Key Points
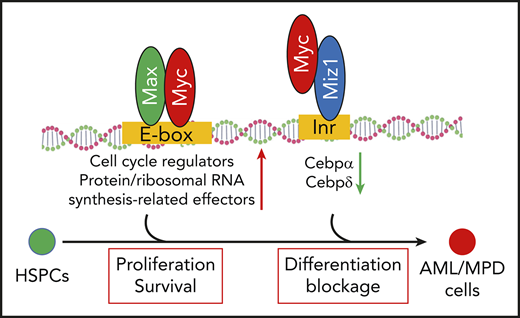
Myc regulates LSC self-renewal through Miz1-mediated transrepressional activity.
Myc plays such a role by repressing the expression of Cebpα and Cebpδ.

Abstract
c-Myc (Myc hereafter) is found to be deregulated and/or amplified in most acute myeloid leukemias (AMLs). Almost all AML cells are dependent upon Myc for their proliferation and survival. Thus, Myc has been proposed as a critical anti-AML target. Myc has Max-mediated transactivational and Myc-interacting zinc finger protein 1 (Miz1)-mediated transrepressional activities. The role of Myc-Max–mediated transactivation in the pathogenesis of AML has been well studied; however, the role of Myc-Miz1–mediated transrepression in AML is still somewhat obscure. Myc protein harboring a V394D mutation (MycV394D) is a mutant form of Myc that lacks transrepressional activity due to a defect in its ability to interact with Miz1. We found that, compared with Myc, the oncogenic function of MycV394D is significantly impaired. The AML/myeloproliferative disorder that develops in mice receiving MycV394D-transduced hematopoietic stem/progenitor cells (HSPCs) is significantly delayed compared with mice receiving Myc-transduced HSPCs. Using a murine MLL-AF9 AML model, we found that AML cells expressing MycV394D (intrinsic Myc deleted) are partially differentiated and show reductions in both colony-forming ability in vitro and leukemogenic capacity in vivo. The reduced frequency of leukemia stem cells (LSCs) among MycV394D-AML cells and their reduced leukemogenic capacity during serial transplantation suggest that Myc-Miz1 interaction is required for the self-renewal of LSCs. In addition, we found that MycV394D-AML cells are more sensitive to chemotherapy than are Myc-AML cells. Mechanistically, we found that Myc represses Miz1-mediated expression of CCAAT/enhancer-binding protein α (Cebpα) and Cebpδ, thus playing an important role in the pathogenesis of AML by maintaining the undifferentiated state and self-renewal capacity of LSCs.
Introduction
Myc protein is encoded by the proto-oncogene Myc, which plays a central role in almost every aspect of the oncogenic process, orchestrating proliferation, apoptosis, differentiation, ribosome biogenesis, protein synthesis, and metabolism.1 Deregulation and amplification of Myc are commonly detected in many types of cancer and are associated with aggressive disease and poor prognosis.2-4 The role of Myc in the development of leukemia has been confirmed by transgenic and knockout animal studies.5-8 Genetic inactivation of Myc represses the proliferation and survival of almost all types of leukemia cells.9,10 Thus, many studies have led to the conclusion that Myc is a critical molecular target for antileukemia therapy.11 Despite significant efforts, targeting Myc to treat leukemia has yet to be successful. We believe that a more detailed understanding of the role of Myc in the pathogenesis of leukemia will allow us to develop more effective and specific anti-Myc chemotherapeutic approaches.
Myc is a pleiotropic transcription factor that functions primarily by regulating the expression of its target genes.12-15 Myc upregulates genes through its transactivational mechanisms. Early studies demonstrated that Myc, in complex with Max, upregulates a group of specific target genes through binding to E-box motifs in the promotors of such genes.16,17 These genes encode positive regulators of the cell cycle, as well as protein and ribosomal RNA synthesis-related effector molecules that are critical for cell proliferation, growth, and differentiation.18 Recent studies suggested that elevated levels of Myc induce the expression of 10% to 15% of all human genes, far more than previously known Myc/Max target genes.14,19,20 These studies suggested that high levels of Myc promote a universal upregulation of transcriptional-activation genes by occupying the core promoters through binding to noncanonical E-box motifs independent of Max.17,21 Myc promotes the expression of such genes in a cell type–specific manner by relieving a transcriptional pause and amplifying the activated transcriptional process.20,22
Myc also represses a large group of target genes. The transrepressive activity of Myc is primarily mediated by its interaction with Myc-interacting zinc finger protein 1 (Miz1).23 Miz1 is a transcription factor that binds to the initiator elements of target genes. Myc represses the expression of Miz1 target genes, which account for 25% to 40% of Myc-repressed genes,24 including Cdk inhibitors (p15Ink4b and p21Cip1) and key transcription factors (CCAAT/enhancer-binding protein α [Cebpα] and Cebpδ).23,25-31 Myc binds to Miz1 by replacing the coactivators P300 and nucleophosmin, and recruiting the DNA methyltransferase Dnmt3a, polycomb-repressive complex, and/or the histone methyltransferase G9a32-34 ; this yields a closed chromatin state and represses many of Miz1’s target genes.
The role of Myc-mediated transactivating activity in the pathogenesis of leukemia has been well studied.2 However, the biological relevance of Myc-induced transrepressive activity in leukemogenesis is not currently well understood. Myc protein harboring a V394D mutation (MycV394D) no longer retains the ability to bind to Miz1 but still has Max-binding ability. As a consequence, MycV394D shows defects in transrepressive activity while preserving all of its other activities.23,35 To study the role of Myc-induced transrepressive activity on leukemia development, we compared the leukemogenic capacity of MycV394D- and Myc-transduced hematopoietic stem/progenitor cells (HSPCs). We also transduced Myc-knockout (Myc−/−) acute myeloid leukemia (AML) cells with MycV394D to determine whether this mutant form of Myc can restore leukemogenic capacity to Myc−/− AML cells.
Materials and methods
Mice
Most of the experiments using HSPCs for transduction and transplantation studies were conducted at Shanghai Normal University, whereas all of the experiments for MLL-AF9 AML cell studies were conducted at Loyola University Chicago. C57BL6/J mice were purchased from either Shanghai Slac Laboratory Animal Co Ltd or The Jackson Laboratory. Rosa26-CreERTMycfx/fx mice were maintained by the Department of Comparative Medicine of Loyola University Chicago in a C57Bl6/J background.9 All mice were housed under a 12-hour light/dark cycle in microisolator cages contained within a laminar flow ventilation system. All procedures were conducted in accordance with the approved guidelines provided by the Laboratory Animal Resource Center of Shanghai Normal University and the National Institutes of Health guidelines for the care and use of laboratory animals for research purposes, and were approved by Loyola University Chicago’s Institutional Animal Care and Use Committee (AU#513380).
Bone marrow HSPC isolation, in vitro culturing, and retroviral gene transduction
c-Kit+ HSPCs were isolated from 8- to 10-week-old C57Bl6/J mice using the EasySep Mouse c-Kit Positive Selection kit (StemCell Technologies). c-Kit+ HSPCs were incubated in stem cell culture medium (RPMI 1640 medium containing 10% fetal blood serum supplemented with 100 ng/mL murine stem cell factor, 50 ng/mL murine interleukin 6, and 20 ng/mL murine interleukin 3 overnight at 37°C in 5% CO2) and 100% humidity to stimulate proliferation. The cells were then transduced with virally expressed genes of interest by spinoculation at 32°C, 2000 rpm for 4 hours. The green fluorescent protein–positive (GFP+) fraction of transduced HSPCs was purified by fluorescence-activated cell sorting (FACS) on day 2 posttransduction and used for in vitro culture studies and in vivo transplantation studies.
