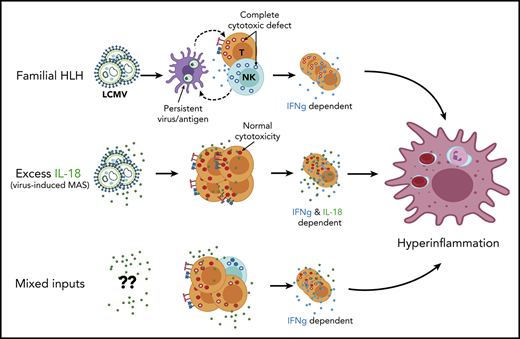
Key Points
Excess IL-18 directs virus-induced hyperinflammation via autoinflammatory amplification of CD8 T-cell responses and IFNg overproduction.
Synergy between (even partial) cytotoxic impairment and excess IL-18 leads to spontaneous hyperinflammation.

Abstract
Hemophagocytic lymphohistiocytosis (HLH) and macrophage activation syndrome (MAS) are life-threatening hyperinflammatory syndromes typically associated with underlying hematologic and rheumatic diseases, respectively. Familial HLH is associated with genetic cytotoxic impairment and thereby to excessive antigen presentation. Extreme elevation of serum interleukin-18 (IL-18) has been observed specifically in patients with MAS, making it a promising therapeutic target, but how IL-18 promotes hyperinflammation remains unknown. In an adjuvant-induced MAS model, excess IL-18 promoted immunopathology, whereas perforin deficiency had no effect. To determine the effects of excess IL-18 on virus-induced immunopathology, we infected Il18-transgenic (Il18tg) mice with lymphocytic choriomeningitis virus (LCMV; strain Armstrong). LCMV infection is self-limited in wild-type mice, but Prf1−/− mice develop prolonged viremia and fatal HLH. LCMV-infected Il18-transgenic (Il18tg) mice developed cachexia and hyperinflammation comparable to Prf1−/− mice, albeit with minimal mortality. Like Prf1−/− mice, immunopathology was largely rescued by CD8 depletion or interferon-γ (IFNg) blockade. Unlike Prf1−/− mice, they showed normal target cell killing and normal clearance of viral RNA and antigens. Rather than impairing cytotoxicity, excess IL-18 acted on T lymphocytes to amplify their inflammatory responses. Surprisingly, combined perforin deficiency and transgenic IL-18 production caused spontaneous hyperinflammation specifically characterized by CD8 T-cell expansion and improved by IFNg blockade. Even Il18tg;Prf1-haplosufficient mice demonstrated hyperinflammatory features. Thus, excess IL-18 promotes hyperinflammation via an autoinflammatory mechanism distinct from, and synergistic with, cytotoxic impairment. These data establish IL-18 as a potent, independent, and modifiable driver of life-threatening innate and adaptive hyperinflammation and support the rationale for an IL-18–driven subclass of hyperinflammation.
Introduction
Hyperinflammation describes life-threatening systemic inflammation arising in a variety of contexts including familial-, malignancy-, bacteria-, and virus- (including coronavirus disease 2019) associated hemophagocytic lymphohistiocytosis (HLH), as well as macrophage activation syndrome (MAS), chimeric antigen receptor T-cell cytokine release syndrome, hyperferritinemic sepsis, and hemorrhagic fever viruses.1,2 Distinguishing between hyperinflammatory syndromes is currently based on their clinical/environmental context, but often this context is not apparent during early stages when precise treatment can prevent organ damage. Even if clinicians are able to identify a specific trigger or context, rarely does this indicate a specific disease mechanism.
Cytotoxic impairment is an important exception wherein profound genetic defects in granule-mediated cytotoxicity help define familial HLH (FHL). Careful genetic, biomarker, and mechanistic studies, largely using knockout mice infected with lymphocytic choriomeningitis virus (LCMV), demonstrate that innate-immune priming mixes with persistent viremia to initiate excessive antigen presentation.3-6 The resulting unrestrained CD8 T cell activation drives immunopathology due, in large part, to the effects of interferon-γ (IFNg).7 Supportive human studies have identified CXCL9 as a sensitive biomarker of IFNg activity,8,9 and a recent clinical trial led to the approval of IFNg neutralizing therapy (emapalumab) for refractory FHL.10,11 Typically, biallelic mutations are required to cause FHL, and single-copy mutations are present at high frequency in the general population.12 However, partial impairment of cytotoxic function could prolong the immune synapse, amplify cytokine production,4 and help explain the enrichment of single-copy FHL mutations in MAS and virus-induced HLH.13-15 Nevertheless, the only available therapy specifically targeting cytotoxic impairment remains hematopoietic stem cell transplantation.
MAS is distinguished by its occurrence in rheumatic diseases such as systemic juvenile idiopathic arthritis (SJIA). Although many biomarkers have been associated with MAS and underlying SJIA,16 multiple studies have confirmed the specificity of very high total interleukin-18 (IL-18) and detectable free IL-18 (that which is unbound by IL-18 binding protein), in MAS.9,17,18 The association of NLRC4 inflammasome gain-of-function mutations, overproduction of IL-18, and hyperinflammation suggested IL-18 may be an upstream susceptibility factor specific for MAS.19,20 In mice, excess free IL-18 drives more severe immunopathology during repeated stimulation through Toll-like receptor 9 (TLR)9, a stimulus that drives inflammation independently of adaptive immunity.9,21 Canonically, IL-18 enhances lymphocyte cytokine production and cytotoxicity,22 but recent evidence suggests chronic IL-18 exposure may actually impair natural killer (NK) cell function.23,24 Thus, it is not clear how IL-18 promotes MAS or how this pathway interacts with cytotoxic impairment.
Viral infections are a common trigger for hyperinflammation in patients with cytotoxic impairment, MAS, or without known susceptibility (most notably Epstein-Barr virus, hemorrhagic viruses, influenza, and possibly SARS-CoV-2).2,25 Together, the ubiquity of IL-18 in MAS and the partial cytotoxic impairments in some patients with MAS/virus-induced HLH underscore the importance of understanding how infection, IL-18, and graded cytotoxic impairments interact. We sought to better understand (1) how excess IL-18 might affect pathogen clearance and immunopathology after viral infection, (2) how cytotoxic impairment might contribute to innate-driven hyperinflammation, and (3) whether these 2 susceptibility factors work by overlapping or complementary pathways. We found that, after LCMV infection, Il18tg mice developed immunopathology consistent with hyperinflammation. However, this immunopathology was caused by direct amplification of T-cell responses rather than via cytotoxic impairment. Contrasting with the effects of excess IL-18,9,21 mice lacking cytotoxicity show no increase in severity of TLR9-MAS. Finally, combined perforin deficiency and excess IL-18 resulted in a spontaneous and fatal hyperinflammatory phenotype responsive to IFNg blockade. These data implicate excess IL-18 as an independent susceptibility factor for hyperinflammation regardless of inciting trigger.
