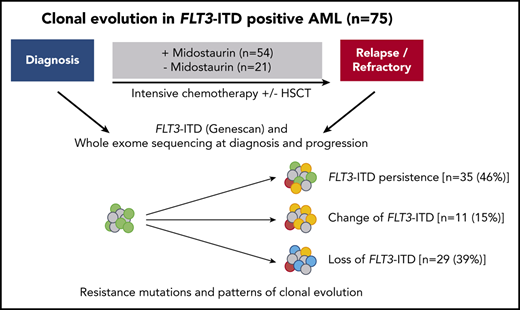
Key Points
At the time of disease resistance or progression, ∼50% of patients treated with midostaurin become FLT3-ITD negative.
FLT3-ITD persistence can be caused by selection of resistant FLT3 clones, mechanisms bypassing FLT3 inhibition, or insufficient drug activity.

Abstract
In the international randomized phase 3 RATIFY (Randomized AML Trial In FLT3 in patients less than 60 Years old) trial, the multikinase inhibitor midostaurin significantly improved overall and event-free survival in patients 18 to 59 years of age with FLT3-mutated acute myeloid leukemia (AML). However, only 59% of patients in the midostaurin arm achieved protocol-specified complete remission (CR), and almost half of patients achieving CR relapsed. To explore underlying mechanisms of resistance, we studied patterns of clonal evolution in patients with FLT3-internal tandem duplications (ITD)-positive AML who were entered in the RATIFY or German-Austrian Acute Myeloid Leukemia Study Group 16-10 trial and received treatment with midostaurin. To this end, paired samples from 54 patients obtained at time of diagnosis and at time of either relapsed or refractory disease were analyzed using conventional Genescan-based testing for FLT3-ITD and whole exome sequencing. At the time of disease resistance or progression, almost half of the patients (46%) became FLT3-ITD negative but acquired mutations in signaling pathways (eg, MAPK), thereby providing a new proliferative advantage. In cases with FLT3-ITD persistence, the selection of resistant ITD clones was found in 11% as potential drivers of disease. In 32% of cases, no FLT3-ITD mutational change was observed, suggesting either resistance mechanisms bypassing FLT3 inhibition or loss of midostaurin inhibitory activity because of inadequate drug levels. In summary, our study provides novel insights into the clonal evolution and resistance mechanisms of FLT3-ITD–mutated AML under treatment with midostaurin in combination with intensive chemotherapy.
Introduction
Internal tandem duplications (ITD) in the fms related tyrosine kinase 3 (FLT3) gene are found in approximately 25% of younger adult patients with acute myeloid leukemia (AML).1 Multiple studies reported an inferior outcome for FLT3-ITD–positive AML,2-4 in particular in patients with a high mutation to wild-type (WT) allelic ratio (AR)2,5 and patients with WT nucleophosmin 1 (NPM1).6 Furthermore, the FLT3-ITD insertion site in the beta1-sheet of the tyrosine kinase domain-1 (TKD1) of the FLT3 receptor has been shown to be associated with an unfavorable prognosis in FLT3-ITD–positive AML.7
Activating mutations in FLT3 can be targeted with small-molecule tyrosine kinase inhibitors (TKIs) and several compounds are currently under investigation in clinical trials.8 Midostaurin, a first-generation TKI, targets not only FLT3 but multiple kinases including protein kinase C, platelet-derived growth factor receptors α and β, cyclin-dependent kinase 1, and KIT proto-oncogene receptor tyrosine kinase.9,10 Demonstrating some activity as single agent,11,12 the addition of midostaurin to intensive chemotherapy significantly improved overall survival and event-free survival compared with placebo in the phase 3 RATIFY (Randomized AML Trial In FLT3 in patients less than 60 Years old; Cancer and Leukemia Group B [CALGB]10603) trial.13 However, even in the midostaurin treatment arm, only 59% of patients achieved a complete remission (CR), and relapses occurred in more than 40% of patients. These data are in line with our recently performed phase 2 trial German-Austrian Acute Myeloid Leukemia Study Group (AMLSG) 16-10 (NCT014776060) to determine whether the addition of midostaurin to intensive chemotherapy followed by allogeneic hematopoietic cell transplantation (HSCT) and single-agent maintenance therapy of 12 months is feasible and impacts on outcome.14 Event-free survival and overall survival at 2 years were 39% and 53% in younger patients, respectively.
Failure of TKI treatment can be caused by primary and secondary resistance mutations, such as point mutations at positions D835, F691, and N676 in the FLT3 gene.15,16 In addition to on-target resistance mutations in the FLT3 receptor, FLT3-independent mechanisms of resistance can occur such as the dysregulation of antiapoptotic proteins MCL1,17 BIRC5,18 and BCL219 or the activation of compensatory signaling pathways.20
In general, accumulation of genetic alterations is a common mechanism in the clonal evolution of AML.21-23 Although preleukemic founder mutations in epigenetic regulating genes like DNA methyltransferase 3A (DNMT3A), tet oncogene family member 2 (TET2), and additional sex combs-like 1 (ASXL1) do often persist during remission and disease progression,24-26 secondary mutations affecting signaling and transcription related genes are less stable at disease progression.25,27 In accordance, relapse can either be driven by the acquisition of mutations in the founder clone or by the selection of resistant subclones during chemotherapy.21
Whole exome sequencing (WES) of FLT3-ITD–positive AML emphasized the interaction of FLT3-ITD with other genetic drivers at disease progression under conventional treatment,23 but between 13% and 25% also lose the ITD at relapse.23,26-28 Studies evaluating the impact of TKIs on clonal evolution showed resistant FLT3 mutations and the accumulation of FLT3-independent mutations after therapy with quizartinib, crenolanib, and gilteritinib.29-31 This supports the hypothesis that, besides site-specific mutations, the expansion of resistant clones might play an important role for the development of TKI resistance in AML.