In vitro CFC assay and in vivo transplantation of transduced HSPCs
Immediately after purification, the transduced HSPCs were seeded into MethoCult GF M3434 medium for colony-forming capacity (CFC) assay following instructions provided by the manufacturer (StemCell Technologies). Colony-forming units (CFUs) were read on day 7 of culturing and then collected for serial replating. For in vivo transplantation, the transduced HSPCs were transplanted into lethally irradiated (9.5 Gy) recipient mice together with support bone marrow (BM) cells. Each mouse received 1 × 105 transduced HSPCs plus 2 × 105 support BM cells. Mice were monitored for leukemia development. For secondary transplantation, AML or myeloid proliferative disorder (MPD) BM cells were collected from the first transplantation and transplanted into sublethally irradiated (5 Gy) mice (5 × 106 cells transplanted per mouse).
MLL-AF9 AML generation and infection
c-kit+ HSPCs were collected from Rosa26-CreERTMycfx/fx mice and transduced with MLL-AF9–expressing virus. The transduced HSPCs were transplanted into lethally-irradiated recipient mice. After the development of leukemia, the CreERTMycfx/fx AML cells were collected from BM and spleens of the mice and transduced with MSCV-GFP (empty vector), Myc-GFP, or MycV394D-GFP–expressing virus to generate MSCV/CreERTMycfx/fx, Myc/CreERTMycfx/fx, and MycV394D/CreERTMycfx/fx AML cells. After purification by FACS for GFP+ cells, the transduced cells were collected for in vitro culture and in vivo transplantation studies.
Induced deletion of Myc gene in AML cells in vitro and in vivo
4-Hydroxytamoxifen (4-OHT) and tamoxifen (TAM) were purchased from Sigma-Aldrich (St. Louis, MO) and dissolved in 100% ethanol. TAM was dissolved in ethanol, then further dissolved in corn oil (Sigma-Aldrich) in a 9:1 oil-to-ethanol ratio. To induce the deletion of endogenous Myc in in vitro culture, MSCV/CreERTMycfx/fx, Myc/CreERTMycfx/fx, and MycV394D/CreERTMycfx/fx AML cells were treated with 4-OHT at a final concentration of 1 μM. To induce the deletion of endogenous Myc in vivo, MSCV/CreERTMycfx/fx, Myc/CreERTMycfx/fx, or MycV394D/CreERTMycfx/fx AML cells were transplanted into C57Bl6/J mice without preconditioning. Each mouse received 106 cells. Three days posttransplantation, mice from each of these 3 groups were randomly divided into 2 subgroups and treated with either 100 mg/kg per day TAM by intraperitoneal injection for 5 consecutive days, or with an equal volume of corn oil on the same schedule.
Statistical analysis
Data are expressed as means plus or minus standard deviation. Two-way analysis of variance (multiple groups) and the Student t test (2 groups) were performed to determine the statistical significance of differences among and between experimental groups at a significance level of P < .05.
Results
MycV394D overexpression maintains colony-forming HSPCs in a less undifferentiated state than Myc overexpression in in vitro culture
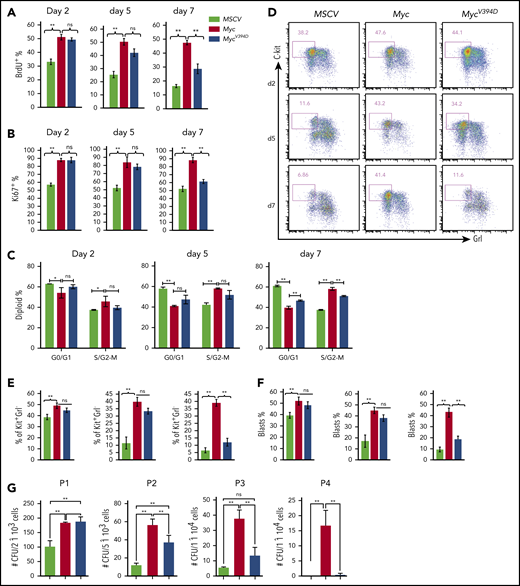
Myc overexpression promotes the proliferation and CFC of HSPCs in in vitro culture.36,37 To study the role of Myc-Miz1–mediated transrepressive activity in Myc-induced proliferation and the CFC of HSPCs, we isolated c-Kit+ HSPCs from mouse BM. The HSPCs were transduced with MSCV-IRES-GFP (MSCV), MSCV-Myc-IRES-GFP (Myc), or MSCV-MycV394D-IRES-GFP (MycV394D). The transduced HSPCs were purified by FACS and seeded into liquid culture for proliferation analysis and seeded in methylcellulose medium for CFC assay (supplemental Figure 1a, available on the Blood Web site). We found that Myc protein levels were comparable between Myc and MycV394D HSPCs (supplemental Figure 1b). Myc-Miz1 interaction in HSPCs was significantly disrupted by the V394D mutation (supplemental Figure 1c). Compared with MSCV HSPCs, the proliferation of Myc and MycV394D HSPCs was significantly increased as determined by 5-bromo-2′-deoxyuridine (BrdU) pulse-labeling assay (Figure 1A; supplemental Figure 2a), Ki67 staining (Figure 1B; supplemental Figure 2b), and cell-cycle analysis (Figure 1C; supplemental Figure 2c). However, there was no difference between Myc and MycV394D HSPCs during the first 5 days of culturing (Figure 1A-C; supplemental Figure 2a-c), this despite a slight increase in apoptosis that was detected in Myc HSPCs due to repressed Bcl2 expression (supplemental Figure 3). Reduced proliferation and cell-cycle arrest were observed by day 7 of culturing for MycV394D HSPCs compared with Myc HSPCs (Figure 1A-C; supplemental Figure 2a-c), observation of which was correlated to increased myeloid differentiation in the transduced HSPCs (Figure 1D-F; supplemental Figure 2d). The Kit+Gr1low/− population comprised undifferentiated myeloid blasts that were more highly proliferative than differentiated Kit−Gr1+ cells for all 3 types of transductions. Significantly higher proliferation of both Kit+Gr1low/− and Kit−Gr1+ cells was observed in Myc and MycV394D transductions when compared with the corresponding types of cells in MSCV transduction (supplemental Figure 2e). During the first 5 days of culture, more Kit+Gr1low/− blasts were maintained in both Myc and MycV394D HSPCs compared with MSCV HSPCs. However, on day 7 of culturing, only Myc HSPCs maintained a higher percentage of Kit+Gr1low/− blasts (Figure 1D-F). Consistent with these observations, we found that the CFCs of Myc and MycV394D HSPCs are significantly increased compared with MSCV HSPCs. Again, there was no difference between Myc and MycV394D HSPCs in CFC during the first round of plating. However, during serial replating, the CFC was significantly reduced for all 3 types of transduced HSPCs, but Myc HSPCs maintained a significantly higher CFC (Figure 1G) and Kit+Gr1− blast percentage (supplemental Figure 4a) compared with MycV394D and MSCV HSPCs. In addition, the colonies in the first round of plating were primarily CFU–granulocyte macrophage (CFU-GM) and burst-forming unit erythroid with a small percentage of CFU–granulocyte, erythroid, macrophage, megakaryocyte (supplemental Figure 4b), whereas the colonies for the second, third, and fourth platings were exclusively CFU-GM (supplemental Figure 4c). There was no significant difference in the percentage of the subcolonies among the 3 types of transductions; however, the size of CFU-GM generated from Myc HSPCs after the second replating was bigger compared with MycV394D and MSCV HSPCs (supplemental Figure 4c). Taken together, it is suggested that Myc overexpression maintains more undifferentiated colony-forming HSPCs compared with MycV394 overexpression.