Materials and methods
Murine models and stimuli
All animals were kept in specific pathogen-free conditions under an animal protocol approved by the University of Pittsburgh. Animals originated from Jackson Laboratories with the following exceptions: Il18tg mice were a kind gift of Tomoaki Hoshino (Kurume University), Nlrc4-T337S mice were generated.9 Il18bp−/− mice were obtained from the Knockout Mouse Project.21 Splenocytes from P14 mice were a kind gift from Larry Kane (University of Pittsburgh). CD4Cre-Il18r1fl/fl mice were generated in the laboratory of Giorgio Trinchieri (National Cancer Institute) by R.S. and C.A.S. as follows: 2 LoxP sites were introduced into introns 2 and 3 of the Il18r1 gene, such that Cre-mediated deletion would result in excision of exon 3. A targeting construct was generated containing loxP sites in introns 2 and 3 and a neomycin cassette flanked by flippase recognition target (FRT) sequences in intron 3. The construct was linearized and electroporated into Bruce 4 C57BL/6 ES cells. G418-selected clones were screened for homologous recombination, injected into C57BL/6-albino blastocysts, and transferred to pseudo-pregnant females. Chimeric offspring were bred with C57BL/6-Albino partners to generate F1 mice that were screened for integration by Southern analysis. Resulting mice were crossed with C57BL/6-FLPe mice (Jackson Laboratories) to facilitate recombination between FRT sequences and generate the Il18r1flox allele. Il18r1flox mice were bred to homozygosity and then to CD4Cre-expressing strains. Specificity of deletion was assured by quantitative polymerase chain reaction (qPCR; data not shown) and on an ongoing basis by flow cytometry.
Assessment of systemic hyperinflammation
Complete blood counts were obtained using a HemaVet950 (Drew Scientific). Aspartate transaminase (AST) activity was assessed by colorimetric assay per the manufacturer’s instructions (Sigma Aldrich). Mouse serum cytokines were measured by Cytometric Bead Array (BD).9 An offset of 1/4 the lowest concentration of each analyte’s standard curve was added to each experimental value to minimize floor effects. To measure viral load, splenic tissue was placed in Trizol immediately after euthanasia. cDNA was reverse transcribed (Bio-Rad iScript cDNA Synthesis Kit), and qPCR was performed using LCMV-specific primers27 and normalized to eukaryotic 18s (Taqman, Applied Biosystems).
Antibodies
To deplete CD8- or NK1.1-expressing cells, the following antibodies were injected intraperitoneally on days −3, 0, 3, and 6 of infection: YTS169 (0.5 mg, CD8), PK136 (0.2 mg, NK1.1). Depletion was confirmed by flow cytometry of spleens and/or livers. IFNg was neutralized with XMG1.2 (0.5 mg) on days −1, 3 and 5 after LCMV per Buatois et al,28 or 0.5 mg on day 1 and 0.25 mg every 3 days in Prf1−/−;Il18tg mice.
Flow cytometry, tetramers, assays, and stimulations
Murine tissues were prepared,9 and flow cytometry was performed on a BD LSRII or LSR Fortessa and analyzed using FlowJo v9.9.4 (Treestar). T-distributed Stochastic Neighbor Embedding (tSNE) analysis was carried out using the EXCYT2 program on Matlab R2020a.29 LCMV-Armstrong specific tetramers (GP33 and NP396) were obtained from the National Institutes of Health tetramer core facility. Peptide and phorbol myristate acetate/ionomycin stimulations were performed.30 In vivo antigen presentation assay performed by magnetic isolation of CD8+ T lymphocytes (Mojosort, Biolegend) from uninfected P14 transgenic mice, labeling with carboxyfluorescein diacetate succinimidyl ester (CFSE) per the manufacturer’s instructions (ThermoFischer), and injecting 750 000 cells per recipient mouse on day 7 after LCMV infection. In vivo killing assay was performed on day 7 of LCMV infection31 using Tag-It Violet and CFSE per the manufacturer’s protocol (Biolegend and ThermoFischer).
RNASeq analysis
On day 8 after LCMV-Arm infection, splenic GP33 tetramer + CD8 T lymphocytes were isolated by fluorescence-activated cell sorting directly into lysis/stabilization media. Messenger RNA purification and fragmentation, complementary DNA synthesis, and target amplification were performed with the Smart-Seq v4 ultra-low-input RNA sequencing preparation kit. Pooled complementary DNA libraries were sequenced on the Illumina NextSeq500, mapped to Mus_musculus_ensembl_v80 reference sequence, and quantified using CLC Genomics Workbench software (V8.1.6, Qiagen). Differentially expressed genes were those defined as having a maximum group mean reads per kilobase million > 5, |fold change| > 2, and false discovery rate of P < .05. Fold changes were not calculated directly from reads per kilobase million values but from the generalized linear model, which corrects for differences in library size between the samples and the effects of confounding factors. Gene Set Enrichment Analysis was performed on untrimmed transcripts per million data comparing Prf1−/− vs Il18tg cells using GSEAv4.0.3 with the following parameters: Gene sets database = MSigDB C7.all.V7.1 (Immunologic Signatures); permutations = 1000; permutation type = gene_set. The raw data will be available on the Gene Expression Omnibus at the time of publication. A principal component analysis plot with scaling by covariance was generated on Partek Genomics Suite v7.18.0723 using unfiltered transcripts per million values.
Histology
Blinded necropsy of Prf1−/−Il18tg mice and littermate controls and review of hematoxylin and eosin–stained sections were performed by the University of Pittsburgh Division of Laboratory Animal Resources Veterinary Pathology (E.K.) and selected slide analysis by a pediatric pathologist (J.P.).
Statistical analysis
Analyses were carried out as indicated in the figure legends using GraphPad Prism v8.
Results
LCMV induces hyperinflammation in IL-18 transgenic mice
Profound cytotoxic impairment permits viral persistence and FHL in both mice and humans. Recent evidence suggests that intact cytotoxicity also restricts LCMV-induced T-cell and macrophage activation.32 The role of cytotoxicity in controlling noninfectious hyperinflammation has not been well characterized. Thus, we investigated the effects of complete cytotoxic impairment on TLR9-driven MAS, an innate, infection-free model. Prf1−/− mice showed comparable TLR9-MAS severity to wild type (WT), even with concomitant IL-10 blockade26 (see supplemental Figure 1, available on the Blood Web site), suggesting cytotoxic impairment has a limited role in TLR9-driven hyperinflammation.