In our study, we comprehensively investigated the clonal evolution of FLT3-ITD–positive AML in patients with relapsed and refractory disease who were treated with intensive chemotherapy in combination with the multikinase inhibitor midostaurin. To elucidate the molecular basis of therapy resistance, we performed WES of samples collected at the time of diagnosis, in CR, and at disease progression. In particular, we were interested to determine how FLT3 mutations and FLT3-independent mutations contributed to disease resistance or progression and to follow-up the clonal evolution mutation pattern during the course of treatment.
Methods
Patients and treatment
We performed Genescan-based fragment-length analysis for characterization of FLT3-ITD and WES in 75 patients that were FLT3-ITD positive, who were enrolled within the RATIFY13 (NCT00651261; n = 25), AMLSG 16-1014 (NCT01477606; n = 41), AMLSG 07-0432 (NCT00151242; n = 7), and AMLSG 11-0833 (NCT00850382; n = 2) trials and received intensive chemotherapy-based treatment in combination with midostaurin (n = 54) or without midostaurin (n = 21). All patients gave informed consent according to the Declaration of Helsinki.
Of 54 patients treated with midostaurin, 19 (35%) had primary refractory disease and 35 (65%) achieved a CR after induction chemotherapy (first cycle, n = 25; second cycle, n = 10). Consolidation consisted of up to 4 cycles of high-dose cytarabine (n = 26) or allogeneic HSCT (n = 9). A total of 23 patients entered midostaurin maintenance and 7 patients completed 12 cycles before disease progression. The main reasons for no midostaurin maintenance in 12 patients were relapse during consolidation or toxicity. The control group consisted of a total of 21 relapsed patients, of which 12 were treated within the RATIFY trial in the placebo arm with intensive chemotherapy only and 9 relapsed patients from the AMLSG 07-04 and 11-08 trials. Further details on the treatment regimens and pretreatment cytogenetics are given in supplemental Table 1, available on the Blood Web site. Clinical characteristics of midostaurin-treated and control group patients were comparable (Table 1). In the group of midostaurin-treated patients who relapsed, more patients were male (24 of 35; 69%) compared with patients with refractory disease (6 of 19; 32%; P = .01) and compared with the control group (7 of 21; 33%; P = .01; supplemental Table 2).
Characterization of FLT3-ITD at diagnosis and disease progression
At the time of diagnosis and at disease progression (relapse or refractory disease), FLT3-ITDs were analyzed using genomic DNA (gDNA)-based polymerase chain reaction derived from available bone marrow (BM; n = 130) or peripheral blood (PB; n = 20) samples followed by capillary electrophoresis (Genescan-based fragment-length analysis) as previously described.13 FLT3-ITDs were characterized with regard to the AR, the number of ITD clones, the nucleotide (nt) length of FLT3-ITD, and the ITD insertion sites according to functional regions of the FLT3 receptor (supplemental Methods).7
WES in paired diagnosis/disease progression samples
WES was performed in paired samples of n = 75 patients using the Nextera Rapid Capture Exome Kit (Illumina) for library preparation (input, 50 ng gDNA). Library preparation was followed by sequencing on an Illumina HiSeq 2000 platform using the 200-cycle TruSeq SBS v3 kit (Illumina) according to the manufacturer’s instructions. In relapsed patients, diagnostic and relapse samples were compared with samples collected at CR (BM, n = 8; PB, n = 48). In patients with refractory disease, samples collected at diagnosis and refractory disease were compared with CD3+-sorted T cells from PB (n = 17) and BM (n = 2) samples obtained at diagnosis (supplemental Methods). WES data analysis and visualization of the clonal evolution with fish plots was performed using an inhouse pipeline as described in the supplemental Methods.
Validation of recurrent gene mutations
FLT3-TKD and NPM1 mutations were determined in all diagnostic samples enrolled by the AMLSG within our standard workup using established methods.34 In patients with the NPM1mutation, NPM1 transcript levels were determined for monitoring of measurable residual disease at diagnosis and disease progression using a sensitive quantitative real-time polymerase chain reaction.35 Furthermore, we validated the presence of 42 AML-associated gene mutations (supplemental Table 6) in selected samples with ultra-deep targeted resequencing on an Illumina MiSeq using a custom-designed HaloPlexHS panel (Agilent; supplemental Methods).
Pathway analysis
To determine pathways that might be affected during clonal evolution, we performed pathway analysis using gene sets from the Molecular Signatures Database (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). We compared mutations between diagnosis and relapse/refractory disease in all patients with respect to midostaurin treatment and FLT3-ITD status (+/−) at the time of disease progression.
Plasma inhibitory activity assay
Plasma inhibitory activity (PIA)36 was determined in serum samples collected at the time of diagnosis, after first induction and at the time of relapse/refractory disease. Data were available for n = 12 patients (n = 7 relapsed and n = 5 refractory), of which n = 6 were FLT3-ITD positive and n = 6 were FLT3-ITD negative at relapse/refractory disease. Further details are given in the supplemental Material.
Results
Molecular characterization of FLT3-ITD clones at diagnosis and disease progression in patients treated with midostaurin
The first aim of our study was to determine molecular characteristics based on conventional Genescan analysis of FLT3-ITDs at diagnosis and disease progression that might associate with resistance to treatment. Thus, we compared FLT3-ITD characteristics between diagnosis and relapsed/refractory disease in 54 patients treated with midostaurin and compared findings to the control group.
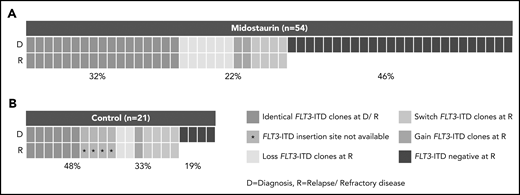
In patients treated with midostaurin, more patients had >1 FLT3-ITD at diagnosis than at disease progression (15 of 54; 28% vs 5 of 54; 9%; P = .02), whereas FLT3-ITD AR, length, and insertion sites were comparable between both time points (Table 2). However, we observed that FLT3-ITD mutation status became negative at the time of disease progression in almost 50% of the patients (25 of 54; 46%; Figure 1A; Table 2), especially in patients who entered midostaurin maintenance and later relapsed (15 of 23; 65% vs 2 of 12; 17%; P = .01).
