3+/− mice (based on mean z scores of 3 independent mice). (C) Venn diagram comparing the significantly more abundant (false discovery rate < 0.1) cytokines in either V617F-Stk4+/+Stk3+/+ group (left) or V617F-Stk4+/−Stk3+/− group (right) when compared against 6 vector-transduced mice. (D) Table depicting the results of Ingenuity Pathway Analysis of top predicted upstream regulators, based on submission of a ranked list of relative cytokine abundance z scores in V617F-Stk4+/−Stk3+/− against a reference background of the entire set of measured cytokines in the array. Upstream regulator name, predicted activation state (activated or inhibited), and adjusted P-values are indicated.
Hippo kinase deletion cooperates with JAK2-V617F to activate innate immune response and proinflammatory cytokine production. (A) Experimental schematic for array-based serum cytokine profiling in JAK2-V617F MPN model in vivo. (B) Heatmap displaying the relative abundance of the most uniquely enriched (top) or depleted (bottom) cytokines in V617F-Stk4+/−Stk3+/− mice (based on mean z scores of 3 independent mice). (C) Venn diagram comparing the significantly more abundant (false discovery rate < 0.1) cytokines in either V617F-Stk4+/+Stk3+/+ group (left) or V617F-Stk4+/−Stk3+/− group (right) when compared against 6 vector-transduced mice. (D) Table depicting the results of Ingenuity Pathway Analysis of top predicted upstream regulators, based on submission of a ranked list of relative cytokine abundance z scores in V617F-Stk4+/−Stk3+/− against a reference background of the entire set of measured cytokines in the array. Upstream regulator name, predicted activation state (activated or inhibited), and adjusted P-values are indicated.
MST1 negatively regulates IRAK1/TRAF6-mediated innate immune activation
The in vivo analysis of cytokine profiles in our JAK2-V617F model led us to hypothesize that del(20q)-associated loss of STK4 contributes to hematologic malignancy through chronic innate immune activation. To explore the relevance of MST1-mediated negative regulation of innate immune signaling through TLR/IL-1R signaling pathways we first confirmed a recently reported interaction between MST1 and IRAK1 (Figure 7A).53 To test whether MST1 negatively regulates NF-κB activation in myeloid malignancy, we performed STK4 knockdown in human myeloid THP-1 cells. STK4 knockdown was sufficient to induce an NF-κB transcriptional response including upregulation of proinflammatory cytokine genes IL6, IL1B, and IL15 that were enriched in the mouse model, and are elevated in MF patients (Figure 7B).54 Consistent with a negative regulatory role through interaction with IRAK1, STK4 knockdown resulted in enhanced phosphorylation of innate immune signaling components IκBα, NF-κB p65, and IRAK1, following LPS stimulation (Figure 7C). Increased activation of innate immune signaling through IRAK1/TRAF6 is well-established to induce MDS phenotypes and bone marrow failure in mice.3,55,56 We therefore performed coimmunoprecipitation experiments with hemoagglutinin-tagged ubiquitin (HA-Ub) to measure TRAF6 E3 ubiquitin ligase activity and self-ubiquitin chain formation, which facilitate downstream assembly and activation of the TAK1-TAB and IκB kinase complexes in innate immune signaling.57 Coexpression of MST1, but not a kinase-dead mutant MST1-K59R, was sufficient to reduce ubiquitin chain formation on TRAF6 (Figure 7D). Coexpression of MST1 also significantly reduced TRAF6-mediated activation of a NF-κB luciferase reporter construct to ∼60% of levels observed in controls, confirming its negative effect on downstream activation of NF-κB (Figure 7E). Taken together, these data demonstrate that MST1 has a biological role in suppression of NF-κB activation in innate immune signaling through IRAK1/TRAF6.