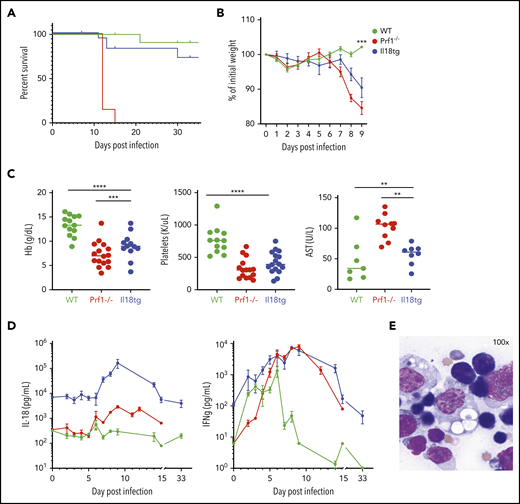
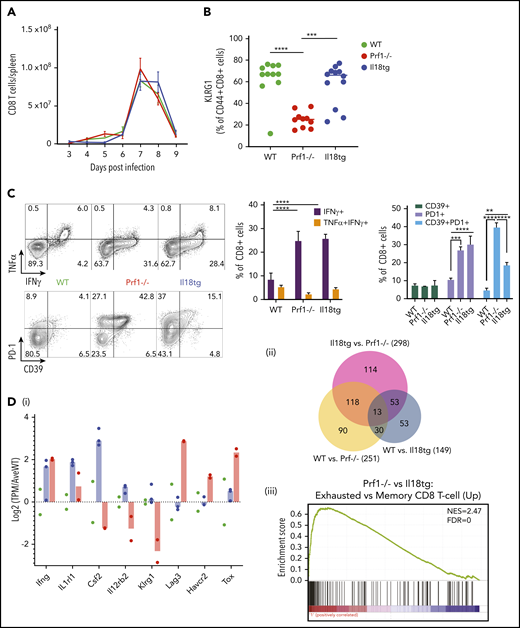
Conversely, excess IL-18 exacerbates TLR9-MAS,9,21 but its role in adaptive immune hyperinflammation has not be assessed. Because IL-18 can promote cytotoxic function,24,33 we hypothesized that (unlike perforin deficiency6 ) excess IL-18 would result in normal or enhanced clearance of LCMV-Armstrong (Arm) infection, and minimal immunopathology. However, we found that Il18tg mice developed hyperinflammation in a similar timeline to Prf1−/− mice, but with less mortality. Specifically, Il18tg mice developed significant cachexia, splenomegaly, cytopenias, transaminitis, and hemophagocytosis (Figure 1; supplemental Figure 2A). As expected, serum IL-18 levels were highest in Il18tg mice, although Prf1−/− and Il18tg mice also developed elevated levels of IFNg and tumor necrosis factor α (TNFα; Figure 1D; supplemental 2B).
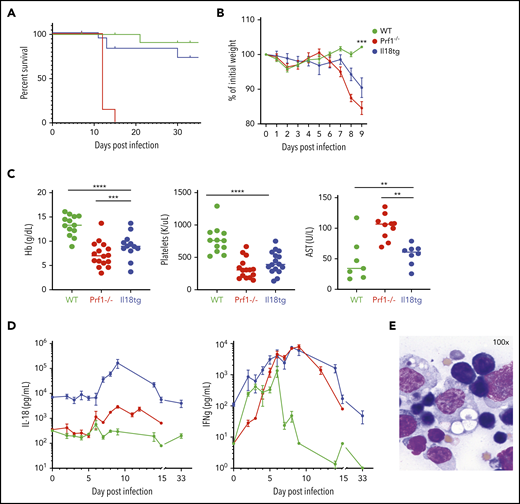
LCMV induces hyperinflammation in Il18tg mice. The indicated genotypes were infected with 200 000 pfu of LCMV Armstrong and assessed for (A) survival and (B) weight (statistical significance for WT vs Il18tg comparison), (C) hemoglobin, platelet count, and serum AST, and (D) serum IFNg and IL-18. (E) Representative splenic touch preparation from Il18tg mouse on day 8 (Wright-Geimsa). Data are composites of at least 3 independent experiments with a minimum of 3 mice per genotype, apart from AST, which is a composite of 2 experiments. Daily cytokine measurements in panel D represent a minimum of 4 mice per genotype. *Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by 1-way analysis of variance (ANOVA) with Tukey post-test on day 8 values. Significance is only shown for comparisons where adjusted P < .05. Error bars represent standard error of the mean (SEM).
LCMV induces hyperinflammation in Il18tg mice. The indicated genotypes were infected with 200 000 pfu of LCMV Armstrong and assessed for (A) survival and (B) weight (statistical significance for WT vs Il18tg comparison), (C) hemoglobin, platelet count, and serum AST, and (D) serum IFNg and IL-18. (E) Representative splenic touch preparation from Il18tg mouse on day 8 (Wright-Geimsa). Data are composites of at least 3 independent experiments with a minimum of 3 mice per genotype, apart from AST, which is a composite of 2 experiments. Daily cytokine measurements in panel D represent a minimum of 4 mice per genotype. *Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by 1-way analysis of variance (ANOVA) with Tukey post-test on day 8 values. Significance is only shown for comparisons where adjusted P < .05. Error bars represent standard error of the mean (SEM).
To assess viral-induced immunopathology in other models of excess IL-18 signaling, we infected mice bearing the Nlrc4T337S gain-of-function inflammasome mutation (who have increased total IL-18 but not free IL-18)9 and Il18bp−/− mice (in whom all mature IL-18 is free). We observed that only mice with combined Nlrc4T337S and IL-18BP deficiency developed viral immunopathology and increased IFNg (supplemental Figure 3). These data confirm the correlation of chronic free IL-18 and severe LCMV-induced immunopathology.
Excess IL-18 acts on T cells to drive an exaggerated, CD8-dependent inflammatory response
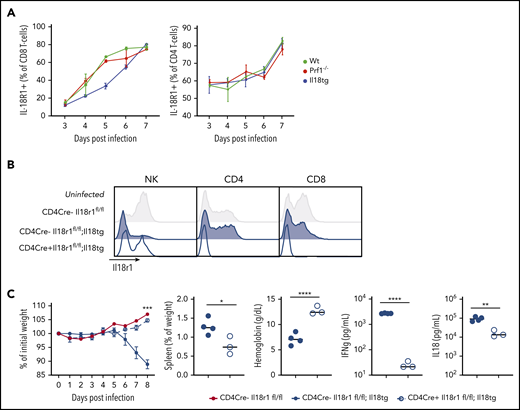
We next evaluated the cellular composition and activation of WT, Il18tg, and Prf1−/− mice spleens after infection of LCMV-Arm. Infected WT, Il18tg, and Prf1−/− mice showed no quantitative difference in T cells or monocytes (supplemental Figure 4). In all genotypes, there were very few NK cells in LCMV-infected spleens relative to T cells (supplemental Figure 4), consistent with previous findings.6 To identify which cells are capable of responding to IL-18, we assessed expression of IL-18R1 in splenocytes. All genotypes dramatically upregulated IL-18R1 on both their CD4 and CD8 T cells to a similar degree during viral infection (Figure 2A). Under homeostatic conditions, IL-18R1 was downregulated in NK and memory T lymphocytes from uninfected Il18tg mice (Figure 2B and data not shown). Because type 1 innate lymphoid and NK cells constitutively express the IL-18 receptor, we assessed their contribution to immunopathology by depleting NK1.1+ cells. NK1.1 depletion of infected Prf1−/− or Il18tg mice resulted in no observed alteration of immunopathology or cytokine production (supplemental Figure 5).
