

Childhood AML is a heterogeneous hematologic disease with a multitude of subtypes characterized by varying morphology, structural alterations, and recurrent mutations. Such heterogeneity and staggering number of genomic and transcriptional alterations has precluded appropriate risk classification. We investigated the expression of long non-coding RNAs (lncRNA) in childhood AML and explored its potential utility for more precise risk characterization at diagnosis.
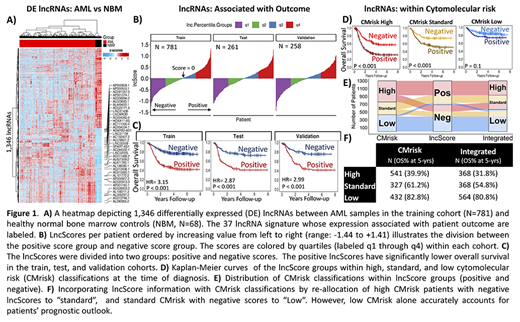
We inquired whether lncRNAs aberrantly expressed in AML compared to healthy normal bone marrows may be utilized for predicting outcome without knowledge of somatic variants, creating a variant-agnostic prognostic indicator. Ribodepleted RNA-sequencing data from normal bone marrows (N=68), as well as diagnostic primary samples (N=1300) from patients with detailed clinical annotations and outcome were included for study. To this end, the study population was divided into training (N=781), test (N=261), and validation (N=258) cohorts using blocked randomization. Upregulated lncRNAs compared to normal marrows in the training set (fold-change > 2, FDR < 0.05, Fig. 1A) were input for a LASSO cox proportional hazards regression which identified a set of 37 lncRNAs whose expression (log2 scale, TMM normalized) associated with patient outcome. The trained model coefficients were applied to the test and validation cohorts to produce a weighted sum of the 37 lncRNAs expression per patient (lncScores, range: -1.44 to +1.41). The distribution of lncScores revealed approximately equal numbers of patients with positive and negative scores in the training, test, and validation cohorts (Fig. 1B).
In the training set, those with positive lncScores had an overall survival (OS) of 42.7% at 5-years from diagnosis compared to 76.3% for those with negative scores (hazard ratio (HR) = 3.15, p < 0.001). Positive lncScores were also associated with adverse event-free survival (EFS, HR = 2.47, p < 0.001). LncScore based outcome analysis in the independent test and validation cohorts showed nearly identical outcome results for OS (HR = 2.86 and 2.99 resp., p < 0.001) and EFS (HR = 2.37 and 2.35, p < 0.001, Fig. 1C) as was seen in the training set, demonstrating stability of the predictive power of the lncScore across independent cohorts. Next, the lncScore's performance was evaluated in relation to cyto/molecular risk groups (CMrisk: high, standard, and low) defined by karyotype, clinically relevant mutations, and cryptic fusions as presented previously (Cooper et al. ASH 2017). A multivariable cox regression model (N = 1300) that included lncScore, CMrisk, and white blood cell count revealed that both lncScore and CMrisk groups remained independent prognostic factors for OS (p < 0.001) and EFS (p ≤ 0.001), suggesting the lncRNA signature may provide additional prognostic information to that defined by CMrisk status. Application of the lncScore to the CMrisk groups demonstrated that 173 of the 541 of patients classified as CMrisk high (32%) would be reallocated to the standard risk arm based on negative lncScores (OS 57.3% vs 31.8%, p < 0.001). Similarly, 40% of patients with CMrisk standard (132/327) would be reclassified as low risk by the lncScore system (OS 73.8% vs 52.4%, p < 0.001). lncScores did not revise risk status in CMrisk low patients. The ribbon plot (Fig. 1E) demonstrates reallocation of patients using the integrated risk stratification system.
Integration of the CMrisk and lncScore demonstrated that in combination the two risk classification systems can provide a more precise assessment of risk status, since both CMrisk high and CMrisk standard cohorts can be further stratified by the lncScore (Fig. 1D). The integrated approach provided a comprehensive risk classifier that incorporates both cytogenetic data, as well as transcriptional evidence, which benefits more than 1 out of 5 patients (23%, 305/1300) with a more accurate determination of risk at the time of diagnosis (Fig. 1F). This study demonstrates development and validation (in two independent cohorts) of a lncRNA based, variant-independent prognostic signature (p < 0.001) that can effectively partition patients into high and low risk groups at diagnosis. It can also significantly enhance the predictive power of conventional cyto/molecular markers for more precise prediction of outcome prior to the start of therapy.
Farrar:Novartis: Research Funding.