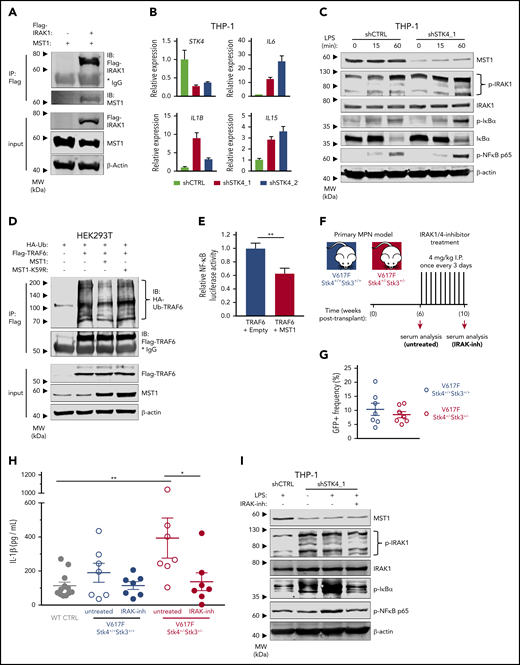
MST1 negatively regulates IRAK1-dependent innate immune activation of NF-κB and proinflammatory cytokine production in MPN. (A) Interaction between IRAK1 and MST1 is assessed in HEK293T cell lysates by immunoblot analysis for MST1 and Flag-IRAK1 after Flag-IP. Input is shown below by immunoblot analysis for Flag-IRAK1, MST1, and β-actin as a loading control. (B) Gene expression measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in THP-1 cells 72 hours after transduction with control short hairpin RNA (shRNA; shCTRL, gray) or 2 independent STK4-targeting shRNAs (shSTK4_1/2, red/pink). Data are means ± SEM for 3 independent biological replicates. (C) Western blot showing the indicated protein and phospho-protein abundance in THP-1 cells 72 hours after transduction with shCTRL or shSTK4_1 vectors and stimulated with LPS (100 ng/mL) for the times indicated. Data are representative of 2 independent experiments. (D) Relative ubiquitination of TRAF6 in HEK293T cells transfected to express HA-ubiquitin (HA-Ub), Flag-TRAF6 and empty vector (−), MST1, or the kinase-dead mutant MST1-K59R, assessed by immunoblot analysis for HA-Ub after Flag-IP (HA-Ub-TRAF6). Input is shown by immunoblot for Flag, MST1, and β-actin as a loading control. Data are representative of 3 independent experiments. (E) Relative luciferase activity of a pNF-κB luciferase reporter construct (4× NF-κB–responsive elements upstream of minimal promoter and firefly luciferase) cotransfected in HEK293T cells with Flag-TRAF6 and either empty vector or MST1 expression vector. pRL-TK (Renilla luciferase reporter) is cotransfected as loading control, and firefly luciferase signal is normalized relative to Renilla. Data are means ± SEM for 3 independent biological replicates. Statistical significance was determined by 2-tailed Student t test. (F) Experimental schematic depicting strategy for generation of JAK2-V617F MPN model in 2 genotypes (Stk4+/+Stk3+/+ and Stk4+/−Stk3+/−) followed by IRAK1/4-inhibitor treatment, 4 mg/kg by intraperitoneal injection once every 3 days for 4 weeks. Wild-type controls, n = 12; V617F-Stk4+/+Stk3+/+, n = 7; and V617F-Stk4+/−Stk3+/−, n = 7. (G) GFP+ cell frequencies in peripheral blood measured by flow cytometry at 6 weeks after transplantation with JAK2-V617F–expressing cells of the indicated genotypes. (H) IL-1β abundance measured by ELISA in peripheral blood serum from mice of the indicated genotypes, before (untreated) or after (IRAK-inh) IRAK1/4-inhibitor treatment, as described in panel F. Statistical significance was determined by 1-way ANOVA followed by the post hoc Tukey test for multiple comparisons. (I) Western blot showing indicated protein and phospho-protein abundance in THP-1 cells 72 hours after transduction with shCTRL or shSTK4_1 vectors and stimulated with LPS (100 ng/mL) for 2 hours where indicated. Cells are pretreated with IRAK1/4-inhibitor (IRAK-inh) overnight at the indicated concentration, and this concentration was maintained during the LPS stimulation period. Data are representative of 2 independent experiments. *P < .05, **P < .01.
