

Key Points
Aberrant glycosylation of anti-SARS-CoV-2 spike IgG immune complexes increases platelet thrombus formation on VWF.
Inhibition of Syk, Btk, P2Y12, or FcγRIIA reverses enhancement of thrombus formation mediated by anti-SARS-CoV-2 spike immune complexes.

Abstract
A subset of patients with coronavirus disease 2019 (COVID-19) become critically ill, suffering from severe respiratory problems and also increased rates of thrombosis. The causes of thrombosis in severely ill patients with COVID-19 are still emerging, but the coincidence of critical illness with the timing of the onset of adaptive immunity could implicate an excessive immune response. We hypothesized that platelets might be susceptible to activation by anti–severe acute respiratory syndrome coronavirus 2 (anti-SARS-CoV-2) antibodies and might contribute to thrombosis. We found that immune complexes containing recombinant SARS-CoV-2 spike protein and anti-spike immunoglobulin G enhanced platelet-mediated thrombosis on von Willebrand factor in vitro, but only when the glycosylation state of the Fc domain was modified to correspond with the aberrant glycosylation previously identified in patients with severe COVID-19. Furthermore, we found that activation was dependent on FcγRIIA, and we provide in vitro evidence that this pathogenic platelet activation can be counteracted by the therapeutic small molecules R406 (fostamatinib) and ibrutinib, which inhibit tyrosine kinases Syk and Btk, respectively, or by the P2Y12 antagonist cangrelor.
Introduction
Coronavirus disease 2019 (COVID-19) i s more likely to progress to a severe, life-threatening condition in patients with preexisting cardiovascular disease and is associated with dysregulated hemostasis and a high incidence of venous and arterial thromboembolism.1-3 Emboli in the pulmonary arteries and microthrombi containing fibrin and platelets in the pulmonary microvasculature of patients with COVID-19 have been identified postmortem4 and are thought to contribute toward development of acute respiratory distress syndrome. It is now believed that multiple factors contribute to the thromboinflammatory state that results in high rates of thrombotic complications. Evidence has indicated the presence of activated vascular endothelial cells, macrophages, platelets, neutrophils, and an activated coagulation system in patients critically ill with COVID-19. The mechanistic trigger that causes the changes that accompany an increase in severity in a subset of patients is still the subject of intense research. However, the disparity between the time of peak viral load at 5 to 6 days after the onset of symptoms and occurrence of acute respiratory distress syndrome after 8 to 9 days imply an excessive immune response rather than direct actions of the virus itself.5
Further evidence that the adaptive immune response is disturbed in critically ill patients who have COVID-19 has been provided by a study that found high levels of extrafollicular B-cell activation in critically ill patients, which correlates with increased morbidity, antibody titers, and levels of inflammatory biomarkers.6 Other studies have also noted the strong association between high antibody titers and disease severity and survival.7,8 However, antibodies in severely ill patients who have COVID-19 have qualitative as well as quantitative differences compared with those whose illness is mild. Anti-spike immunoglobulin G (IgG) in serum samples from severely ill patients who have COVID-19 were found to have low levels of fucosylation and increased galactosylation in the Fc domain.9,10
Platelets express the antibody receptor FcγRIIA, but it is not known whether immune complexes containing afucosylated IgG might activate platelet FcγRIIA. Clustering of FcγRIIA from platelets, induced by ligand binding, triggers intracellular signaling via Syk and Bruton tyrosine kinase (Btk) activation and promotes granule secretion and integrin αIIbβ3 activation.11,12 Therefore, activation of FcγRIIA by afucosylated anti-spike IgG might further exacerbate thromboinflammation in critically ill patients who have COVID-19.