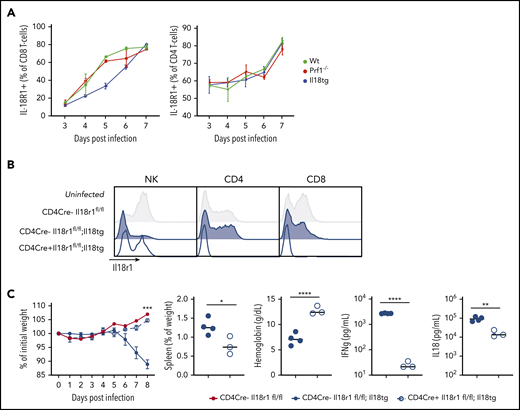
Excess IL-18 acts on expanding T cells to drive virus-induced hyperinflammation. (A) Mice were infected with LCMV-Arm and assessed for IL-18R1 expression on splenic CD8+ and CD4+ T cells. (B) Representative histograms of IL-18R1 expression on splenic NK, CD4+CD44+, and CD8+CD44+ T lymphocytes of uninfected mice of the indicated genotypes. (C) Mice of the indicated genotypes were infected with LCMV-Arm and assessed for weight loss, splenomegaly, anemia, and serum IFNg and IL-18. Dashed horizontal lines represent the median values of infected CD4Cre-Il18r1fl/fl mice. Weight statistical significance compares Il18tg;CD4Cre-Il18r1fl/fl; and Il18tg;CD4Cre+Il18r1fl/fl mice. Data are representative of 2 independent experiments with a minimum of 3 mice per genotype. *Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by unpaired t test on Day 8 values. Significance is only shown for comparisons where adjusted P < .05. Error bars represent SEM.
Excess IL-18 acts on expanding T cells to drive virus-induced hyperinflammation. (A) Mice were infected with LCMV-Arm and assessed for IL-18R1 expression on splenic CD8+ and CD4+ T cells. (B) Representative histograms of IL-18R1 expression on splenic NK, CD4+CD44+, and CD8+CD44+ T lymphocytes of uninfected mice of the indicated genotypes. (C) Mice of the indicated genotypes were infected with LCMV-Arm and assessed for weight loss, splenomegaly, anemia, and serum IFNg and IL-18. Dashed horizontal lines represent the median values of infected CD4Cre-Il18r1fl/fl mice. Weight statistical significance compares Il18tg;CD4Cre-Il18r1fl/fl; and Il18tg;CD4Cre+Il18r1fl/fl mice. Data are representative of 2 independent experiments with a minimum of 3 mice per genotype. *Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by unpaired t test on Day 8 values. Significance is only shown for comparisons where adjusted P < .05. Error bars represent SEM.
Given the dramatic expansion of (especially CD8) T-cells with LCMV infection, and their upregulation of IL-18R1 (Figure 2A), we hypothesized that T-cells were responding to IL-18 to drive immunopathology in this model. To test this, we developed Il18tg mice lacking the IL-18 receptor specifically on T lymphocytes (CD4Cre+;Il18tg;Il18r1flox/flox, Figure 2B). Interestingly, downregulation of the IL-18R1 was less profound in CD4Cre+;Il18tg;Il18r1flox/flox than Il18tg mice, suggesting T-cell responses to IL-18 result in diminished NK-cell IL-18R1 expression under homeostatic conditions. Upon LCMV infection, CD4Cre+;Il18tg;Il18r1flox/flox mice behaved like WT mice, whereas CD4Cre- controls developed immunopathology similarly to Il18tg mice (Figure 2C). Specifically, infected CD4Cre+;Il18tg;Il18r1flox/flox mice, despite retaining elevated serum IL-18, did not lose weight, develop splenomegaly or anemia, or exhibit elevated serum IFNg. These data demonstrate that LCMV-induced hyperinflammation in Il18tg mice is dependent on IL-18 sensing by T lymphocytes.
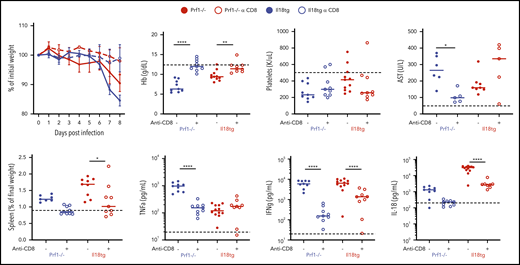
LCMV induces a profound virus-specific effector CD8+ T-cell response, and CD8 depletion rescues Prf1−/− mice from LCMV-induced immunopathology.6 Given the necessity of IL-18 signaling in T cells for immunopathology in Il18tg mice, we specifically depleted CD8 T lymphocytes before and during LCMV infection. We found that CD8 depletion prevented weight loss, splenomegaly, anemia, and IL-18 and IFNg production in both Il18tg and Prf1−/− mice (Figure 3), whereas transaminitis and TNFα production were not improved with CD8 depletion.
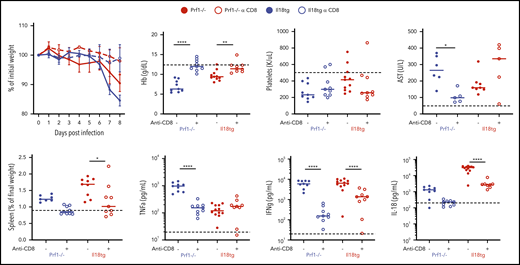
Virus-induced hyperinflammation in Il18tg mice is largely rescued by CD8 T-cell depletion. Mice of the indicated genotypes were treated with CD8-depleting (clone YTS169) or control antibody, infected with LCMV-Arm, and assessed at day 8 for weight loss, spleen weight, hemoglobin (Hb), serum AST, serum TNFα, IFNg, and IL-18. Horizontal dashed lines indicate the median value of uninfected WT control mice. Results are composites of at least 2 independent experiments with a minimum of 3 mice per group.*Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by 1-way ANOVA with Tukey post-test on day 8 values. Significance is only shown for comparisons where adjusted P < .05. Error bars represent SEM.
Virus-induced hyperinflammation in Il18tg mice is largely rescued by CD8 T-cell depletion. Mice of the indicated genotypes were treated with CD8-depleting (clone YTS169) or control antibody, infected with LCMV-Arm, and assessed at day 8 for weight loss, spleen weight, hemoglobin (Hb), serum AST, serum TNFα, IFNg, and IL-18. Horizontal dashed lines indicate the median value of uninfected WT control mice. Results are composites of at least 2 independent experiments with a minimum of 3 mice per group.*Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by 1-way ANOVA with Tukey post-test on day 8 values. Significance is only shown for comparisons where adjusted P < .05. Error bars represent SEM.