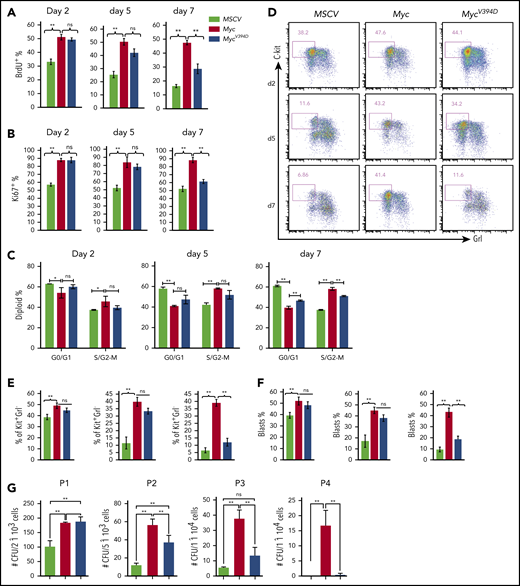
HSPC differentiation is blocked to a greater degree by Myc transduction than by MycV394Dtransduction. c-Kit+ HSPCs were transduced with MSCV-GFP, Myc-GFP, or MycV394D-GFP, respectively. Transduced Kit+ HSPCs were purified by FACS 2 days posttransduction and incubated in HSPC culture medium with medium change every other day (A-F) or seeded into methylcellulose for serial replating clone-forming assay (G). Proliferation was examined by BrdU pulse-labeling assay (A), Ki67 staining assay (B), and cell-cycle analysis (C) at the indicated number of days in culture. Cell differentiation was analyzed by flow cytometry to examine c-Kit and Gr1 expression (D-E) and by morphology to count the percentage of leukemic blasts on indicated days (F). The numbers of CFUs were counted 7 days after each plating (G). *P < .05; **P < .01. ns, not significant; P1/P2/P3/P4, plating 1/plating 2/plating 3/plating 4.
HSPC differentiation is blocked to a greater degree by Myc transduction than by MycV394Dtransduction. c-Kit+ HSPCs were transduced with MSCV-GFP, Myc-GFP, or MycV394D-GFP, respectively. Transduced Kit+ HSPCs were purified by FACS 2 days posttransduction and incubated in HSPC culture medium with medium change every other day (A-F) or seeded into methylcellulose for serial replating clone-forming assay (G). Proliferation was examined by BrdU pulse-labeling assay (A), Ki67 staining assay (B), and cell-cycle analysis (C) at the indicated number of days in culture. Cell differentiation was analyzed by flow cytometry to examine c-Kit and Gr1 expression (D-E) and by morphology to count the percentage of leukemic blasts on indicated days (F). The numbers of CFUs were counted 7 days after each plating (G). *P < .05; **P < .01. ns, not significant; P1/P2/P3/P4, plating 1/plating 2/plating 3/plating 4.
Reduced leukemogenic capacity of MycV394D-transduced HSPCs compared with Myc-transduced HSPCs
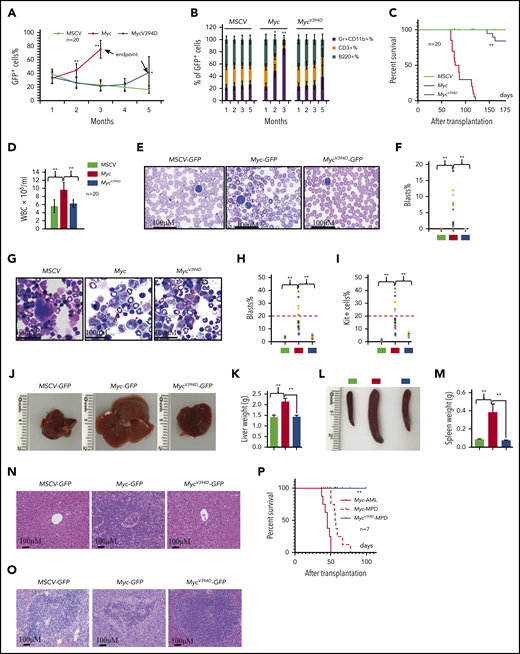
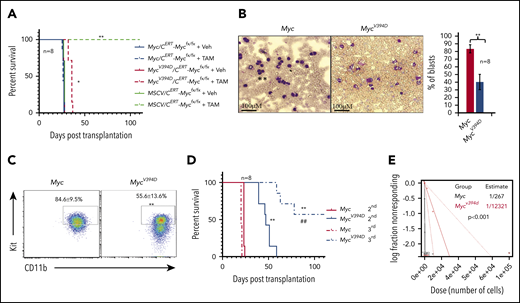
Overexpression of Myc in HSPCs by virus-mediated transduction induces MPDs and/or AML in different systems due to the significantly increased production of myeloid progenitors and excessive generation of mature myeloid cells.37,38 To study the role of Myc-Miz1–mediated transrepressive activity in the development of Myc-induced leukemia, we transplanted MSCV, Myc, or MycV394D HSPCs, together with support BM cells, into different groups of lethally-irradiated recipient mice. To ensure that the experimental data were comparable and biologically reproducible among all transplanted groups, all recipient mice were 7 to 8 weeks old at the time of transplantation and equal numbers of male and female mice were used in each group. The mice were monitored for leukemia/MPD development by examining the percentages of GFP+ cells in their peripheral blood (PB) and the percentages of Gr1+CD11b+ myeloid cells in the GFP+ population (Figure 2A-B) as well as leukemia-related death (Figure 2C). A significant increase in the percentage of PB GFP+ cells was observed in almost all Myc-HSPC–transplanted mice 2 months after transplantation, whereas the same was only observed 145 to 170 days after transplantation and in only 6 of 20 mice (30%) that had received MycV394D HSPCs (Figure 2A). The increased percentage of GFP+ cells in PB was associated with an increased percentage of Gr1+CD11b+ myeloid cells and a reduction of CD3+ T-lymphocyte percentage and B220+ B-lymphocyte percentage (Figure 2B). All 20 mice that received Myc HSPCs died within 70 to 120 days of transplantation (Figure 2C). However, all mice that had received MycV394D-GFP HSPCs survived for >145 days (Figure 2C). Unfortunately, we were only permitted to monitor the mice up to 170 days because they developed radiation-related skin lesions that increased in severity. Immediately after euthanizing the mice, leukemia was verified by examining white blood cell counts (WBCs) (Figure 2D), leukemic blasts in PB (Figure 2E-F) and BM (Figure 2G-I) as well as leukemic cell infiltration into livers (Figure 2J-K) and spleens (Figure 2L-M), and histologic assessment (Figure 2N-O). We found that 30% of Myc mice (6 of 20) developed AML as demonstrated by increased circulating blasts and >20% blasts in BM (Figure 2E-I; supplemental Figure 5) whereas the remaining 70% of mice (14 of 20) developed MPD as demonstrated by blood leukocytosis and hepatosplenomegaly (Figure 2D). Only 3 of 20 mice that had received MycV394D HSPCs developed MPD. Furthermore, both Myc-AML and Myc-MPD cells were able to generate similar AML and MPD in the second round of recipients; however, MycV394D-MPD cells failed to do so (Figure 2P), suggesting that MPD stem cell number was reduced. Our study suggests that Myc-Miz1 signaling plays a critical role in the development of Myc-induced MPD/leukemia.