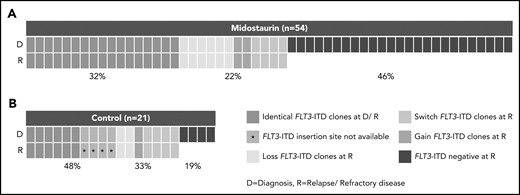
Presence of FLT3-ITD clones at diagnosis and disease progression based on a Genescan-based routine diagnostic assay. Each column represents 1 patient. (A) Presence of FLT3-ITD clones at diagnosis (D) and relapse/refractory disease (R) in patients treated with midostaurin. (B) Presence of FLT3-ITD clones between D and R in 21 intensively treated patients without the addition of midostaurin. Green indicates that the same ITDs were detected at both time points. A change of ITDs between D and R is color-coded in orange (light, loss of ITDs at progression with at least 1 persistent FLT3-ITD between both time points; medium, switch of ITD insertion site or ITD length; dark, gain of ITD at R). Blue indicates that no FLT3-ITDs were detected at R.
Presence of FLT3-ITD clones at diagnosis and disease progression based on a Genescan-based routine diagnostic assay. Each column represents 1 patient. (A) Presence of FLT3-ITD clones at diagnosis (D) and relapse/refractory disease (R) in patients treated with midostaurin. (B) Presence of FLT3-ITD clones between D and R in 21 intensively treated patients without the addition of midostaurin. Green indicates that the same ITDs were detected at both time points. A change of ITDs between D and R is color-coded in orange (light, loss of ITDs at progression with at least 1 persistent FLT3-ITD between both time points; medium, switch of ITD insertion site or ITD length; dark, gain of ITD at R). Blue indicates that no FLT3-ITDs were detected at R.
Between patients with persistence or loss of FLT3-ITD at disease progression, we found that the FLT3-ITD AR at diagnosis was significantly lower in the loss cases (median, 0.39 [0.06-0.96] vs 0.62 [0.10-4.32]; P = .03), whereas ITD length and insertion sites were comparable (Table 3). A change of FLT3-ITD clones (loss/gain of ITD clones, switch of ITD insertion sites/nt length) between diagnosis and disease progression was observed in 12 of 54 (22%) patients (Figure 1; Table 2). Although 6 of 54 (11%) patients lost FLT3-ITD clones, previously undetected FLT3-ITDs were found in 6 of 54 (11%) patients. These changes were mainly seen in relapsed cases with only 2 patients with refractory disease showing loss of FLT3-ITD clones (supplemental Figure 1). In patients with refractory disease, we observed a higher percentage of FLT3-ITD loss cases in females (8 of 13; 62% vs 0 of 6; 0%; P = .02; supplemental Table 3).
Between patients treated with midostaurin (n = 54) and the control group (n = 21), we observed that the percentage of patients who were FLT3-ITD negative was significantly higher at disease progression in patients treated with midostaurin compared with the control group (25 of 54; 46% vs 4 of 21; 19%; P = .04; Figure 1; Table 2); this was also observed when we compared patients treated with midostaurin who relapsed (n = 35) to the control group (17 of 35; 49% vs 4 of 21; 19%; P = .04; supplemental Figure 1; supplemental Table 4).
Distribution of somatic mutations detected with WES at diagnosis and disease progression
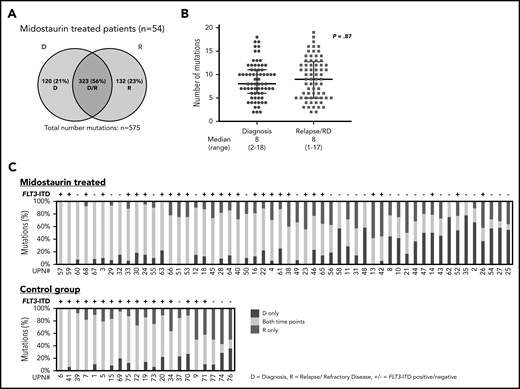
With a median coverage of 110× by WES, we identified a total of 900 mutations in all diagnostic and disease progression samples (midostaurin-treated relapsed/refractory disease, n = 575; control group, n = 325). In 54 patients treated with midostaurin, 323 of 575 (56%) mutations were shared between diagnosis and disease progression (only diagnosis, 120 of 575 [21%]; only relapsed/refractory disease, 132 of 575 [23%]; Figure 2A; supplemental Figure 2A). The average number of mutations was comparable between diagnosis and disease progression (8 [2-18] vs 8 [1-17]; P = .87; Figure 2B; supplemental Figure 2B). Besides FLT3-ITD, at least 1 mutation was shared between both time points (median, 5 [1–15]); 30 of 35 (86%) patients gained additional mutations at relapse (median, 2 [0–10]), whereas only 13 of 19 (68%) patients with refractory disease gained additional mutations (median, 1 [0-5]). Between patients, the stability of mutations that persisted between diagnosis and disease progression was highly variable, with a wide range of persistent somatic variants (median, 61% [9%-100%]; Figure 2C; supplemental Figure 2C).