In this study, we investigated the effects of low fucosylation and high galactosylation of anti-spike IgG on platelet activation to determine the significance of aberrant IgG glycosylation on platelet-mediated thrombus formation, which has been identified in critically ill patients who have COVID-19. We found that potent activation of platelets by immune complexes containing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike and anti-spike IgG occurs only when the IgG expresses both low fucosylation and high galactosylation in the Fc domain and when an additional prothrombotic signal (we used von Willibrand factor [VWF]) is also present. Enhanced platelet activation and thrombus formation, measured in vitro, were sensitive to FcγRIIA inhibition and to small molecule inhibitors of Syk, Btk, and P2Y12, suggesting that these therapeutic strategies might reduce platelet-mediated thrombosis in critically ill patients who have COVID-19.
Materials and methods
Spike protein
The sequence of SARS-CoV-2 S1 was obtained from the cloned full-length S sequence and was cloned into the expression vector pTriEx1.1 (EMD Millipore, Gillingham, United Kingdom) and characterized as previously described.13 Sf9 cells were transfected with the baculovirus expression vector flashBAC Gold (Oxford Expression Technologies, Oxford, United Kingdom) and with the SARS-CoV-2-S1 transfer vector to produce recombinant baculovirus. Large-scale protein expression was performed by infecting 1 L of T.nao38 cells with a high-titer stock of the recombinant baculovirus and incubating it for 3 to 5 days at 27°C. After incubation, the supernatant containing the secreted protein was harvested, clarified by centrifugation at 4300g for 20 minutes, and filtered through a 0.45-μm filter. The clear supernatant was supplemented with 0.5 nM nickel sulfate before being loaded onto the Bio-Scale Mini Profinity IMAC Cartridge (Bio-Rad, Watford, United Kingdom). The elution was carried out at a flow rate of 2.5 mL/min with a gradient elution of 0.05 to 0.25 M imidazole over 60 minutes. Characterization of the spike protein by western blot and enzyme-linked immunosorbent assay (ELISA) showed that the protein is not cleaved at the furin site (no S2 was detected), preventing the conformation change required for the postfusion form. This was confirmed by ELISA with human CV30 monoclonal anti-spike protein antibody and CR3022 anti-COVID-19 and SARS-CoV-2-S glycoprotein antibody (Absolute Antibody, Redcar, United Kingdom) whose differential binding was consistent with the prefusion trimer.
Recombinant anti-spike IgG
Blood preparation
Blood samples were obtained from healthy donors who had given informed consent and by using procedures approved by the University of Reading Research Ethics Committee. Samples were collected into vacutainers containing 3.8% (w/v) sodium citrate. Platelet-rich plasma was prepared by centrifuging whole blood at 100g for 20 minutes. Washed platelets were prepared by adding acid citrate dextrose to platelet-rich plasma, centrifuging at 350g for 20 minutes, and resuspending the platelet pellet at 4 × 108 cells/mL in Tyrode’s buffer (134 mM NaCl, 2.68 mM KCl, 1.80 mM CaCl2, 1.05 mM MgCl2, 417 μM NaH2PO4, 11.9 mM NaHCO3, 5.56 mM glucose [pH 7.4]).
Platelet adhesion assay
Glass 8-well microslides (Ibidi, Munich, Germany) were coated with 5 μg/mL recombinant SARS-COV-2 spike protein, incubated for 60 minutes at 37°C, washed, and then blocked with 10% fetal calf serum (FCS) for 1 hour at 37°C. The slides were then washed and treated with 10 μg/mL COVA1-18 antibodies for 1 hour at 37°C. Washed platelets at 2 × 107 cells per mL were incubated on the slides for 1 hour at 37°C. Nonadherent platelets were washed off with Tyrode’s buffer, and slides were fixed with 10% formyl saline for 10 minutes. Wells were then washed, and the platelets were labeled with 2 μM DiOC6. Fluorescence images of adherent platelets were captured with the 20× objective lens of a confocal Ti2 microscope (Nikon, Surbiton, United Kingdom).