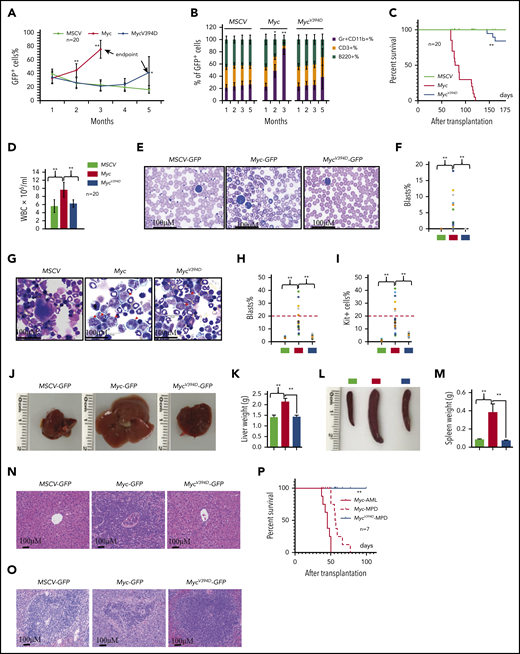
Reduced leukemogenic capacity of MycV394D-transduced HSPCs compared with Myc-transduced HSPCs. c-Kit+ HSPCs were transduced with MSCV-GFP, Myc-GFP, or MycV394D-GFP, respectively. The transduced HSPCs were purified by FACS 2 days posttransduction and transplanted into lethally-irradiated recipient mice, which were observed for leukemia development. Twenty mice were transplanted for each group. The blood cell engraftments of the transduced HSPCs were analyzed over time by examining the percentage of GFP+ cells in PB of the recipient mice (A). The data for the last time point in both Myc and Mycv394d groups were collected when the mice were euthanized (end point). The lineage commitments of the transduced HSPCs were evaluated by examining the percentages of Gr1+CD11b+ myeloid cells, B220+ B lymphocytes, and CD3+ T lymphocytes within the GFP+ cells (B). Survival of the recipient mice is demonstrated by Kaplan-Meier analysis (C). Leukemia/MPD was verified immediately after the death of the mice by examining the WBCs in PB (D), leukemic blasts in PB (E-F), leukemic blasts in BM (G-I), liver size (J-K), spleen size (L-M), as well as leukemic cell infiltration in livers (N) and spleens (O). *P < .05; **P < .01. (P) BM cells were collected from Myc-AML, Myc-MPD, and MycV394D-MPD mice and transplanted into second-round recipient mice, respectively. Mice were observed for AML or MPD-related death. The survival of the recipient mice is demonstrated by Kaplan-Meier analysis.
Reduced leukemogenic capacity of MycV394D-transduced HSPCs compared with Myc-transduced HSPCs. c-Kit+ HSPCs were transduced with MSCV-GFP, Myc-GFP, or MycV394D-GFP, respectively. The transduced HSPCs were purified by FACS 2 days posttransduction and transplanted into lethally-irradiated recipient mice, which were observed for leukemia development. Twenty mice were transplanted for each group. The blood cell engraftments of the transduced HSPCs were analyzed over time by examining the percentage of GFP+ cells in PB of the recipient mice (A). The data for the last time point in both Myc and Mycv394d groups were collected when the mice were euthanized (end point). The lineage commitments of the transduced HSPCs were evaluated by examining the percentages of Gr1+CD11b+ myeloid cells, B220+ B lymphocytes, and CD3+ T lymphocytes within the GFP+ cells (B). Survival of the recipient mice is demonstrated by Kaplan-Meier analysis (C). Leukemia/MPD was verified immediately after the death of the mice by examining the WBCs in PB (D), leukemic blasts in PB (E-F), leukemic blasts in BM (G-I), liver size (J-K), spleen size (L-M), as well as leukemic cell infiltration in livers (N) and spleens (O). *P < .05; **P < .01. (P) BM cells were collected from Myc-AML, Myc-MPD, and MycV394D-MPD mice and transplanted into second-round recipient mice, respectively. Mice were observed for AML or MPD-related death. The survival of the recipient mice is demonstrated by Kaplan-Meier analysis.
Reduced leukemogenic capacity of MycV394D-AML cells compared with Myc-AML cells
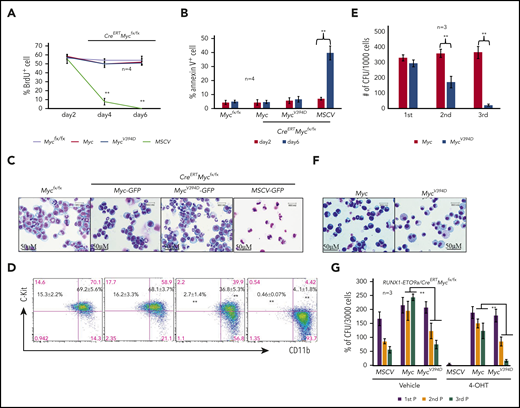
Using MLL-AF9–transduced murine AML models, we have demonstrated that induced deletion of Myc represses proliferation and induces complete differentiation of AML cells.39 To establish that Myc-Miz1–mediated transrepressive activity is required to maintain leukemogenic capacity after AML has developed, we transduced c-Kit+ HSPCs isolated from CreERTMycfx/fx mice with an MLL-AF9 fusion gene. The transduced HSPCs were transplanted into recipient mice. After development of leukemia, AML cells were collected from the BM and spleens of the recipient mice and were used in this study. We transduced CreERTMycfx/fx AML cells with MSCV-GFP, Myc-GFP, or MycV394D-GFP to generate MSCV/CreERTMycfx/fx, Myc/CreERTMycfx/fx, and MycV394D/CreERTMycfx/fx AML cells, respectively (supplemental Figure 6a). Slight increases in proliferation and CFC were observed in both Myc/CreERTMycfx/fx and MycV394D/CreERTMycfx/fx AML cells when compared with MSCV/CreERTMycfx/fx AML cells (supplemental Figure 6b-c). The transduced cells were purified by FACS for GFP+ cells and were treated with 4-OHT to generate MSCV-, Myc-, and MycV394D-AML cells by inducing the deletion of endogenous Myc. Successful deletion of endogenous Myc was confirmed by polymerase chain reaction (PCR) (supplemental Figure 6d) and the ectopic expression of Myc and MycV394D was evaluated by western blotting (supplemental Figure 6e). One day after induction with 4-OHT, cells were cultured in liquid medium for proliferative, differentiation, and apoptotic analysis, or seeded into methylcellulose medium for CFC assay. We found that in the first 2 days of culturing, there was no significant difference in proliferation (Figure 3A) nor in survival (Figure 3B) among the 3 types of transduced AML cells. A significant reduction in proliferation and increased apoptosis was observed only in MSCV-AML cells after 4 days in culture (Figure 3A-B). The reduced proliferative capacity and increased apoptosis in MSCV-AML cells are associated with the full differentiation of these cells as demonstrated by morphologic analysis (Figure 3C) and c-Kit expression (Figure 3D). Consistent with findings from our previous studies, we found that MSCV-AML cells lost CFC.39 Interestingly, both Myc and MycV394D transductions fully restored survival, proliferative capacity, and CFC to Myc-deficient AML cells, suggesting that Myc-Miz1 signaling is not required for the proliferation and survival of AML cells (Figure 3). However, compared with Myc-AML cells, MycV394D-AML cells were partially differentiated as demonstrated by morphology (Figure 3C) and c-Kit expression (Figure 3D). Despite the increase in the differentiated Kit− population (which was cycling slowly) in MycV394D-AML cells compared with Myc-AML cells, the reduction of Kit+CD11blow quiescent leukemia stem cells (LSCs) (Figure 3D; supplemental Figure 7) might explain why we did not detect a significant reduction of proliferation in MycV394D-AML cells (Figure 3A).39 Myc- and MycV394D-AML cells generated comparable numbers of CFUs in the first plating. However, during serial replating, Myc-AML cells maintained consistent numbers of CFUs whereas the numbers of CFUs were gradually reduced for MycV394D- AML cells (Figure 3E), correlating with their partially differentiated morphology (Figure 3F) and increased phagocytosis (supplemental Figure 8). We also found that Myc-Miz1 signaling played a similar role in leukemic genes RUNX1-ETO9a– and PML-RARα–induced leukemia, as demonstrated by serial replating CFC assay (Figure 3G; supplemental Figure 9).