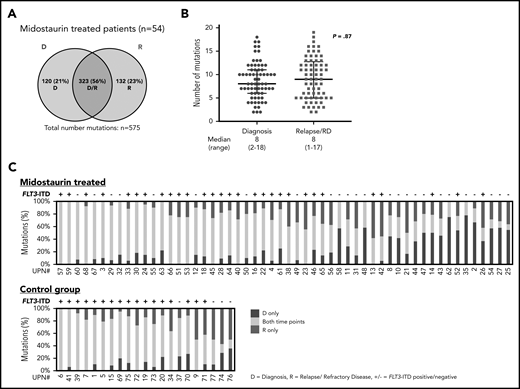
Distribution of somatic variants detected with WES and stability of mutations at D and R. (A) Number of all mutations detected with WES in patients treated with midostaurin (n = 54), which were either present only at D (blue), at R (red), or detectable at both time points (purple intersection). (B) Median number of mutations in patients treated with midostaurin present at the time of diagnosis (blue) or relapse/refractory disease (RD, red). Each dot represents the number of mutations found in a given patient at a given time point. P value was calculated using the Mann-Whitney test. (C) Stability of mutations between D and R in patients treated with midostaurin and the control group. Each bar represents 1 patient. Given is the percentage of mutations present only at D (blue), at R (red), and present at both time points (yellow). Bars are arranged in descending order of stably detectable mutations (yellow). Information on the presence of FLT3-ITD (+ = positive; − = negative) at R is given on the top of each bar.
Distribution of somatic variants detected with WES and stability of mutations at D and R. (A) Number of all mutations detected with WES in patients treated with midostaurin (n = 54), which were either present only at D (blue), at R (red), or detectable at both time points (purple intersection). (B) Median number of mutations in patients treated with midostaurin present at the time of diagnosis (blue) or relapse/refractory disease (RD, red). Each dot represents the number of mutations found in a given patient at a given time point. P value was calculated using the Mann-Whitney test. (C) Stability of mutations between D and R in patients treated with midostaurin and the control group. Each bar represents 1 patient. Given is the percentage of mutations present only at D (blue), at R (red), and present at both time points (yellow). Bars are arranged in descending order of stably detectable mutations (yellow). Information on the presence of FLT3-ITD (+ = positive; − = negative) at R is given on the top of each bar.
Between patients treated with midostaurin (n = 54) and the control group (n = 21), a significant finding was that the number of persistent mutations was higher in the control group (212 of 325; 65% vs 323 of 575; 56%; P = .009; Figure 2A; supplemental Figure 2A), which was also reflected by a high percentage of persistent mutations in each individual patient (median, 71% [14%-100%]; Figure 2C). A higher percentage of persistent mutations in the control group was also observed when we analyzed only relapsed patients (control, n = 21, midostaurin-treated, n = 35; 212 of 325; 65% vs 219 of 418; 52%; P = .009).
Mutational landscape of FLT3-ITD positive AML at diagnosis and disease progression
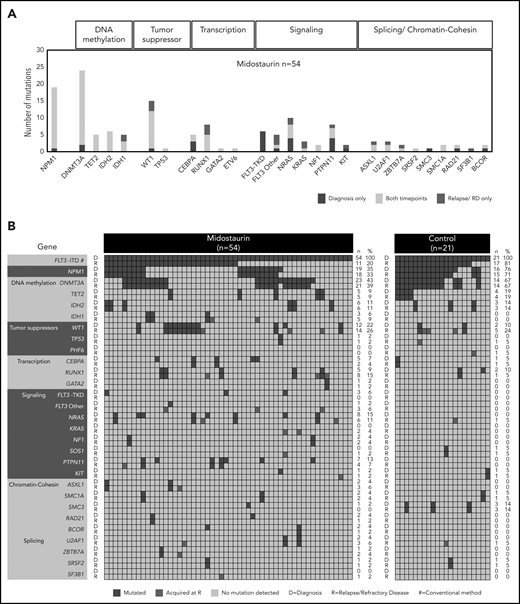
The largest class-defining genomic alterations37 at diagnosis in patients treated with midostaurin were NPM1 (19 of 54; 35%) mutations followed by mutations in chromatin and RNA-splicing genes (7 of 54; 13%). In 16 of 54 (29%) patients, no class-defining driver was present at diagnosis in addition to FLT3-ITD (supplemental Figure 3).
The most frequent comutational patterns were FLT3-ITD+/DNMT3A+/NPM1+ (11 of 54; 20%) and FLT3-ITD+/WT1+ (12 of 54; 22%; Figure 3A-B; supplemental Figure 4A-B), which were both previously associated with an adverse outcome.37-39 Additional mutations in the FLT3 gene at diagnosis were found at positions D835Y (n = 3) and V592A (n = 1) in 4 of 54 midostaurin-treated cases.
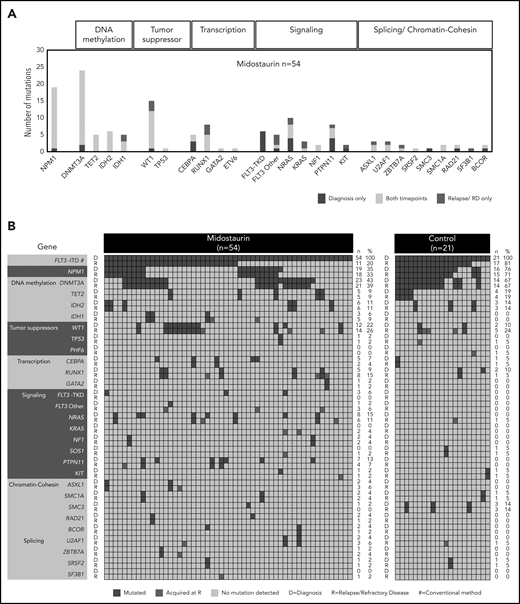
Recurrently mutated genes at D and at R. (A) Frequencies of recurrent gene mutations in all patients treated with midostaurin (n = 54) present only at D (blue), only at R (red), and at both time points (yellow). Genes are arranged according to functional groups as described at the top of the graph. (B) Presence of gene mutations in patients treated with midostaurin and the control group at D and R. Genes are arranged with regard to functional groups. Each column represents a single patient. For each patient, the presence of mutations is given at both time points in subsequent rows. Blue, presence of a mutation; gray, wild type. Mutations present only at R are highlighted in red. The number (n) and percentage (%) of mutations at the respective time points are given next to the figures.