Il18tg mice have enhanced effector CD8 T-cell activation and IFNg production
Given the improvement in immunopathology with CD8 depletion, we investigated the activation state of CD8 T cells in infected Il18tg mice. All genotypes had equivalent proliferation and contraction of splenic CD8 T cells (Figure 4A). KLRG1 is upregulated during terminal CD8 T-effector differentiation but can then be downregulated with persistent antigen presentation.34 WT and Il18tg CD8 T cells showed high expression of KLRG1, but Prf1−/− cells were lower (Figure 4B). On ex vivo stimulation with an LCMV-specific peptide (GP33-41), the proportion of CD8 T cells producing IFNg in Il18tg and Prf1−/− mice was increased over WT (Figure 4C). The same pattern was also seen after GP61-80 peptide stimulation of CD4 T cells (data not shown).
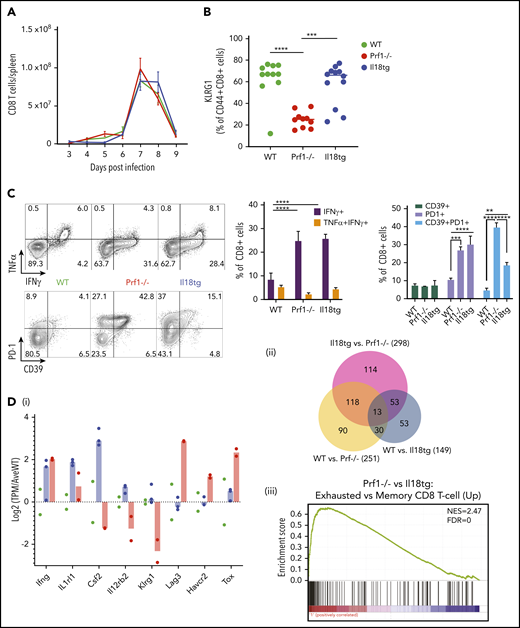
Il18tg mice have enhanced CD8 T-cell activation and IFNg production but gene expression similar to WT mice. Mice of the indicated genotypes were infected with LCMV-Arm and assessed for (A) absolute number of splenic CD8+ T lymphocytes. On day 8 of infection, mice were further evaluated for (B) the percentage of splenic CD8+ T lymphocytes expressing KLRG1+, (C) cytokine expression after stimulation with GP33 peptide, and the percentage of effector CD8 T lymphocytes expressing inhibitory markers. *Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by 1-way ANOVA with Tukey post-test. Significance is only shown for comparisons where adjusted P < .05. Error bars represent SEM. Results are representative of at least 2 independent experiments with a minimum of 3 mice per genotype. (D) RNASeq analysis of GP33-antigen specific CD8 T lymphocytes including (i) relative expression of selected genes (normalized to the average expression of WT), (ii) Venn diagram of differentially expressed genes, and (iii) Gene Set Enrichment Analysis (GSEA) of the Prf1−/− vs Il18tg comparison showing the most highly enriched MSigDb C7 (immunologic signature) CD8 T-cell upregulation gene set (from GSE9650). See supplemental Data for complete differential expression and GSEA data. FDR, false discovery rate; NES, normalized enrichment score.
Il18tg mice have enhanced CD8 T-cell activation and IFNg production but gene expression similar to WT mice. Mice of the indicated genotypes were infected with LCMV-Arm and assessed for (A) absolute number of splenic CD8+ T lymphocytes. On day 8 of infection, mice were further evaluated for (B) the percentage of splenic CD8+ T lymphocytes expressing KLRG1+, (C) cytokine expression after stimulation with GP33 peptide, and the percentage of effector CD8 T lymphocytes expressing inhibitory markers. *Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by 1-way ANOVA with Tukey post-test. Significance is only shown for comparisons where adjusted P < .05. Error bars represent SEM. Results are representative of at least 2 independent experiments with a minimum of 3 mice per genotype. (D) RNASeq analysis of GP33-antigen specific CD8 T lymphocytes including (i) relative expression of selected genes (normalized to the average expression of WT), (ii) Venn diagram of differentially expressed genes, and (iii) Gene Set Enrichment Analysis (GSEA) of the Prf1−/− vs Il18tg comparison showing the most highly enriched MSigDb C7 (immunologic signature) CD8 T-cell upregulation gene set (from GSE9650). See supplemental Data for complete differential expression and GSEA data. FDR, false discovery rate; NES, normalized enrichment score.
We hypothesized that excess IL-18 may promote immunopathology by preventing the upregulation of inhibitory receptors. However, we saw similar upregulation of PD-1 and more modest upregulation of CD39 in both Il18tg and Prf1−/− mice at day 8 (Figure 4C). Unlike LCMV-infected Prf1−/− mice, most Il18tg mice did not succumb to immunopathology when followed to 30+ days after infection (Figure 1A; supplemental Figure 6A). At more than 30 days after infection, Il18tg mice had fewer LCMV-specific CD8 T cells of both effector- and central-memory phenotypes than WT mice (supplemental Figure 6B). Nevertheless, these LCMV-specific CD8 T cells showed higher expression of activation markers like PD-1 (supplemental Figure 6C). Neither Il18tg nor WT mice reinfected with LCMV more than 30 days after initial infection showed weight loss or other evidence of inflammation, consistent with development of neutralizing immunity (data not shown).
To further characterize the nature of CD8 T-cell activation in Il18tg mice, we performed RNA sequencing of GP33-antigen specific CD8 T cells from LCMV-infected WT, Prf1−/−, and Il18tg mice on day 8 after infection. We saw expression of Ifng was elevated about fourfold in both Prf1−/− and Il18tg mice. Genes specifically upregulated in Il18tg CD8 T-cells included Csf2, Il1rl1, and Il12rb2 (encoding GM-CSF, ST2 subunit of the IL-33 receptor, and IL-12 receptor, respectively; Figure 4; supplemental Figure 7). However, we observed the greatest differences between Prf1−/− and either WT or Il18tg mice, with relatively few differentially expressed genes between WT and Il18tg CD8 T cells. These differences between Prf1−/− and WT/Il18tg CD8 T cells included Klrg1 and transcripts associated with CD8 T-cell exhaustion, including the inhibitory receptors Lag3, Havcr2 (Tim-3), and Tigit and the transcription factors Tox and Prdm1 (Figure 4D; supplemental Figure 7; supplemental Data). Overall, these data suggest CD8 T-cell hyperactivation in Il18tg mice, persisting to memory time points, but not indicative of chronic antigen stimulation or exhaustion.