Partial differentiation of MycV394D-AML cells compared with Myc-AML cells. MSCV-GFP/CreERTMycfx/fx, Myc-GFP/CreERTMycfx/fx, and MycV394D-GFP/CreERTMycfx/fx AML cells were treated with 1 μM 4-OHT to induce the deletion of endogenous Myc. Beginning on day 2 following 4-OHT treatment, cells were collected for analysis at the indicated time points. Mycfx/fx AML cells were studied in parallel as controls. (A) BrdU (final concentration 10 µM) was added to the culture to label the cells 2 hours before they were to be collected. The percentages of BrdU+ cells were analyzed by intracellular antibody staining and flow cytometric analysis. (B) Apoptosis was examined by annexin V staining. (C-D) Cells were collected on day 6 for morphologic (C) and phenotypic analysis (D). (E-F) One day after 4-OHT treatment, cells were collected and seeded for serial replating CFU assay (E). CFUs were counted on day 7 after plating. Cells were collected from the colonies of the third replating for morphologic study (F). (G) c-Kit+ HSPCs were collected from CreERTMycfx/fx mice and cotransduced with RUNX1-ETO9a-GFP plus MSCV-mCherry, Myc-mCherry, or MycV394D-mCherry, respectively. The transduced HSPCs were purified by FACS and seeded into the methylcellulose with or without 4-OHT-induction. CFUs were counted 7 days after plating. Cells from the CFUs were collected for second and third replatings. **P < .01. 1st P/2nd P/3rd P, first plating/second plating/third plating.
Partial differentiation of MycV394D-AML cells compared with Myc-AML cells. MSCV-GFP/CreERTMycfx/fx, Myc-GFP/CreERTMycfx/fx, and MycV394D-GFP/CreERTMycfx/fx AML cells were treated with 1 μM 4-OHT to induce the deletion of endogenous Myc. Beginning on day 2 following 4-OHT treatment, cells were collected for analysis at the indicated time points. Mycfx/fx AML cells were studied in parallel as controls. (A) BrdU (final concentration 10 µM) was added to the culture to label the cells 2 hours before they were to be collected. The percentages of BrdU+ cells were analyzed by intracellular antibody staining and flow cytometric analysis. (B) Apoptosis was examined by annexin V staining. (C-D) Cells were collected on day 6 for morphologic (C) and phenotypic analysis (D). (E-F) One day after 4-OHT treatment, cells were collected and seeded for serial replating CFU assay (E). CFUs were counted on day 7 after plating. Cells were collected from the colonies of the third replating for morphologic study (F). (G) c-Kit+ HSPCs were collected from CreERTMycfx/fx mice and cotransduced with RUNX1-ETO9a-GFP plus MSCV-mCherry, Myc-mCherry, or MycV394D-mCherry, respectively. The transduced HSPCs were purified by FACS and seeded into the methylcellulose with or without 4-OHT-induction. CFUs were counted 7 days after plating. Cells from the CFUs were collected for second and third replatings. **P < .01. 1st P/2nd P/3rd P, first plating/second plating/third plating.
MSCV-AML cells failed to generate leukemia in recipient mice upon transplantation.39 To study the effects of Myc-Miz1–mediated transrepressive activity on leukemogenic capacity, we transplanted MSCV/CreERTMycfx/fx, Myc/CreERTMycfx/fx, or MycV394D/CreERTMycfx/fx AML cells into different groups of recipient mice (supplemental Figure 6). Three days posttransplantation, the recipient mice were randomly divided into 2 groups and injected with either TAM to induce the deletion of endogenous Myc or with vehicle. As expected, all mice injected with vehicle died of leukemia 21 to 23 days after transplantation regardless of the genotype of the transplanted AML cells. All mice that received MSCV/CreERTMycfx/fx AML cells and TAM were completely healthy even 100 days posttransplantation. Finally, mice that received Myc/CreERTMycfx/fx AML cells and TAM died of leukemia 21 to 23 days posttransplantation, just as was observed in vehicle control-treated animals. However, mice that had received MycV394D/CreERTMycfx/fx AML cells and TAM died of leukemia 32 to 37 days posttransplantation, a delay of 10 days compared with the vehicle-only group (Figure 4A). Consistent with our in vitro data, MycV394D-AML cells collected from the BM of transplanted mice showed a partially differentiated phenotype compared with cells from mice transplanted with Myc AML (Figure 4B-C). During serial transplantation, the development of leukemia was further delayed in mice that had received MycV394D-AML cells compared with mice that had received Myc-AML cells (Figure 4D). To study the role of Myc-Miz1–mediated transrepressive activity in LSC self-renewal, we collected leukemic cells from recipient mice and examined the frequencies of LSCs by serial dilution and competitive transplantation. We found that the frequencies of LSCs in MycV394D-GFP AML-transplanted mice were significantly lower than those among Myc-GFP AML-transplanted mice (Figure 4E). These data suggested that Myc-Miz1–mediated transrepressive activity is required for maintaining the undifferentiated self-renewal state of LSCs.
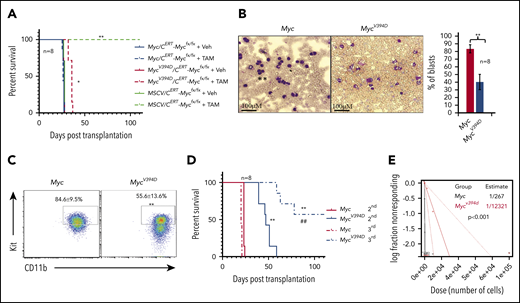
Reduced leukemogenic capacity of MycV394D-AML cells compared with Myc-AML cells. (A) MSCV/CreERTMycfx/fx, Myc/CreERTMycfx/fx, or MycV394D/CreERTMycfx/fx AML cells were transplanted into different groups of recipient mice. Starting on day 3 after transplantation, mice were treated with vehicle (Veh) or TAM daily for 5 consecutive days to induce the deletion of endogenous Myc. The mice were monitored for leukemia development and leukemia-related death. (B-E) Myc-GFP and MycV394D-GFP AML cells were collected from the mice that had leukemia (above) and the differentiation status of these cells was assessed by morphologic study for leukemic blasts (B) and c-Kit expression (C); the self-renewal capacity of LSCs was analyzed by serial transplantation (D). The frequency of LSCs was examined by serial dilution and competitive transplantation assay (E). *P < .05; **P < ..01 compared with vehicle groups in panel A, and Myc groups in panels B-D. ##P < .01 compared with MycV394D second transplantation.