Recurrently mutated genes at D and at R. (A) Frequencies of recurrent gene mutations in all patients treated with midostaurin (n = 54) present only at D (blue), only at R (red), and at both time points (yellow). Genes are arranged according to functional groups as described at the top of the graph. (B) Presence of gene mutations in patients treated with midostaurin and the control group at D and R. Genes are arranged with regard to functional groups. Each column represents a single patient. For each patient, the presence of mutations is given at both time points in subsequent rows. Blue, presence of a mutation; gray, wild type. Mutations present only at R are highlighted in red. The number (n) and percentage (%) of mutations at the respective time points are given next to the figures.
In the entire group of n = 75 patients with the FLT3-ITD mutation, we further detected mutations in genes not described as recurrently mutated in AML thus far (AMPD2, ERF, GALNT9, GLI1, HERC2, MYH4, NRXN1, PEG10, PPP1R3A, SLCO3A1, TMC6, and VWA8, each mutated in 2 patients at diagnosis).
Besides common pathways related to cancer and leukemia, in this FLT3-ITD cohort, mutations at diagnosis were enriched for genes related to cell cycle regulation, signaling pathways (MAPK, neurotrophin, GnRH), and RAS-related cancer types (bladder cancer, non–small-cell lung cancer). Although patients in the NPM1 genomic group37 less often had refractory disease (3 of 19; 16% vs 16 of 35; 46%; P = .04; supplemental Figure 3), genes related to MAPK signaling were enriched at diagnosis in patients who later relapsed (supplemental Figure 5A).
In patients treated with midostaurin, we found a high stability of mutations in genes related to epigenetic modification and NPM1, which persisted in most patients between diagnosis and disease progression. In contrast, mutations in all other functional groups were more variable between both time points (Figure 3A-B). Driver mutations that were newly acquired at relapse/refractory disease most commonly affected WT1 (n = 3), RUNX1 (n = 3), RAS (n = 4), and IDH1 (n = 2), as well as genes related to chromatin/splicing (ASXL1, U2AF1, ZBTB7A, and SF3B1; n = 4; Figure 3A-B). Site-specific resistance mutations at position N676 in the FLT3 gene16 were detected only in 2 relapsed patients with low variant allele frequencies (VAFs; <5%).
Gene enrichment analysis revealed similar functional clusters between diagnosis and disease progression (supplemental Figure 5B). However, in patients with persistence or loss of FLT3-ITD at disease progression, gene mutations clustered in different pathways. In patients with persistent FLT3-ITD, relapse/refractory disease associated mutations were enriched for genes related to cell cycle regulation (CCND3, SMC1A, RAD21, CDKN1C). In patients with loss of FLT3-ITD at relapse/refractory disease, we observed an enrichment of mutations in genes related to RAS-associated cancer types (bladder, endometrial, non–small-lung cancer, melanoma) and MAPK signaling-related pathways (supplemental Figure 5C).
Clonal evolution of FLT3-ITD–positive AML at diagnosis and disease progression
Analyzing the VAFs of mutations at diagnosis, CR and relapse/refractory disease, we could assess the clonal evolution over the course of treatment and distinguish 3 major patterns considering FLT3-ITD status and comutations at diagnosis and disease progression during midostaurin treatment.
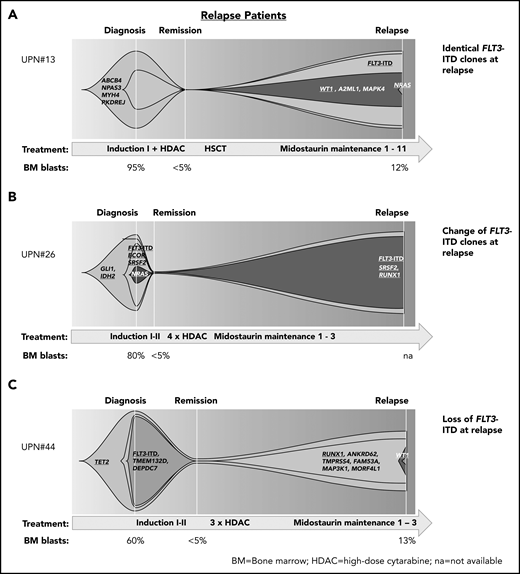
In patients who had received midostaurin during therapy and relapsed, we found patients with (1) persistence of the diagnostic FLT3-ITD clones at relapsed disease, thereby suggesting that FLT3-ITD clones were either insufficiently inhibited by midostaurin (eg, because of noncompliance or poor pharmacokinetics) or that resistance mechanisms bypassing FLT3 inhibition contributed to treatment failure (Figure 4A); (2) change of the composition of FLT3-ITD clones between diagnosis and relapse, suggesting that the initial clones were sensitive to treatment and other FLT3-ITD clones were involved in disease recurrence (Figure 4B); and (3) loss of FLT3-ITD at relapse, pointing to other resistance mechanisms (Figure 4C; supplemental Figure 6).