LCMV-induced hyperinflammation is not associated with delayed viral clearance, impaired cytotoxicity, or persistent antigen presentation
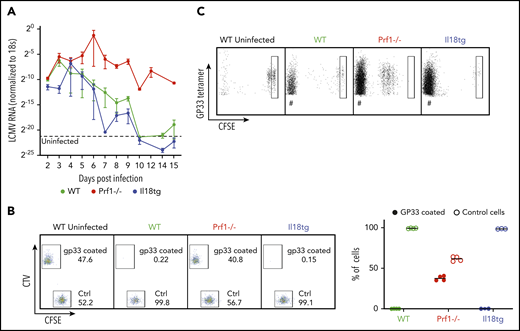
Cytotoxic impairment promotes hyperinflammation via persistent antigen presentation (because of prolonged viremia) and prolongation of individual immune synapses, both greatly enhancing T-cell cytokine production.3,4,6 We postulated that excess IL-18, potentially through chronic exposure, may also cause hyperinflammation through an acquired cytotoxic defect. However, Il18tg mice cleared LCMV in a kinetic comparable to WT mice (Figure 5A). Using a more sensitive assessment of cytotoxic function (an in vivo killing assay35 ) we found complete and specific clearance of target cells coated with LCMV-specific GP33-41 peptide by WT and Il18tg mice but persistence of these cells in Prf1−/− mice (Figure 5B). It remained possible that excess IL-18 nevertheless caused persistent antigen presentation independently of cytotoxic function. To test this, we transferred CFSE-labeled LCMV-specific CD8 (P14) T cells into mice 7 days after infection and evaluated the survival and division of transferred cells on day 10. Only in infected Prf1−/− mice did we observe persistence and proliferation of transferred T cells (Figure 5C). Together, these data suggest that virus-triggered immunopathology in Il18tg mice occurs via autoinflammation rather than persistent antigen-mediated activation.
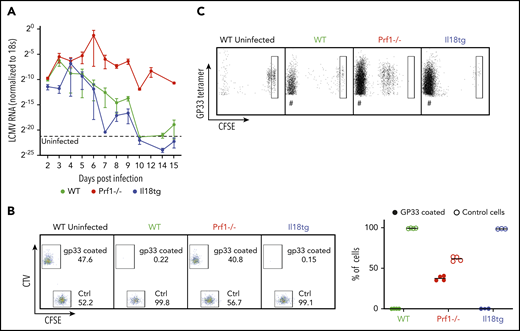
Virus-induced hyperinflammation in Il18tg mice is not associated with immunodeficiency or prolonged antigen presentation. After LCMV-Arm infection, mice of the indicated genotypes were assessed for (A) the presence of LCMV RNA in the spleen by qPCR. (B) In vivo killing assay: WT splenocytes loaded with GP33 peptide were labeled with TagIT Violet, and control WT splenocytes were labeled with CFSE. Equal proportions of both were then adoptively transferred into mice of the indicated genotypes on day 8 of LCMV-Arm infection and remaining cells were assessed 24 hours later in recipient spleens. (C) Equal numbers of CFSE-labeled P14 CD8 T cells were transferred into mice of the indicated genotypes on day 7 of infection. Spleens were assessed 72 hours later for the persistence/proliferation of transferred cells. Boxes indicate CFSE fluorescence of undivided cells. # indicates non-CFSE labeled host GP33 tetramer + CD8 T cells.
Virus-induced hyperinflammation in Il18tg mice is not associated with immunodeficiency or prolonged antigen presentation. After LCMV-Arm infection, mice of the indicated genotypes were assessed for (A) the presence of LCMV RNA in the spleen by qPCR. (B) In vivo killing assay: WT splenocytes loaded with GP33 peptide were labeled with TagIT Violet, and control WT splenocytes were labeled with CFSE. Equal proportions of both were then adoptively transferred into mice of the indicated genotypes on day 8 of LCMV-Arm infection and remaining cells were assessed 24 hours later in recipient spleens. (C) Equal numbers of CFSE-labeled P14 CD8 T cells were transferred into mice of the indicated genotypes on day 7 of infection. Spleens were assessed 72 hours later for the persistence/proliferation of transferred cells. Boxes indicate CFSE fluorescence of undivided cells. # indicates non-CFSE labeled host GP33 tetramer + CD8 T cells.
LCMV-induced hyperinflammation in Il18tg mice is partially IFNg dependent
IFNg contributes to the pathogenesis of both TLR9- and LCMV-induced immunopathology,6,26 and IFNg blockade is a viable therapeutic strategy in MAS36 (NCT03311854), HLH,10 and severe coronavirus disease 2019 (NCT04324021). Nevertheless, in certain settings, hyperinflammation proceeds in the absence of IFNg, and in other settings, IFNg activity may even be protective.37-40 Il18tg mice given IFNg-neutralizing antibody and infected with LCMV were largely prevented from developing immunopathology, with improvement in splenomegaly, anemia, and IL-18 and promising trends in transaminitis and other cytokinemia (supplemental Figure 8). Although protection from immunopathology was perhaps not as complete in Il18tg mice as Prf1−/− mice, these data suggest that IL-18 amplifies the canonical CD8 T-cell/IFNg-driven response to LCMV.
Combined cytotoxic impairment and excess IL-18 drive spontaneous hyperinflammation
Host susceptibility to hyperinflammation is multifactorial. For example, heterozygous/monoallelic variants in FHL-associated genes (ie, those that cause FHL when homozygous/biallelic) are enriched in SJIA-MAS patients.13,14 Meanwhile, nearly all MAS patients have extremely elevated serum IL-18.9,18 If acting by independent mechanisms, we hypothesized that excess IL-18 and cytotoxic impairment would demonstrate at least additive effects on LCMV-induced immunopathology. To test this, we generated Prf1−/−;Il18tg mice with the intention of assessing their responses to LCMV. We were unable to ethically infect Prf1−/−;Il18tg mice because they spontaneously developed lethal immunopathology. They demonstrated cachexia, splenomegaly, hepatitis, and cytokinemia indicative of hyperinflammation in the absence of an exogenous trigger, dying at around 7 weeks of age (Figure 6A-C). Compared with Prf1−/− and Il18tg controls, 4-week-old Prf1−/−;Il18tg mice were found to have severe splenic and hepatic extramedullary hematopoiesis, obscuring all normal splenic architecture (data not shown). Liver histology revealed marked hepatic sinusoidal distension with modest hepatitis (supplemental Figure 9A). Histologic analyses of brain, lung, skin, bone marrow, heart, and small and large intestine showed no remarkable differences (data not shown). Notably, even Prf1+/−;Il18tg mice developed an intermediate hyperinflammatory phenotype across all parameters (Figure 6) but did not show appreciably more severe LCMV-induced immunopathology than Il18tg mice (data not shown).