Reduced leukemogenic capacity of MycV394D-AML cells compared with Myc-AML cells. (A) MSCV/CreERTMycfx/fx, Myc/CreERTMycfx/fx, or MycV394D/CreERTMycfx/fx AML cells were transplanted into different groups of recipient mice. Starting on day 3 after transplantation, mice were treated with vehicle (Veh) or TAM daily for 5 consecutive days to induce the deletion of endogenous Myc. The mice were monitored for leukemia development and leukemia-related death. (B-E) Myc-GFP and MycV394D-GFP AML cells were collected from the mice that had leukemia (above) and the differentiation status of these cells was assessed by morphologic study for leukemic blasts (B) and c-Kit expression (C); the self-renewal capacity of LSCs was analyzed by serial transplantation (D). The frequency of LSCs was examined by serial dilution and competitive transplantation assay (E). *P < .05; **P < ..01 compared with vehicle groups in panel A, and Myc groups in panels B-D. ##P < .01 compared with MycV394D second transplantation.
Increased sensitivity of MycV394D-AML cells to chemotherapy compared with Myc-AML cells
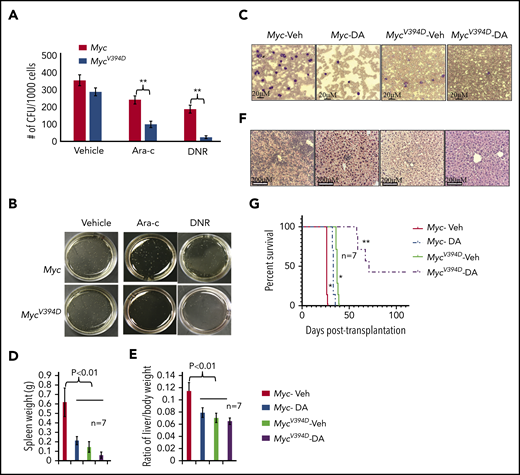
Because MycV394D-AML cells have a lower frequency of LSCs, we tested whether Myc-Miz1–mediated signaling regulates drug sensitivity in AML cells. We found that MycV394D-AML cells were more sensitive to both daunorubicin (DNR) and cytarabine (Ara-C) treatment compared with Myc-AML cells, as demonstrated by in vitro CFC assay (Figure 5A-B). To study whether MycV394D-GFP AML cells are also sensitive to chemotherapy in vivo, we transplanted Myc/CreERTMycfx/fx and MycV394D/CreERTMycfx/fx AML cells into 2 groups of recipient mice. All mice were injected with TAM daily on days 3 to 7 to induce the deletion of endogenous Myc. Beginning on day 8, mice in each group were randomly divided into 2 subgroups and treated with either 3 days of DNR plus 5 days of Ara-C (D3+A5) or vehicle. A panel of mice was examined on day 24 for leukemia development (Figure 5C-E) and another panel of mice was monitored for leukemia-related death (Figure 5G). We found that in the vehicle-only control groups, mice that had received Myc-AML cells developed severe leukemia by day 24 as demonstrated by leukemic blasts in PB (Figure 5C) and hepatosplenomegaly (Figure 5D-E) due to leukemic cell infiltration (Figure 5F), and died within 26 to 27 days after transplantation (Figure 5G). Mice that had received MycV394D-AML cells did not show any signs of leukemia by day 24 (Figure 5C-F) and died of leukemia 36 to 37 days following transplantation (Figure 5G). D3+A5 treatment delayed leukemia development among Myc-AML animals for only 6 days (Figure 5G) (small leukemic cell infiltrations can be detected in livers; Figure 5F), whereas disease development among animals transplanted with MycV394D-AML was delayed by >20 days (Figure 5G). Three of 7 mice in the D3+A5-treated MycV394D-AML group survived over 100 days without leukemia.

Increased sensitivity of MycV394DAML cells to chemotherapy compared with Myc AML cells. (A-B) Myc- and MycV394D-AML cells were seeded into methylcellulose medium for CFU assay with or without Ara-C and DNR treatment. CFUs were counted (A) and imaged (B) on day 7 of culturing. (C-G) Myc/CreERTMycfx/fx and MycV394D/CreERTMycfx/fx AML cells were transplanted into recipient mice. Beginning on day 3 after transplantation, mice were treated with TAM every day for 5 consecutive days to induce the deletion of endogenous Myc. Beginning on day 8 after transplantation, mice were treated with D3+A5 regimen of chemotherapy. A panel of mice was euthanized on day 24 posttransplantation to assess the development of leukemia by examining leukemic blasts in PB smears (C), spleen size (D), liver size (E), and leukemic cell infiltration into livers (F). The other panel of mice was monitored for leukemia-related death (G). *P < .05 compared to Myc-Veh group; **P < .01 compared to Myc-DA group. DA, daunorubicin and cytarabine.
Increased sensitivity of MycV394DAML cells to chemotherapy compared with Myc AML cells. (A-B) Myc- and MycV394D-AML cells were seeded into methylcellulose medium for CFU assay with or without Ara-C and DNR treatment. CFUs were counted (A) and imaged (B) on day 7 of culturing. (C-G) Myc/CreERTMycfx/fx and MycV394D/CreERTMycfx/fx AML cells were transplanted into recipient mice. Beginning on day 3 after transplantation, mice were treated with TAM every day for 5 consecutive days to induce the deletion of endogenous Myc. Beginning on day 8 after transplantation, mice were treated with D3+A5 regimen of chemotherapy. A panel of mice was euthanized on day 24 posttransplantation to assess the development of leukemia by examining leukemic blasts in PB smears (C), spleen size (D), liver size (E), and leukemic cell infiltration into livers (F). The other panel of mice was monitored for leukemia-related death (G). *P < .05 compared to Myc-Veh group; **P < .01 compared to Myc-DA group. DA, daunorubicin and cytarabine.
Myc-Miz1 signaling is required for maintaining the undifferentiated status of AML cells by inhibiting the expression of Cebpα and Cebpδ
To study how the interaction of Myc-Miz1 promotes leukemogenic capacity and drug resistance, we compared the gene-expression profiles of MycV394D- and Myc-AML cells 4 days after deletion of the endogenous Myc gene. It was surprising to see that 497 genes were downregulated whereas only 211 genes were upregulated in MycV394D-AML cells compared with Myc-AML cells. (Figure 6A-B). Interestingly, many of the downregulated genes are associated with LSC self-renewal and survival, such as Flt3, Kit, Ptpn3, Il1r1, Il3ra, Gata2, and Evi (Figure 6C-D). Several Myc-Miz1–targeted genes, including Bcl2, p15INK4, and p27KIP1 were also downregulated (Figure 6E). We reported that p27KIP1 is highly expressed in Kit+CD11blow LSCs, which is required for maintaining the slow cycling and self-renewal phenotypic features of quiescent LSCs.39 We also found that the expression of Bcl2, p15INK4, Flt3, Kit, Il1r1, Il3ra, Gata2, and Evi were higher in Kit+CD11blow LSCs compared with Kit+CD11bhigh AML blasts (Figure 6F). Further study demonstrated that the downregulation of these genes was correlated to a reduction of Kit+CD11blow LSCs, suggesting a secondary effect of LSC differentiation (Figure 6G-H).