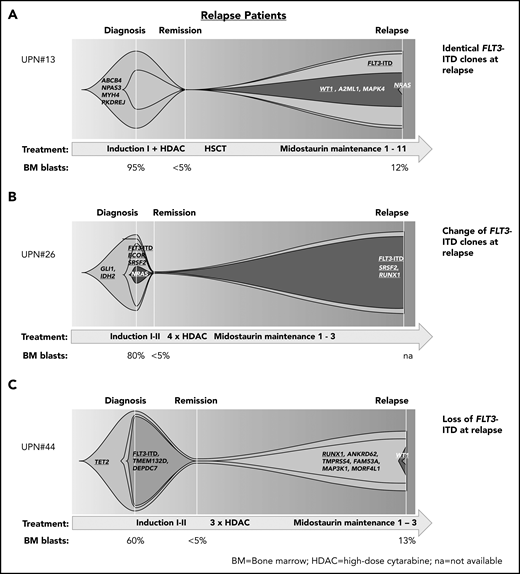
Patterns of clonal evolution in relapse patients. Fish plots visualizing patterns of clonal evolution in relapse patients according to the variant allele frequencies at diagnosis, remission, and relapse in patients with persistent FLT3-ITD (relapse occurred after midostaurin maintenance cycle 11; A), change of FLT3-ITD (relapse after cycle 3; B), and loss of FLT3-ITD (relapse after cycle 3; C). Information on treatment and BM blast count is given below each plot. HDAC, high-dose cytarabine; HSCT, hematopoietic stem cell transplantation; NA, not available.
Patterns of clonal evolution in relapse patients. Fish plots visualizing patterns of clonal evolution in relapse patients according to the variant allele frequencies at diagnosis, remission, and relapse in patients with persistent FLT3-ITD (relapse occurred after midostaurin maintenance cycle 11; A), change of FLT3-ITD (relapse after cycle 3; B), and loss of FLT3-ITD (relapse after cycle 3; C). Information on treatment and BM blast count is given below each plot. HDAC, high-dose cytarabine; HSCT, hematopoietic stem cell transplantation; NA, not available.
To explore whether mutations, which were gained at relapse, were already present as subclones at diagnosis, we additionally compared the VAFs determined with our ultra-deep sequencing approach for recurrent gene mutations in selected samples. For all 3 patients with sufficient material for both time points (diagnosis/relapse), mutations that emerged at relapse were not detected at diagnosis (supplemental Table 7).
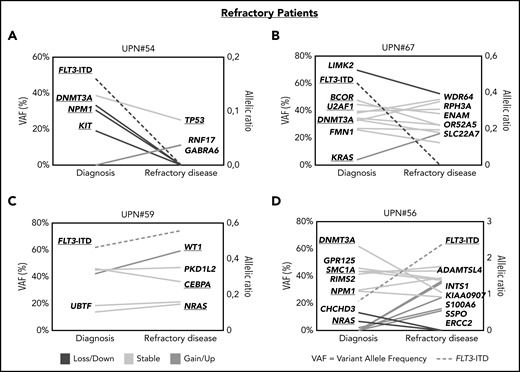
In patients with refractory disease, we observed a similar picture that either (1) the FLT3-ITD–harboring clones were successfully suppressed during treatment and other mutations persisted (Figure 5A-B; supplemental Figure 7A-B) or that (2) FLT3-ITD escaped treatment together with co-occurring mutations, which were already present at diagnosis (Figure 5C; supplemental Figure 7C) or grew out at refractory disease (Figure 5D; supplemental Figure 7D).
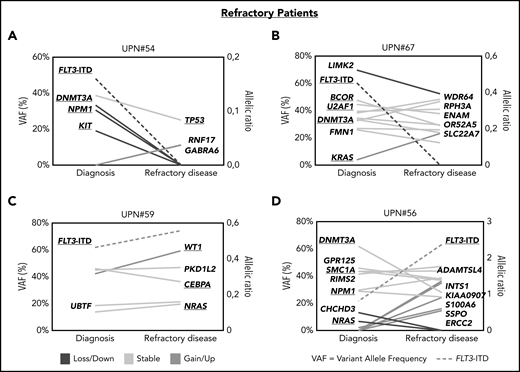
Patterns of clonal evolution in patients with refractory disease. Patterns of clonal evolution in refractory (RD) patients with loss of FLT3-ITD at RD (A-B) or persistent FLT3-ITD at RD (C-D). VAFs are given on the left of each graph and the allelic ratios of FLT3-ITDs on the right. Persistent mutations are color-coded in yellow, lost mutations in blue, and gained or upregulated mutations in green. FLT3-ITDs are indicated by dotted lines.
Patterns of clonal evolution in patients with refractory disease. Patterns of clonal evolution in refractory (RD) patients with loss of FLT3-ITD at RD (A-B) or persistent FLT3-ITD at RD (C-D). VAFs are given on the left of each graph and the allelic ratios of FLT3-ITDs on the right. Persistent mutations are color-coded in yellow, lost mutations in blue, and gained or upregulated mutations in green. FLT3-ITDs are indicated by dotted lines.
PIA analysis36,40 from n = 12 patients with relapse/refractory disease showed FLT3 inhibition in 10 of 12 (83%) patients at the time of disease progression; however, levels of phosphorylated FLT3 were varying between 20% and 100% (supplemental Table 5; supplemental Figure 8).
With regard to the percentages of VAFs at diagnosis, we observed functional group-related differences: Mutations in epigenetic regulating genes and transcription factors were found with higher median VAFs compared with mutations in NPM1, WT1, and signaling-related genes, suggesting an early acquisition during leukemogenesis (supplemental Figure 9). We also found that pre-leukemic mutations persisted in 7 patients with VAFs >10% in CR, showing that these mutations alone were not sufficient to cause a leukemic phenotype; however, they probably supported the outgrowth of other resistance mutations (supplemental Figure 6). Although NPM1 mutations were not detected in CR in most patients (13 of 15; 87%), most of the mutations in WT1 (7 of 11; 64%) persisted as subclones at CR (VAF range, 0.3% to 4.6%), which implies that they were more resistant to therapy.
Discussion
In FLT3-mutated AML, the addition of midostaurin to intensive standard treatment regimens has widely entered clinical practice. Thus, it is important to understand mechanisms of resistance that might impact treatment outcome. We studied paired samples from 54 patients with FLT3-ITD AML who either were refractory to or relapsed after intensive chemotherapy in combination with midostaurin using diagnostic FLT3-ITD Genescan analysis and WES.