MST1 negatively regulates IRAK1-dependent innate immune activation of NF-κB and proinflammatory cytokine production in MPN. (A) Interaction between IRAK1 and MST1 is assessed in HEK293T cell lysates by immunoblot analysis for MST1 and Flag-IRAK1 after Flag-IP. Input is shown below by immunoblot analysis for Flag-IRAK1, MST1, and β-actin as a loading control. (B) Gene expression measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in THP-1 cells 72 hours after transduction with control short hairpin RNA (shRNA; shCTRL, gray) or 2 independent STK4-targeting shRNAs (shSTK4_1/2, red/pink). Data are means ± SEM for 3 independent biological replicates. (C) Western blot showing the indicated protein and phospho-protein abundance in THP-1 cells 72 hours after transduction with shCTRL or shSTK4_1 vectors and stimulated with LPS (100 ng/mL) for the times indicated. Data are representative of 2 independent experiments. (D) Relative ubiquitination of TRAF6 in HEK293T cells transfected to express HA-ubiquitin (HA-Ub), Flag-TRAF6 and empty vector (−), MST1, or the kinase-dead mutant MST1-K59R, assessed by immunoblot analysis for HA-Ub after Flag-IP (HA-Ub-TRAF6). Input is shown by immunoblot for Flag, MST1, and β-actin as a loading control. Data are representative of 3 independent experiments. (E) Relative luciferase activity of a pNF-κB luciferase reporter construct (4× NF-κB–responsive elements upstream of minimal promoter and firefly luciferase) cotransfected in HEK293T cells with Flag-TRAF6 and either empty vector or MST1 expression vector. pRL-TK (Renilla luciferase reporter) is cotransfected as loading control, and firefly luciferase signal is normalized relative to Renilla. Data are means ± SEM for 3 independent biological replicates. Statistical significance was determined by 2-tailed Student t test. (F) Experimental schematic depicting strategy for generation of JAK2-V617F MPN model in 2 genotypes (Stk4+/+Stk3+/+ and Stk4+/−Stk3+/−) followed by IRAK1/4-inhibitor treatment, 4 mg/kg by intraperitoneal injection once every 3 days for 4 weeks. Wild-type controls, n = 12; V617F-Stk4+/+Stk3+/+, n = 7; and V617F-Stk4+/−Stk3+/−, n = 7. (G) GFP+ cell frequencies in peripheral blood measured by flow cytometry at 6 weeks after transplantation with JAK2-V617F–expressing cells of the indicated genotypes. (H) IL-1β abundance measured by ELISA in peripheral blood serum from mice of the indicated genotypes, before (untreated) or after (IRAK-inh) IRAK1/4-inhibitor treatment, as described in panel F. Statistical significance was determined by 1-way ANOVA followed by the post hoc Tukey test for multiple comparisons. (I) Western blot showing indicated protein and phospho-protein abundance in THP-1 cells 72 hours after transduction with shCTRL or shSTK4_1 vectors and stimulated with LPS (100 ng/mL) for 2 hours where indicated. Cells are pretreated with IRAK1/4-inhibitor (IRAK-inh) overnight at the indicated concentration, and this concentration was maintained during the LPS stimulation period. Data are representative of 2 independent experiments. *P < .05, **P < .01.