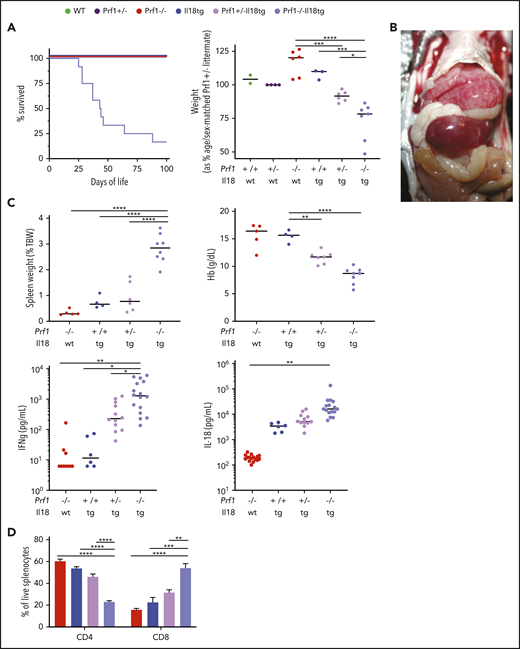
Combined cytotoxic impairment and excess IL-18 leads to spontaneous hyperinflammation. Mice of the indicated genotypes were bred and assessed for (A) survival and body weight. Prf1−/−, Prf1+/−, Prf1+/−;Il18tg, and Prf1−/−Il18tg littermates were compared with littermate sex-matched controls. Similar to published data,3,7 naïve Prf1−/− and WT mice demonstrate no difference in spleen weight, Hb, serum IFNg, or IL-18 (data not shown). WT and Il18tg mice were littermates, and their weights were compared with age- and sex-matched Prf+/− controls. (B) hepatosplenomegaly in a 5-week-old Prf1−/−Il18tg mouse. (C) Spleen weight, Hb, serum IFNg, and IL-18. (D) percentage of splenic T cells in the indicated genotypes. *Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by 1-way ANOVA with Tukey post-test. Significance is only shown for comparisons where adjusted P < .05. Error bars represent SEM. Results are a composite of 2 independent experiments (weight, spleen weight, and Hb) and 3 independent experiments (serum cytokine measurements).
Combined cytotoxic impairment and excess IL-18 leads to spontaneous hyperinflammation. Mice of the indicated genotypes were bred and assessed for (A) survival and body weight. Prf1−/−, Prf1+/−, Prf1+/−;Il18tg, and Prf1−/−Il18tg littermates were compared with littermate sex-matched controls. Similar to published data,3,7 naïve Prf1−/− and WT mice demonstrate no difference in spleen weight, Hb, serum IFNg, or IL-18 (data not shown). WT and Il18tg mice were littermates, and their weights were compared with age- and sex-matched Prf+/− controls. (B) hepatosplenomegaly in a 5-week-old Prf1−/−Il18tg mouse. (C) Spleen weight, Hb, serum IFNg, and IL-18. (D) percentage of splenic T cells in the indicated genotypes. *Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by 1-way ANOVA with Tukey post-test. Significance is only shown for comparisons where adjusted P < .05. Error bars represent SEM. Results are a composite of 2 independent experiments (weight, spleen weight, and Hb) and 3 independent experiments (serum cytokine measurements).
Reminiscent of CD8 expansion in LCMV-infected Il18tg mice, spleens and livers of Prf1−/−;Il18tg mice showed spontaneous increases in CD8 T cells (Figure 6D; data not shown). Splenic CD8 T cells from Prf1−/−;Il18tg mice demonstrated evidence of profound T-effector activation, including expression of inhibitory receptors and production of IFNg (Figures 7A-B). Given this spontaneous hyperinflammation, we wondered to what extent this phenotype may also be IFNg dependent. After 14 days of IFNg neutralization, adult Prf1−/−;Il18tg mice showed improved weight gain, anemia, and thrombocytopenia compared with isotype-treated controls (Figure 7C). Quantitatively, there were no differences in splenic and hepatic T cells, but there was a small increase in peripheral and splenic neutrophils (supplemental Figure 9B). These data suggest the combination of excess IL-18 and cytotoxic impairment results in spontaneous systemic and cellular hyperinflammation consistent with that seen in experimental HLH and MAS.
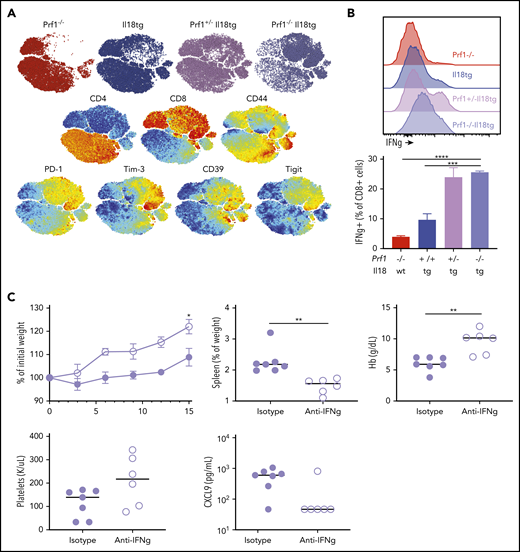
Combined cytotoxic impairment and excess IL-18 leads to spontaneous CD8 T-cell activation and IFNg-dependent hyperinflammation. (A) tSNE plot of live splenic T cells from the indicated genotypes. (B) IFNγ production by CD8+ T cells following ex vivo stimulation. (C) Five-week-old Prf1−/−;Il18tg mice were treated with IFNg-neutralizing (clone XMG1.2) or control antibody for 2 weeks and then assessed for weight gain, spleen weight, Hb, platelet count, and serum CXCL9. *Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by (B) 1-way ANOVA with Tukey post-test and (C) unpaired t test. Significance is only shown for comparisons where adjusted P < .05. Error bars represent SEM. Results are composites of 2 independent experiments with a minimum of 3 mice per genotype.
Combined cytotoxic impairment and excess IL-18 leads to spontaneous CD8 T-cell activation and IFNg-dependent hyperinflammation. (A) tSNE plot of live splenic T cells from the indicated genotypes. (B) IFNγ production by CD8+ T cells following ex vivo stimulation. (C) Five-week-old Prf1−/−;Il18tg mice were treated with IFNg-neutralizing (clone XMG1.2) or control antibody for 2 weeks and then assessed for weight gain, spleen weight, Hb, platelet count, and serum CXCL9. *Adjusted P < .05, **P < .01, ***P < .001, ***P < .0001 by (B) 1-way ANOVA with Tukey post-test and (C) unpaired t test. Significance is only shown for comparisons where adjusted P < .05. Error bars represent SEM. Results are composites of 2 independent experiments with a minimum of 3 mice per genotype.
Discussion
Over the last 20 years, studying genetic cytotoxic impairments has propelled research on HLH susceptibility.41 These studies have yielded clinical genetic testing panels, a variety of functional tests, iterative clinical trials, and successful drug development. Nevertheless, only a minority of hyperinflammatory patients have even partial genetic defects in cytotoxicity. Meanwhile, the evidence connecting IL-18 to MAS is substantial and grows stronger as more patients’ sera are measured and uniformly show dramatic elevation of total IL-18 and detectable free IL-18.9,18,42 IL-18 is also an independent predictor of mortality in severe sepsis,43 which is increasingly identified to have hyperinflammatory features.44,45 More recent genetic discoveries have ushered IL-18 from biomarker to upstream driver: the association of hyperinflammation and excess IL-18 in NLRC4-MAS was followed by similar associations in XIAP deficiency,46 C-terminal mutations in CDC42,47,48 and possibly IL-18BP deficiency.49 Therapeutically, a few such patients responded to IL-18 or IFNg blockade.48,50 Recombinant IL-18BP showed promise in a small study of Stills disease and in 2 children with MAS,18,50 and more definitive trials are ongoing (NCT03113760). These data suggest a family of IL-18opathies, diseases associated with and caused by chronic excess of IL-18, and corroborate the links between IL-18 and MAS.