An important finding of our study was that FLT3-ITD was no longer detected in 46% of patients treated with midostaurin at the time of relapsed or refractory disease. With the limitation of low numbers, loss of the FLT3-ITD clone was only seen in 4 of 21 (19%) patients who did not receive midostaurin. Although FLT3-ITD was described as a relatively unstable genetic alteration during disease progression, the number of FLT3-ITD–negative cases in the patients treated with midostaurin was higher than reported in the literature.26,27,41 Also, the median AR of FLT3-ITDs at relapse was higher in the control group compared with the AR in patients treated with midostaurin, suggesting that midostaurin inhibited the expansion of FLT3-ITD clones. Interestingly, loss of FLT3-ITD was also found in almost half of patients (42%) with refractory disease, which provides evidence that FLT3-ITD inhibition was achieved at an early stage of treatment in many patients and resistance to treatment was driven by other persisting mutations. In line, we recently showed that midostaurin achieves the strongest FLT3-inhibition (indicated by the lowest level of FLT3 phosphorylation levels) at the end of the first induction cycle.40 This was also observed in n = 12 patients with available PIA analysis data in our cohort.
However, loss of FLT3-ITD must be interpreted with caution because of the rather low sensitivity of the diagnostic Genescan-based assay (∼5%) that was also used as clinical trial assay in the RATIFY and AMLSG 16-10 trials. Nevertheless, our data suggest that in these loss cases, the FLT3-ITD subclone was sensitive to the combination therapy, whereas other mutations contributed to disease progression. In 11% of patients treated with midostaurin, we detected novel FLT3-ITD clones that were not detected at diagnosis or expanded during treatment. This pattern of clonal evolution was only found in patients with relapse and not with refractory disease, which supports the hypothesis that clonal selection through midostaurin inhibition only occurs after a prolonged exposure.
As many patients in the RATIFY and AMLSG 16-10 trials stopped or reduced midostaurin treatment because of side effects,13,14 inadequate drug levels caused by nonadherence or poor pharmacokinetics should also be considered as causes for treatment failure. Although our PIA assays showed ongoing FLT3 inhibitiory activity at disease progression in 10 of 12 analyzed patient samples, a high variability of inhibitory activity was seen. This could explain insufficient FLT3 inhibition in some cases, which could then lead again to an outgrowth of the suppressed but persisting FLT3-ITD–positive clone driving the disease. In cases with high inhibitory activity, these data do also functionally support that downstream mutations that are bypassing the ongoing FLT3 inhibition contributed to disease progression.
Using WES, we could comprehensively assess the clonal evolution of mutations during disease progression in an unbiased way. We identified, on average, 8 and 8 mutations at diagnosis and relapse, respectively, which was comparable to previous studies.21,23 Patients with refractory disease had, on average, a higher number of persistent mutations and acquired less novel mutations compared with relapsing patients. This observation is in line with the assumption that resistance mutations in patients with refractory disease are already present at diagnosis, whereas in relapsing patients, treatment failure is caused by selection and outgrowth of resistant clones and acquisition of novel clones. In contrast to NPM1 mutations, which were more frequent in patients who initially responded and subsequently relapsed compared with patients with primary refractory disease (15 of 35; 43% vs 4 of 19; 21%), the frequency of WT1 mutations at diagnosis was high (20%) in both patient groups, thereby supporting previous studies, which associated WT1 mutations in FLT3-ITD AML with poor outcome.38,39 A recent study provided biological insights for this finding, showing that WT1 mutations cooperate with FLT3-ITD to enhance self-renewal capacities of FLT3-ITD stem and progenitor cells.42
Point mutations in the FLT3 gene that have been shown to confer resistance to midostaurin treatment15,16 were rarely found in patients with refractory disease or those who had relapsed; in our study, only 2 patients acquired a FLT3 N676 albeit with a low VAF (<5%), thereby making it unlikely that this mutation was causative for treatment resistance.
Our results are consistent with those from other genome-wide studies that described a high heterogeneity of mutations at the time of disease progression, especially in FLT3-ITD–positive AML during treatment with chemotherapy-based regimens23 or TKIs.29,30 Of note, all patients in our study shared at least 1 mutation between diagnosis and refractory or relapsed disease, pointing to a common ancestral leukemic clone that persisted at disease progression. Despite the large heterogeneity of mutations, our analysis identified several genes that were enriched at the time of disease progression including WT1, NRAS, KRAS, and IDH1, which were previously associated with resistance to chemotherapy39,43 and midostaurin treatment.20,44 We also observed that mutations in genes related to chromatin-cohesin/splicing (ASXL1, U2AF1, ZBTB7A, and SF3B1) were acquired at the time of resistance.
Pathway enrichment analysis showed that in patients where FLT3-ITD became undetectable, mutations were enriched for genes linked to MAPK-related signaling pathways. This is of interest because a constitutive activation of the downstream pathways AKT (protein kinase B) and extracellular signal-regulated kinase was also linked to resistance mechanisms in cell line models after prolonged exposure to TKIs, although autophosphorylation of FLT3 was completely inhibited.20 We further observed that the median AR of FLT3-ITD at diagnosis was lower in patients with loss of FLT3-ITD during midostaurin treatment. This observation suggests that in patients with a lower AR of FLT3-ITD at diagnosis, the main tumor clone might be less dependent on FLT3 signaling. Based on these observations, the use of MAPK inhibitors in combination with salvage therapy might be a novel approach in patients with a FLT3-ITD–negative relapse.