In vitro thrombus formation
Thrombus formation experiments were performed using microfluidic flow chips (Vena8, Cellix, Dublin, Ireland) coated with 5 μg/mL recombinant SARS-COV-2 spike protein for 60 minutes at 37°C, washed, and then blocked with 10% FCS for 1 hour at 37°C. The slides were then washed and treated with 10 μg/mL COVA1-18 antibodies for 1 hour at 37°C and then 20 μg/mL VWF (Abcam, United Kingdom) for 1 hour. Thrombus formation was measured by perfusing citrated whole blood with 20 μg/mL VWF through the flow chambers at 1000 s−1 for 6 minutes before fixing with 10% formyl saline, staining with 2 μM DiOC6, and then imaged by acquiring z-stacks using the 20× objective lens of a confocal Ti2 fluorescence microscope (Nikon).
Light transmission aggregometry
Aggregation was measured in washed platelets in half-area 96-well plates (Greiner, Stonehouse, United Kingdom) by shaking at 1200 rpm for 5 minutes at 37°C using a plate shaker (Quantifoil Instruments, Jena, Germany) after stimulating with collagen at a range of concentrations. Changes in light transmittance of 405 nm were measured using a FlexStation 3 plate reader (Molecular Devices, Wokingham, United Kingdom).
Flow cytometry measurement of fibrinogen binding
Fibrinogen binding was measured by using washed platelets pretreated with immune complexes that were created by incubating 5 μg/mL recombinant SARS-CoV-2 spike protein with 10 μg/mL COVA1-18 for 60 minutes at 37°C. Platelets were then stimulated with 1 µg/mL collagen-related peptide (CRP), 10 μM adenosine 5′-diphosphate (ADP), or 1 μM thrombin receptor activator peptide 6 (TRAP-6) in the presence of fluorescein isothiocyanate–conjugated polyclonal rabbit anti-fibrinogen antibody (Agilent Technologies, Cheadle, United Kingdom) and incubated for 20 minutes in the dark. Platelets were then fixed by adding filtered formyl saline (0.2% formaldehyde in 0.15 M NaCl), and median fluorescence intensities were measured for 5000 platelets per sample on an Accuri C6 Flow Cytometer (BD Biosciences, Wokingham, United Kingdom).
Statistical methods
Statistical testing as described in figure legends and in “Results” was performed with GraphPad Prism Software (GraphPad, La Jolla, CA).
Results
Aberrant glycosylation of anti-spike IgG enhances in vitro thrombus formation on VWF
Previous studies have found a correlation between the severity of COVID-19 and the glycosylation state of anti-SARS-CoV-2 IgG, with low levels of fucosylation and high levels of galactosylation present predominantly in critically ill patients.9,10,15 To investigate whether anti-spike IgG with aberrant glycosylation activates platelets and to assess the importance of low fucosylation and high galactosylation, we studied adhesion and spreading on coverslips coated with IgG spike immune complexes containing recombinant SARS-CoV-2 spike protein and the anti-spike antibody COVA1-18. COVA1-18 is a monoclonal antibody isolated from a convalescent COVID-19 patient that binds to the receptor-binding domain of the spike protein and inhibits SARS-CoV-2 infection with picomolar efficacy.14 Four subtypes of COVA1-18 with different glycosylation states were used in the study: IgG with normal levels of fucosylation and galactosylation (WT), a low-fucose IgG (Low Fuc), a high-galactose IgG (High Gal), and IgG with both low fucose and high galactose (Low Fuc High Gal). We compared the number of platelets adhered to the 4 different immune complexes relative to spike protein alone and found no significant difference (Figure 1Ai-ii). Levels of platelet adhesion to spike were not significantly greater than adhesion to FCS, which was used as a negative control. Adhesive platelet ligands collagen and cross-linked CRP (CRP-XL) were included as positive controls. These results indicated that the immune complexes, regardless of the glycosylation state of the anti-spike IgG, were poor ligands for platelet adhesion.