Acutely, IL-18 stimulation promotes lymphocyte cytotoxicity and cytokine production. However, SJIA patients often have chronic IL-18 elevation, and their NK cells are deficient in both number and function, specifically lacking responsiveness to IL-18.23,24,51 Thus, there remain dangerous gaps in our understanding of host susceptibility to hyperinflammation, including how IL-18 might confer such susceptibility. We first explored how these putative host susceptibility factors, perforin deficiency, and excess IL-18 might interact with different exogenous triggers. We found that excess IL-18 worsened immunopathology following both innate (TLR9) and adaptive (LCMV) stimulation, whereas perforin deficiency had no effect on innate stimulation. Subsequently, we found that excess IL-18 drove virus-induced hyperinflammation via amplification of T-cell responses and not through an acquired cytotoxic impairment or persistent antigen presentation. Together, these data support excess IL-18 as an autoinflammatory (as opposed to autoimmune or immunodeficient) pathway to hyperinflammation.
In WT mice, LCMV elicits an antigen-specific response characterized by CD8 T-cell expansion and IFNg production.52 Using both genetic and depletion strategies, we observe amplification of CD8 T-cell responses and IFNg overproduction as the main mechanisms by which IL-18 drives LCMV-induced hyperinflammation. This suggests that IL-18 acts by amplifying existing responses rather than recruiting new pathways. We were surprised that excess IL-18 promoted immunopathology (both systemically and in individual CD8 T-cells) consistent with hyperinflammation, albeit with minimal fatality. CD8 T-cells from both Il18tg and Prf1−/− mice showed similar upregulation of some T-cell inhibitory receptors (PD-1, CD39, etc). These cells nevertheless overproduced IFNg, suggesting cytotoxic impairment and excess IL-18 are individually capable of overwhelming the effects of this negative regulation. However, the downregulation of KLRG134 and upregulation of an exhaustion transcriptional program were unique to only Prf1−/− cells, implying chronic antigen presentation was not a feature of the T-cell hyperactivation in Il18tg mice. Overall, these data indicate that both excess IL-18 and cytotoxic impairment, albeit by different paths, affect the amplitude of the inflammatory response to infection.
How IL-18 may affect CD8- or IFNg-independent hyperinflammatory triggers, those that recruit other effector populations (like neutrophils), remains unclear. Humans incapable of responding to IFNg have been reported to develop hyperinflammation,40 and various murine systems including genetic IFNg deficiency, IL-10 blockade, or memory T-cell induction demonstrate IFNg-independent pathways to hyperinflammation.30,37,38 These may proceed via other mediators like IL-6, TNFα, or, as observed in our RNAseq dataset, granulocyte-macrophage colony-stimulating factor (Csf2).30,38 Additionally, IL-18 induces both inflammation and immunodeficiency in neonatal bacterial peritonitis by amplifying an IL-1/IL-17–dependent pathway.53 TLR9 stimulation and LCMV infection are known to elicit type 1 responses and may not represent the gamut of relevant hyperinflammatory triggers. Nevertheless, our data bolster the primary role of type 1 inflammation in both FHL and IL-18–driven hyperinflammation. Likewise, the evidence that type 1 inflammatory responses predominate in human hyperinflammation is substantial: many of the hyperinflammatory disease activity biomarkers in clinical use (eg, neopterin and CXCL9) are downstream of IFNg, and IFNg is a promising target in hyperinflammation beyond FHL. Ongoing clinical trials of IFNg neutralization in both HLH and MAS will help establish the degree to which IFNg is the defining effector cytokine driving hyperinflammation.
Finally, we observed spontaneous hyperinflammation in mice with both perforin deficiency and excess IL-18. These mice bore the hallmarks of hyperinflammation (cachexia, cytopenias, splenomegaly, CD8 T-cell expansion/hyperactivation, IFNg overproduction) in the absence of a specific trigger. This synergy between cytotoxic impairment and IL-18 excess was observed even in perforin haplosufficient mice. As with LCMV-induced disease, spontaneous hyperinflammation in Il18tg;Prf1−/− mice responded to IFNg blockade. Monoallelic, FHL-associated variants in cytotoxicity genes are common in the healthy population. However, their enrichment in MAS and virus-induced HLH may clinically illustrate the dangerous interaction between (even mild) cytotoxic impairment and excess IL-18.13-15,31,54
Hyperinflammatory syndromes remain challenging to categorize and manage. Our findings suggest hyperinflammation results from the summation of dynamic and coexisting inputs, both environmental and endogenous. Among many potential host susceptibility factors, human genetic and biomarker/functional work most strongly implicates cytotoxic impairment and excess IL-18. Our findings suggest these mechanisms make independent and synergistic contributions, and they operate differently based on the type of exogenous stimulus. Future work will uncover novel host and environmental drivers of hyperinflammation and will determine whether these drivers necessarily converge on the ill effects of IFNg. Meanwhile, clinicians must quickly identify these shifting inputs, integrate their contributions, and intervene rationally to prevent the grave effects of unchecked hyperinflammation.
RNA sequencing is available at https://www.ncbi.nlm.nih.gov/geo/, accession #GSE150077.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Larry Kane and Lyndsay Avery for assistance with LCMV Armstrong preparation and for cells from P14 transgenic mice; Giorgio Trinchieri and Tomoaki Hoshino for sharing the Il18r1fl/fl mice and Il18tg mice, respectively; the National Institutes of Health tetramer core facility; and Brian Campfield, Timothy Hand, and Amanda Poholek for advice and critical review.
This work was supported by the Intramural Program of the National Institutes of Health (NIH), National Cancer Institute (R.S., C.A.S.), the NIH, Eunice Kennedy Shriver National Institute of Child Health and Human Development grant R01HD098428 (S.W.C.), the NIH, National Institute of Allergy and Infectious Diseases grants K22AI123366 (S.W.C.) and T32AI089443 (L.V.D.K.), the Rheumatology Research Foundation (S.W.C.), and the Richard King Mellon Institute for Pediatric Research.
Authorship
Contribution: P.T., E.R., and S.W.C. planned the experiments; R.S. and C.A.S. generated key reagents; P.T., E.R., E.S.W., C.S., V.D., and L.V.D.K. performed experiments; J.P. and E.K. conducted the pathology/histology; P.T. and S.W.C. analyzed data; P.T. and S.W.C. wrote the manuscript; and all authors reviewed and approved the submitted manuscript.
Conflict-of-interest disclosure: S.W.C. has a consultancy with AB2Bio Ltd. and Novartis. The remaining authors declare no competing financial interests.
Correspondence: Scott W. Canna, Children’s Hospital of Pittsburgh of UPMC, 8124 Rangos Research Bldg, 4401 Penn Ave, Pittsburgh, PA 15224; e-mail: scott.canna@chp.edu.