In summary, in our cohort of patients with FLT3-ITD–mutated AML treated with midostaurin, on-target mutations in FLT3 as potential drivers of disease progression were not recurrently found, whereas almost half of the patients lost the FLT3-ITD mutation. WES revealed a high heterogeneity of mutations that either persisted or were gained between diagnosis and progressive disease, and pathway enrichment analysis pointed to aberrant MAPK signaling in patients with FLT3-ITD loss at relapse. In accordance, a recent targeted next-generation sequencing study identified mutations activating the RAS/MAPK pathway signaling in AML with disease progression on gilteritinib, whereas secondary resistance mutations such as FLT3-F691 were also less frequently detected.31 In perspective, more comprehensive single-cell targeted DNA sequencing will provide additional insights into the dynamic and complex changes underlying clonal selection and evolution in response and resistance to TKI therapy in AML. Ultimately, an improved understanding of these mechanisms can be used to better monitor treatment response and the selection of resistant subclones, also providing a basis for personalized combination treatment approaches in FLT3-ITD–mutated AML.
Presented in part as an oral presentation (abstract 182) at the 58th annual meeting of the American Society of Hematology, San Diego, CA, 9 December 2017.
Raw data were generated at the University of Ulm. Data are available on request from the corresponding authors (K.D. and L.B.).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank all German-Austrian Acute Myeloid Leukemia Study Group (AMLSG) institutions and investigators who contributed to this study; Karl-Heinz Holzmann (Genomics Core Facility, Ulm University) for analysis and technical support with whole exome sequencing; Veronica Teleanu for information on cytogenetics for samples collected by AMLSG; and Jessica Kohlschmidt for information on clinical and cytogenetic parameters for samples collected by the Cancer and Leukemia Group B (CALGB).
This work was supported, in part, by the Deutsche Forschungsgemeinschaft (SFB 1074 project B3) (K.D., L.B.), and a grant by the Bundesministerium für Bildung und Forschung (BMBF; DRAMA 01KT1603 (H.D.). L.K.S. is a participant in the Berlin Institute of Health–Charité Clinician Scientist Program funded by the Charité–Universitätsmedizin Berlin and the Berlin Institute of Health.
Authorship
Contribution: L.K.S. performed research and interpreted data; A.D., S.C., and T.J.B. designed WES analysis pipeline; L.K.S., H.G.S., and S.M. analyzed and interpreted WES data; E.S. and A.D. designed fish plots; N.J. and E.P. helped with data validation; F.T. performed PIA experiments; J.H., S.S., F.G.R., V.I.G., P.P., W.F., H.R.S., G.W., T.S., M.L., R.F.S., F.T., M.H., R.A.L., A.G., R.M.S., and C.D.B. were involved directly or indirectly in care of patients and sample procurement; L.K.S., K.D., and L.B. designed the research; and L.K.S., L.B., K.D., and H.D. wrote the paper.
Conflict-of-interest disclosure: L.B. was on advisory committees for AbbVie, Amgen, Astellas, Bristol-Myers Squibb, Celgene, Daiichi Sankyo, Gilead, Hexal, Janssen, Jazz Pharmaceuticals, Menarini, Novartis, Pfizer, Sanofi, and Seattle Genetics. K.D. was on advisory committees for Novartis, Janssen Pharmaceutica, Celgene, Bristol-Myers Squibb, and Daiichi Sankyo. H.D. was on consulting committees for AbbVie, Agios, Amgen, Astellas, Astex-Pharmaceuticals, Celgene, Helsinn, Janssen, Jazz Pharmaceuticals, Novartis, Oxford Biomedicals, and Roche and received research funding from Amgen, AROG Pharmaceuticals, Bristol Myers Squibb, Celgene, Jazz Pharmaceuticals, Novartis, Pfizer, and Suneisi. A.G. provided consultancy services to Celgene and Novartis. F.T. was on advisory committees for Celgene, Novartis, Jazz, and AbbVie. R.F.S. consulted for or was on advisory committees for Daiichi Sankyo, and Pfizer; was on the speakers' bureau for Pfizer, Daiichi Sankyo, and Novartis; received research funding from PharmaMar, AstraZeneca, Pfizer, and Daiichi Sankyo; and received travel, accommodations, and expenses from Daiichi Sankyo. H.R.S. was on advisory committees for Synimmune, Novartis, and Pfizer and received research support from Synimmune. W.F. consulted for or was on advisory committees for Amgen, ARIAD/Incyte, Novartis, Pfizer, Celgene, AbbVie, and Jazz Pharmaceuticals; received royalties from Amgen; received support for meeting attendance from Amgen, Gilead, Jazz Pharmaceuticals, and Daiichi Sankyo; and received research funding from Amgen and Pfizer. R.A.L. was a consultant for Novartis, Amgen, Ariad/Takeda, Astellas, Celgene/BMS, CVS/Caremark, Epizyme, and MorphoSys; received clinical research support from Novartis, Astellas, Celgene, Cellectis, Daiichi Sankyo, Forty Seven, and Rafael Pharmaceuticals; and received royalties from UpToDate. R.M.S. reports personal fees from AbbVie, Actinium, Agios, Argenx, Astellas, AstraZeneca, Biolinerx, Celgene, Daiichi Sankyo, Elevate, Gemoab, Janssen, Jazz, Macrogenics, Novartis, Otsuka, Pfizer, Hoffman LaRoche, Stemline, Syndax, Syntrix, Syos, Takeda, and Trovagene and received research support from AbbVie, Agios, Arog, and Novartis. P.P. played an advisory role for Agios, Astellas, Jazz, Novartis, and Pfizer; was on the speakers bureau for Agios, Astellas, Jazz, Novartis, and Pfizer; and received travel support from AbbVie, Celgene, and Janssen. The remaining authors declare no competing financial interests.
Correspondence: Konstanze Döhner, Department of Internal Medicine III, University Hospital of Ulm, Albert-Einstein-Allee 23, 89081 Ulm, Germany; e-mail: konstanze.doehner@uniklinik-ulm.de; and Lars Bullinger, Department of Hematology, Oncology and Tumorimmunology, Charité University Medicine, Augustenburger Platz 1, 13353 Berlin, Germany; e-mail: lars.bullinger@charite.de.