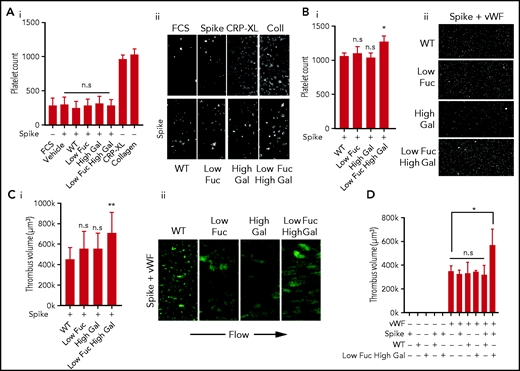
Low fucosylation and high galactosylation of the IgG tail enhances adhesion to VWF. Platelet adhesion to slides coated with immune complexes containing recombinant SARS-CoV-2 spike protein and COVA1-18 recombinant anti-spike IgG with modified glycosylation. (Ai) Numbers of platelets adhered to glass slides coated with FCS (negative control), spike protein only, or spike protein plus unmodified IgG (WT) or IgG modified to have Low Fuc, High Gal, or Low Fuc High Gal and (Aii) representative images (acquired at ×20 original magnification) of adhered platelets stained with DiOC6. (Bi) Numbers of platelets adhered to VWF plus immune complexes containing spike protein and modified IgGs and (Bii) representative images (acquired at ×20 original magnification) of adhered platelets. (Ci) Volume of thrombi formed on VWF with immune complexes containing spike and either WT IgG or IgG with modified glyosylation and (Cii) representative images (acquired at ×20 original magnification) of thrombi stained with DiOC6. (D) Volume of thrombi formed on spike, WT IgG, Low Fuc High Gal IgG, or VWF alone and in combination. Values are mean ± standard error of the mean (SEM). Significant differences were tested by 2-way analysis of variance (ANOVA) with the Tukey multiple comparisons test. *P < .05; **P < .01. n.s., not significant.
Low fucosylation and high galactosylation of the IgG tail enhances adhesion to VWF. Platelet adhesion to slides coated with immune complexes containing recombinant SARS-CoV-2 spike protein and COVA1-18 recombinant anti-spike IgG with modified glycosylation. (Ai) Numbers of platelets adhered to glass slides coated with FCS (negative control), spike protein only, or spike protein plus unmodified IgG (WT) or IgG modified to have Low Fuc, High Gal, or Low Fuc High Gal and (Aii) representative images (acquired at ×20 original magnification) of adhered platelets stained with DiOC6. (Bi) Numbers of platelets adhered to VWF plus immune complexes containing spike protein and modified IgGs and (Bii) representative images (acquired at ×20 original magnification) of adhered platelets. (Ci) Volume of thrombi formed on VWF with immune complexes containing spike and either WT IgG or IgG with modified glyosylation and (Cii) representative images (acquired at ×20 original magnification) of thrombi stained with DiOC6. (D) Volume of thrombi formed on spike, WT IgG, Low Fuc High Gal IgG, or VWF alone and in combination. Values are mean ± standard error of the mean (SEM). Significant differences were tested by 2-way analysis of variance (ANOVA) with the Tukey multiple comparisons test. *P < .05; **P < .01. n.s., not significant.
Severe COVID-19 symptoms include increased levels of many prothrombotic plasma proteins, including VWF, which has been noted as being up to fivefold higher (to ∼5 U/mL) in patients with COVID-19 who are in intensive care.16-18 We combined the immune complex coatings with VWF at 20 μg/mL to simulate a modest increase in plasma VWF levels of ∼2 U/mL. The combined immune complex and VWF coating resulted in significantly more platelet adhesion to the Low Fuc High Gal IgG than the WT IgG (Figure 1Bi-ii). Adhesion to IgG with either Low Fuc or High Gal was not significantly different from that of WT IgG. This suggested that both hypofucosylation and hypergalactosylation are required for platelet activation by anti-spike IgG immune complexes and that prothrombotic conditions in the circulation of severely ill patients who have COVID-19, coupled with dysregulation of IgG glycosylation, may result in increased platelet activation.