IRAK1 inhibition rescues the aberrant inflammatory cytokine production associated with Hippo kinase loss in MPN
IRAK1 is a promising therapeutic target in hematologic malignancies.6,58 Given our identification of Hippo kinase loss resulting in enhanced innate immune signaling and proinflammatory cytokine production, we wondered whether these phenotypes would be amenable to therapeutic targeting of IRAK1. We established JAK2-V617F expressing MPNs in both mouse genotypes (Stk4+/+Stk3+/+ and Stk4+/−Stk3+/−) with similar JAK2-V617F (GFP+) cell engraftment at 6 weeks after transplantation and monitored in vivo IL-1β abundance in serum by ELISA (Figure 7F-G). Consistent with our previous observations, serum IL-1β was significantly increased in a Hippo kinase loss–dependent manner in MPN (Figure 7H). At this point, we administered a course of in vivo treatment using the small-molecule IRAK1/4-inhibitor. IRAK1/4-inhibitor treatment was sufficient to reduce serum IL-1β abundance in V617F-Stk4+/−Stk3+/− mice to comparable amounts, as observed in healthy wild-type controls (Figure 7H). IRAK1/4-inhibitor treatment similarly reduced the excessive innate immune signaling activation in LPS-stimulated THP-1 cells with STK4 knockdown (Figure 7I). We also asked whether a similar course of IRAK1/4-inhibitor administration could rescue additional hematopoietic phenotypes associated with somatic homozygous Hippo kinase inactivation (supplemental Figure 9A; Figure 3). IRAK1 inhibition was not sufficient to rescue these hematopoietic defects, including the peripheral leukopenia and macrocytic thrombocytopenia (supplemental Figure 9B-D). Together, these results demonstrate the importance of IRAK1 signaling in mediating the aberrant proinflammatory phenotypes, but not all hematopoietic defects, associated with Hippo kinase inactivation in hematological malignancy.
Discussion
Chronic innate immune activation of NF-κB and dysregulated inflammatory signaling has become appreciated as a driver of MDS and MPN pathogenesis.50,51,59,60 This immune activation can be derived from both cell-extrinsic and -intrinsic mechanisms,59-61 and gene mutations affecting seemingly disparate pathways have a common underlying mechanism of activated inflammatory signaling in hematologic disease.56,62-64 Here, we have identified a novel mechanistic connection between the hematopoietic-intrinsic functions of the 20q gene STK4 (MST1) and malignancy. Our data demonstrate that MST1 is a negative regulator of innate immune signaling through IRAK1, including the ability to suppress TRAF6 autoubiquitination and dampen downstream NF-κB activation in myeloid cells. These data link the recurring del(20q) chromosome abnormality with a commonly disrupted signaling pathway in myeloid malignancy.
The specific molecular events that cooperate with activated JAK2 to promote MPN disease progression to MF are still incompletely understood, and myelofibrotic transformation remains a significant health burden in patients.65,66 Here, we observed clear evidence of accelerated progression from polycythemia vera to lethal MF upon heterozygous inactivation of the Hippo kinases in a JAK2-V617F MPN model. Furthermore, a significant portion of double-mutant mice in our cohorts experienced disease progression that closely resembles what is typically observed in patients, including a prefibrotic phase dominated by peripheral granulocytosis followed by progressive anemia and thrombocytopenia with extramedullary myeloproliferation in the spleen.65 The genetic interaction between STK4 inactivation and JAK2-V617F mutation is one of the most clinically significant findings, as it indicates that cooperation between these lesions could lead to accelerated myelofibrotic transformation in human disease.