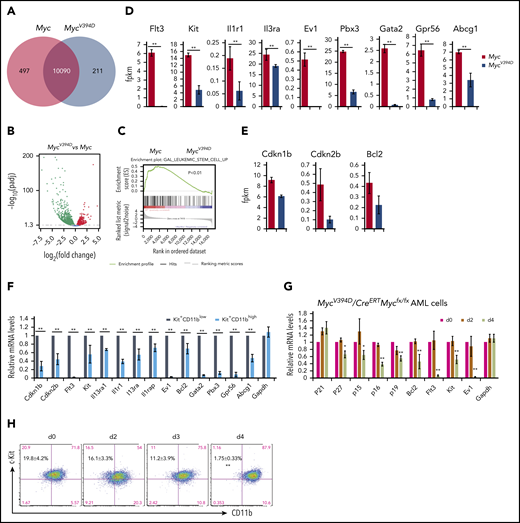
Downregulation of LSC genes in MycV394D-AML cells compared with Myc-AML cells. Myc/CreERTMycfx/fx and MycV394D/CreERTMycfx/fx AML cells were treated with 4-OHT to induce the deletion of endogenous Myc. (A-E) Myc- and MycV394D-AML cells were collected on day 4 of 4-OHT treatment and Myc deletion was confirmed. The differentially expressed genes between Myc- and MycV394D-AML cells are presented as a Venn diagram (A) and a volcano plot (B). The expression of LSC genes was analyzed by gene set enrichment analysis (C). The expression of selected LSC-related genes (D) and Myc-Miz1 target genes (E) were compared between Myc- and MycV394D-AML cells. (F) Kit+CD11blow LSCs and Kit+CD11bhigh leukemic blasts were purified from Myc-AML cells. The expression of selected LSC genes and Myc-Miz1 target genes was examined by quantitative real-time PCR (qRT-PCR) assay and compared. (G-H) MycV394D/CreERTMycfx/fx AML cells were treated with 4-OHT to induce the deletion of endogenous Myc. Cells were collected on day 0 (vehicle), day 2, and day 4 after 4-OHT treatment. The expression of the selected genes was examined by qRT-PCR and normalized to messenger RNA (mRNA) levels for day 0 (G). The percentages of Kit+CD11blow LSCs and Kit+CD11bhigh leukemic blasts were examined by flow cytometry (H). *P < .05; **P < .01. fpkm, fragments per kilobase of transcript per million mapped reads.
Downregulation of LSC genes in MycV394D-AML cells compared with Myc-AML cells. Myc/CreERTMycfx/fx and MycV394D/CreERTMycfx/fx AML cells were treated with 4-OHT to induce the deletion of endogenous Myc. (A-E) Myc- and MycV394D-AML cells were collected on day 4 of 4-OHT treatment and Myc deletion was confirmed. The differentially expressed genes between Myc- and MycV394D-AML cells are presented as a Venn diagram (A) and a volcano plot (B). The expression of LSC genes was analyzed by gene set enrichment analysis (C). The expression of selected LSC-related genes (D) and Myc-Miz1 target genes (E) were compared between Myc- and MycV394D-AML cells. (F) Kit+CD11blow LSCs and Kit+CD11bhigh leukemic blasts were purified from Myc-AML cells. The expression of selected LSC genes and Myc-Miz1 target genes was examined by quantitative real-time PCR (qRT-PCR) assay and compared. (G-H) MycV394D/CreERTMycfx/fx AML cells were treated with 4-OHT to induce the deletion of endogenous Myc. Cells were collected on day 0 (vehicle), day 2, and day 4 after 4-OHT treatment. The expression of the selected genes was examined by qRT-PCR and normalized to messenger RNA (mRNA) levels for day 0 (G). The percentages of Kit+CD11blow LSCs and Kit+CD11bhigh leukemic blasts were examined by flow cytometry (H). *P < .05; **P < .01. fpkm, fragments per kilobase of transcript per million mapped reads.
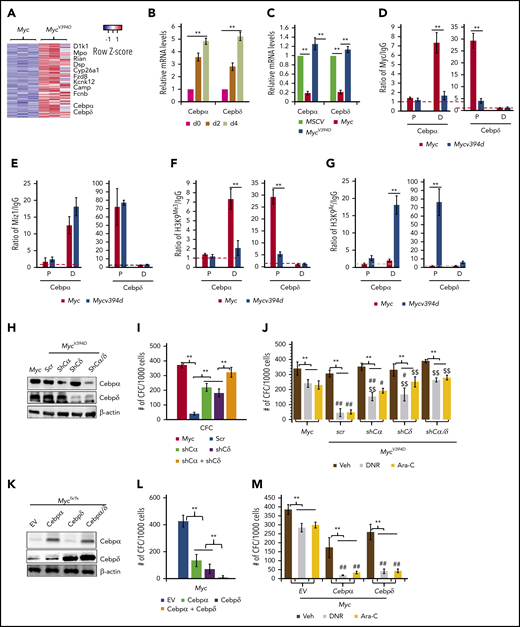
Both Cebpα and Cebpδ were among the upregulated genes in MycV394D-AML cells, in which upregulation can be detected at as early as day 2 after endogenous Myc deletion (Figure 7A-B). Cebpα and Cebpδ are key transcription factors in myeloid cells that are required for granulocyte and macrophage differentiation.40,41 Consistently, Myc overexpression significantly repressed the expression of both Cebpα and Cebpδ in HSPCs but MycV394D overexpression did not (Figure 7C). Chromatin immunoprecipitation (ChIP) assay demonstrated that both Myc and Miz1 bind to the proximal promoter of the Cebpδ gene and to the distal promotor of the Cebpα gene (Figure 7D-E). The binding of MycV394D to both promotors was reduced (Figure 7D), which was associated with increased H3K9Ac and a reduction of H3K9Me3 (Figure 7F-G) on both promoters in MycV394D-AML cells. Hypermethylation of these 2 promoters has been reported in AML patient samples and correlates to low Cebpα/Cebpδ expression and poor patient prognosis.42-45 We assessed whether the partially differentiated phenotype of MycV394D-AML cells is due to the upregulation of Cebpα and Cebpδ. We found that knockdown of either Cebpα or Cebpδ (Figure 7H) can largely restore the undifferentiated status (supplemental Figure 10a-b), serial replating CFC (Figure 7I), and chemoresistance (Figure 7J) to MycV394D-AML cells. We also found that overexpression of either Cebpα or Cebpδ (Figure 7K) can promote partial differentiation (supplemental Figure 10c-d), repress serial replating CFC (Figure 7L), and enhance sensitivity to chemotherapy (Figure 7M) to Myc-AML cells. We also found that simultaneous knockdown or overexpression of both Cebpα and Cebpδ showed additive effects on AML cell differentiation (Figure 7H-M). Our study suggests that Myc-Miz1 signaling is required for maintaining the undifferentiated state of AML cells by repressing Cebpα and Cebpδ expression.