The increase in platelet adhesion to IgG-spike immune complex and VWF-coated surfaces was modest under static conditions; however, VWF facilitates platelet adhesion to the vascular endothelium in the high shear environment found in arteries and arterioles. To replicate these conditions, we performed thrombus formation experiments in perfusion chambers coated with IgG-spike immune complexes and VWF at a shear rate of 1000 s−1 (Figure 1Ci-ii). Under these conditions a small, nonsignificant increase in the average volume of thrombi formed on VWF combined with Low Fuc or High Gal IgG relative to WT IgG accompanied by qualitative alterations in the morphology of thrombi from small microaggregates to larger aggregates. However, there was a significant increase in thrombus volume formed on VWF in the presence of Low Fuc High Gal IgG immune complexes. We validated immune complex formation on the surface of the perfusion chambers by coating with spike alone or in combination with WT or Low Fuc High Gal IgG by immunocytochemistry using total internal reflection fluorescence microscopy (supplemental Figure 1). We identified high levels of coverage with both spike (red) and IgG (green), as well as evidence of clustered immune complexes (yellow).
To understand whether immune complex formation was required for increased thrombus formation on VWF, we also investigated the role of spike, IgG, and VWF alone in stimulating thrombus formation under shear (Figure 1D). We found that spike and IgG alone or combined to form immune complexes were insufficient to stimulate thrombus formation in the absence of VWF. The VWF coating alone supported formation of thrombi, but this was not further increased when combined with spike, IgG (WT or Low Fuc High Gal), or immune complexes containing spike and WT IgG. However, a significant increase in thrombus volume was found when the VWF coating was combined with immune complexes containing spike and Low Fuc High Gal IgG. This suggests that anti-spike IgG with aberrant glycosylation of the Fc domain synergizes with VWF to enhance thrombus formation. This replicates the synergy observed between platelet receptors that predominantly mediate adhesion, such as GPIb and integrin α2β1, with receptors that strongly activate platelet signaling such as GPVI and CLEC-2.19
FcγRIIA is the only Fc receptor expressed in platelets that activates intracellular signaling via Syk activation.20 We hypothesized that FcγRIIA may play the role of a signaling receptor to synergize with the vWF adhesion receptor GPIb to enhance thrombus formation. To test this, whole blood was preincubated with FcγRIIA blocking antibody IV.3 before perfusion through VWF-coated flow chambers with either WT or Low Fuc High Gal immune complexes (Figure 2Ai-ii). Blockade of FcγRIIA resulted in a significant reduction in the volume of thrombi formed on the Low Fuc High Gal IgG-containing immune complexes. A modest, nonsignificant increase in thrombus volume observed with WT IgG was also reversed by IV.3.

Platelet activation by Low Fuc High Gal IgG1 immune complexes is dependent on FcγRIIA and functions at low and high shear. (Ai) Volume of thrombi formed on VWF plus immune complexes containing spike and either WT IgG or IgG with Low Fuc High Gal in the presence or absence of 20 μg/mL IV.3 and (Aii) representative images (acquired at ×20 original magnification) of thrombi stained with DiOC6. (Bi) Volume of thrombi formed on VWF plus WT IgG or IgG with Low Fuc High Gal at a shear rate of 200 s−1 or 1000 s−1 and (Bii) representative images of thrombi (acquired at ×20 original magnification) stained with DiOC6. Values are mean ± SEM. Significant differences were tested by 2-way ANOVA with the Tukey multiple comparisons test. *P < .05; **P < .01. Veh, vehicle.
Platelet activation by Low Fuc High Gal IgG1 immune complexes is dependent on FcγRIIA and functions at low and high shear. (Ai) Volume of thrombi formed on VWF plus immune complexes containing spike and either WT IgG or IgG with Low Fuc High Gal in the presence or absence of 20 μg/mL IV.3 and (Aii) representative images (acquired at ×20 original magnification) of thrombi stained with DiOC6. (Bi) Volume of thrombi formed on VWF plus WT IgG or IgG with Low Fuc High Gal at a shear rate of 200 s−1 or 1000 s−1 and (Bii) representative images of thrombi (acquired at ×20 original magnification) stained with DiOC6. Values are mean ± SEM. Significant differences were tested by 2-way ANOVA with the Tukey multiple comparisons test. *P < .05; **P < .01. Veh, vehicle.