Development of PMF, or post–polycythemia vera MF, is a complex process involving an important role for cytokine secretion and cross talk between hematopoietic cell populations and bone marrow stromal cells.67 Our data suggest an important role for hematopoietic-intrinsic activation of innate immune signaling as contributing to this process. Through quantitative serum protein profiling in our mouse model of MPN, we identified significantly elevated proinflammatory cytokines that contributed to remodeling of the bone marrow microenvironment and myelofibrotic progression. Several of these same cytokines, including IL-6, IL-1β, and IL-15 are elevated in the serum of MF patients,54 highlighting the clinical relevance of our study. We further confirmed that the aberrantly elevated IL-1β production in JAK2-V617F and Stk4+/−Stk3+/− double-mutant mice was apparent at a relatively early time point in MPN progression. Importantly, we found that IRAK1/4-inhibitor treatment was sufficient to inhibit this chronic innate immune activation and reduce IL-1β abundance in serum in MPN. These findings demonstrate the importance of IRAK1 activation in mediating the proinflammatory phenotypes associated with Hippo kinase inactivation and are consistent with another study that found that STK4 downregulation in macrophages could accelerate hepatocellular carcinoma development through increased IRAK1-mediated inflammatory signaling.53
Although Hippo kinase deficiency alone did not promote clonal expansion of hematopoietic stem/progenitor populations, we found that it caused specific myelodysplastic phenotypes associated with del(20q). In MDS, del(20q), as a sole abnormality, frequently presents as isolated thrombocytopenia42,43 and, consistent with this, we reproducibly observed megakaryocytic/platelet abnormalities in our conditional knockout mice. There is strong evidence that abnormal megakaryocyte biology leading to reduced platelet production is an inherent feature of del(20q)-associated malignancy. del(20q) is among the most frequent cooperating mutations in MPN overall, yet it is exceedingly rare in patients with essential thrombocythemia,46 and thrombocytopenia (a consistent phenotype in our models) is a significant phenotypic correlation in PMF patients with del(20q).47 In contrast to proinflammatory signaling in the JAK2-V617F model, however, IRAK1 inhibition was not sufficient to rescue the abnormal platelet phenotypes associated with somatic Hippo kinase inactivation, at least during the course of our observations. Additional signaling pathways that are affected upon Hippo kinase loss in hematopoietic cells most likely contribute to these phenotypes; further studies characterizing megakaryocyte-intrinsic gene inactivation may be important to pursue.
In our genetic models, we have characterized the consequences of loss of both homologous Hippo kinases. There is no evidence that STK3 (MST2) is downregulated in MDS or MPN patient cohorts. However, in contrast to most tissues where MST1 and MST2 are able to compensate for one another, MST1 is functionally dominant in the hematopoietic system owing to its higher (10- to 20-fold) mRNA and protein expression. Therefore, the contribution of Stk3 loss in our described phenotypes is minimal; and where applicable, we used Stk3 allele inactivation to provide a very modest further reduction in total Hippo kinase abundance in attempt to more accurately reflect the total activity in patients with subhaploinsufficient STK4 expression, as was observed in del(20q) MDS and MPN.
In summary, our data suggest that loss of Hippo kinase signaling contributes to pathogenesis of myeloid malignancies with chromosome 20q deletions. Heterozygous Hippo kinase inactivation significantly accelerated myelofibrotic transformation in a JAK2-V617F MPN model through effects on innate immune signaling and proinflammatory cytokine production. Further efforts toward therapies aimed at mitigating chronic innate immune activation through IRAK1 may be beneficial in del(20q)-associated MF.
Plasmids, resources, and data generated in the current study are provided as supplemental tables, or will be made available upon request to the corresponding investigator. The published datasets used for analysis during the current study are available in the NCBI GEO repository.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the La Jolla Institute for Allergy & Immunology Flow Cytometry Core staff for assistance with flow sorting.
This work was supported by National Institutes of Health, National Cancer Institute grants R01CA104509, R01CA192924 (D.E.Z.) and F31CA189452 (S.A.S.). UCSD Moores Cancer Center Tissue Technology Shared Resource is supported by a National Cancer Center Support Grant (CCSG P30CA23100).
Authorship
Contribution: S.A.S. and D.-E.Z. conceived of the study and designed the methodology for the experiments; S.A.S., M.Y., K.T.H.L., K.-I.A., and D.T.J. performed research and investigation, mouse tissue collection, and data collection and analysis; S.A.S., T.S., and H.-Y.W. performed formal analysis of histological staining and MF grading; S.A.S. wrote the original draft of the manuscript; R.B., C.J., and K.-L.G. provided essential resources and critical review and editing of the manuscript; and D.-E.Z. supervised the study and manuscript preparation and acquired the funding to support the study.
Conflict-of-interest disclosure: K.L.G. is a cofounder and has an equity interest in Vivace Therapeutics, Inc. The terms of this arrangement have been reviewed and approved by the USCD, in accordance with its conflict-of-interest policies. The remaining authors declare no competing financial interests.
Correspondence: Dong-Er Zhang, Moores Cancer Center, 3855 Health Sciences Dr, UC San Diego, La Jolla, CA 92037; e-mail: dez@ucsd.edu.