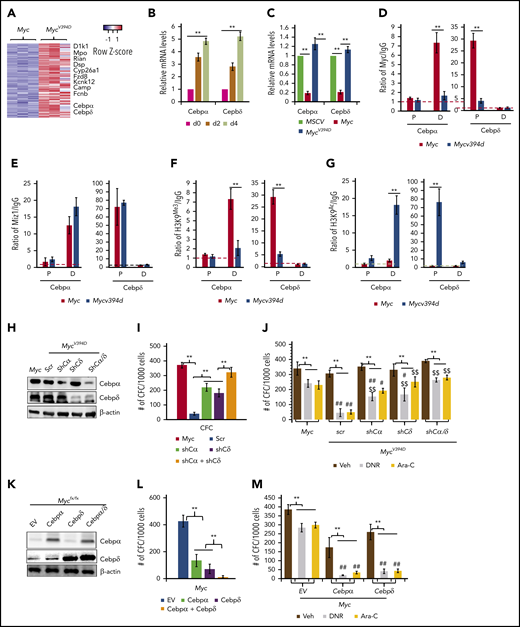
Upregulation of Cebpα and Cebpδ in MycV394DAML cells contributes to the partial differentiation phenotype. (A) Representative upregulated genes in MycV394-AML cells as identified from RNA sequencing are presented in a heatmap. (B) MycV394D-AML cells were collected at the indicated times after 4-OHT treatment as described in Figure 6G-H. The expression of Cebpα and Cebpδ was examined by qRT-PCR. (C) The expression of Cebpα and Cebpδ was examined by qRT-PCR in MSCV-, Myc- and MycV394D-transduced HSPCs on day 4 after transduction. (D-E) The binding of Myc, MycV394D, and Miz1 to the promoters of the Cebpα or Cebpδ genes was examined in AML cells by ChIP/quantitative PCR (qPCR) assay. (F-G) H3K9Me3 and H3K9Ac status of the promoters of Cebpα or Cebpδ genes were examined by ChIP/qPCR assay. (H-J) Cebpα or Cebpδ or both were knocked down in MycV394D AML cells by short hairpin RNA (shRNA) specific for Cebpα (shCα) or Cebpδ (shCδ) (H). The third replating CFC (I) and response to DNR or Ara-C treatment (J) of the gene knockdown cells were compared with scrambled shRNA (scr)-transduced MycV394D AML cells. Myc AML cells were used as controls. (K-M) Cebpα or Cebpδ or both were overexpressed in Myc AML cells by viral transduction (K). The third replating CFC (L) and response to DNR or Ara-C treatment (M) of the gene overexpressed cells were compared with empty vector (EV)-transduced Myc-AML cells. **P < .01, ##P < .01, and #P < .05, compared with corresponding Myc groups in panel J and corresponding EV groups in panel M. $$P < .01 and $P < .05, compared with corresponding scr groups. D, distal promoto; P, proximal promoter.
Upregulation of Cebpα and Cebpδ in MycV394DAML cells contributes to the partial differentiation phenotype. (A) Representative upregulated genes in MycV394-AML cells as identified from RNA sequencing are presented in a heatmap. (B) MycV394D-AML cells were collected at the indicated times after 4-OHT treatment as described in Figure 6G-H. The expression of Cebpα and Cebpδ was examined by qRT-PCR. (C) The expression of Cebpα and Cebpδ was examined by qRT-PCR in MSCV-, Myc- and MycV394D-transduced HSPCs on day 4 after transduction. (D-E) The binding of Myc, MycV394D, and Miz1 to the promoters of the Cebpα or Cebpδ genes was examined in AML cells by ChIP/quantitative PCR (qPCR) assay. (F-G) H3K9Me3 and H3K9Ac status of the promoters of Cebpα or Cebpδ genes were examined by ChIP/qPCR assay. (H-J) Cebpα or Cebpδ or both were knocked down in MycV394D AML cells by short hairpin RNA (shRNA) specific for Cebpα (shCα) or Cebpδ (shCδ) (H). The third replating CFC (I) and response to DNR or Ara-C treatment (J) of the gene knockdown cells were compared with scrambled shRNA (scr)-transduced MycV394D AML cells. Myc AML cells were used as controls. (K-M) Cebpα or Cebpδ or both were overexpressed in Myc AML cells by viral transduction (K). The third replating CFC (L) and response to DNR or Ara-C treatment (M) of the gene overexpressed cells were compared with empty vector (EV)-transduced Myc-AML cells. **P < .01, ##P < .01, and #P < .05, compared with corresponding Myc groups in panel J and corresponding EV groups in panel M. $$P < .01 and $P < .05, compared with corresponding scr groups. D, distal promoto; P, proximal promoter.
Discussion
Deregulation of Myc is detected in almost all types of leukemia, inducing high levels of proliferation and blocking the full differentiation of HSPCs, which are key events in the pathogenesis of most hematopoietic malignancies.1,46 Most previous studies have tried to target Myc by repressing its transactivating activity or by inhibiting the mediator/superelongation complex47-49 ; however, no success has thus far been achieved in this regard. We found that in AML cells, Myc represses Cebpα and Cebpδ expression through partnering with Miz1, and that this is required for maintaining the undifferentiated state of leukemic cells and self-renewal in LSCs. MycV394D-AML cells showed a partial differentiation phenotype due to the upregulation of Cebpα and Cebpδ. MycV394D-AML cells showed reduced leukemogenic ability and increased sensitivity to standard chemotherapy compared with Myc-AML cells, due to a reduction in the frequency of LSCs. Our study suggests that inhibition of Myc-Miz1–mediated transrepressive activity might be a useful strategy by which to treat leukemia when combined with standard chemotherapy.
The mechanism by which Myc promotes proliferation has been very well studied. In most types of cells, Myc promotes proliferation primarily by upregulating positive cell-cycle regulators.1 However, in certain instances, Myc promotes proliferation primarily through Miz1-mediated repression of negative cell-cycle regulators.50-54 However, we found that in both the Myc-induced MPD/AML model and the MLL-AF9 AML model, inhibition of Myc-Miz1 interaction did not affect the proliferation of leukemic cells. Our study suggests that the Myc-Miz1–mediated transrepressive activity involved in the pathogenesis of leukemia operates primarily through maintaining the undifferentiated state of leukemic cells and promoting the self-renewal capacity of LSCs.
Myc promotes proliferation of neural progenitors by inducing Myc/Max-mediated transactivation activity, whereas Myc enhances the self-renewal of neural progenitors by inducing Myc/Miz1-mediated transrepressive activity.55,56 We found that deletion of Myc leads to the complete differentiation of AML cells, which occurs prior to cell-cycle arrest, suggesting that blocking differentiation is the critical role of Myc in the pathogenesis of AML. However, MycV394D can only partially restore the undifferentiated status of AML cells and self-renewal to LSCs, indicating that there are both Miz1 interaction-dependent and -independent roles for Myc in the regulation of AML cell differentiation. The Miz1 interaction-dependent role of Myc in AML cells is primarily mediated by repression of the key granulocytic transcription factors Cebpα and Cebpδ.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the staff of the Department of Comparative Medicine of Loyola University Medical Center for high-quality animal care services, as well as Nancy Zeleznik-Le, Manuel Diaz, and Andrew Dingwall for their ongoing professional collaboration and scientific suggestions and discussions, which improved the present studies. The authors appreciate FACS sorting and analysis assistance contributed by Patricia Simms and her staff at Loyola University Medical Center.
This work was supported by the National Natural Science Foundation of China (projects 81670151 and 81700141), and the 973 National Basic Research Program of China (project 2013CB966803). This work was also supported by a Leukemia Research Foundation New Investigator Award (J.L.) and Loyola Laboratory Research Support (Jiwang Zhang).
Authorship
Contribution: L.Z., J.L., H.X., X.S., L.F., C.H., W.L., and Y.H. conducted most of the in vitro culturing and in vivo transplantation studies; L.Z., H.X., W.W., and P.B. performed PCR, western blotting, and tissue-section studies; Y.X., F.Z., S.D., K.J., P.B., Jiwang Zhang, and Jun Zhang contributed to data collection and interpretation of the results and the conceptual model; L.Z., J.L., and H.X. designed the experiments and analyzed the data; Jiwang Zhang and Jun Zhang supervised the research and wrote and edited the manuscript; and P.B. wrote, edited, and refined the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jiwang Zhang, Oncology Institute, Cardinal Bernardin Cancer Center, Loyola University Chicago, Maywood, IL 60153; e-mail: jzhang@luc.edu; and Jun Zhang, College of Life Sciences, Shanghai Normal University, Shanghai, 200234, People’s Republic of China; e-mail: zhj@shnu.edu.cn.