To understand whether potentiation of thrombus formation on VWF also occurred at lower shear, which is more representative of that found in small veins and venules, we measured thrombus formation at 200 s−1 (Figure 2B). We found that thrombi formed on VWF at a shear rate of 200 s−1 were smaller than those formed at 1000 s-1, but Low Fuc High Gal IgG-containing immune complexes again caused significant potentiation of thrombus volume relative to WT IgG.
Small molecule drugs targeting Syk, Btk, and P2Y12 inhibit IgG-induced potentiation of thrombus formation on VWF in vitro
To understand the signaling processes underpinning the enhancement of thrombus formation on VWF and Low Fuc High Gal IgG and identify potential treatment strategies to counteract pathogenic platelet activation, we studied the effects of small molecule inhibitors (Figure 3). FcγRIIA signals through the tyrosine kinase Syk, so we treated whole blood with the Syk inhibitor R406, which is the active metabolite of the US Food and Drug Administration (FDA)-approved drug fostamatinib. Treatment with R406 significantly reduced the volume of thrombi formed on Low Fuc High Gal IgG (373 000 ± 42 000 μm3) relative to vehicle (820 000 ± 172 000 μm3), indicating that activation of Syk is important to the prothrombotic effects of aberrantly glycosylated anti-spike IgG and that treatment with fostamatinib might be beneficial for patients with severe COVID19 through suppression of IgG-driven platelet activation. The FcγRIIA signaling pathway is also dependent on Btk,12 and therefore we treated platelets with the Btk inhibitor ibrutinib, which is an FDA- and European Medicines Agency–approved drug for treatment of B-cell cancers. We found that ibrutinib treatment reduced the volume of thrombi formed on the Low Fuc High Gal IgG (348 000 ± 68 000 μm3) to levels similar to the WT IgG (478 000 ± 76 000 μm3). Platelet activation stimulated by FcγRIIA triggers secretion of ADP, which activates the P2Y12 receptor and provides positive feedback signaling required for integrin αIIbβ3 activation and aggregation.21 We hypothesized that inhibition of P2Y12 using an antagonist might also help reduce thrombotic tendencies in severely ill patients who have COVID-19, so we treated platelets with the P2Y12 antagonist cangrelor, an active drug molecule that does not require metabolism. We found that cangrelor reduced the volume of the thrombi formed on WT IgG, although this reduction was nonsignificant. Treatment with cangrelor significantly reduced thrombi formed on Low Fuc High Gal IgG (389 000 ± 40 000 μm3) to levels comparable to those observed with WT IgG.
![Prothrombotic activity of Low Fuc High Gal IgG1 immune complexes is inhibited by Syk, Btk, or P2Y12 inhibition. (A) Volume of thrombi formed in perfusion chambers on VWF plus immune complexes containing spike protein plus either WT IgG or IgG modified to have low fuc high gal after treatment with vehicle (dimethyl sulfoxide [DMSO]), 1 μM R406, 1 μM ibrutinib (Ibr), or 1 μM cangrelor (Cang). (B) Representative images (acquired at 20× magnification) of thrombi stained with DiOC6. Values are mean ± SEM. Significant differences were tested by 2-way ANOVA with the Tukey multiple comparisons test. *P < .05; **P < .01.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/16/10.1182_blood.2021011871/2/m_bloodbld2021011871f3.png?Expires=1771261361&Signature=rFpPHNOcZRu7436na97gI254Ti6jsApztUnhPYag7A22KduK0wxCAQo27vScEqwhzfw-5j6GYe6t~XQJybBPCUxAoikLkOyPDfgNbjVcRBOV7fcxlbZEfLi1SJHKS7ii7wo-6~~jG5w6cQgXg0cksuc4JrnZLu0QAw3VEohn7breLvLCU2plDafT0CtQdASdWrOvYxoRpzau4cDY6Eda7fIQXKb9UfsGPNe~bltE9-HlJFbYUWyhR76Yx15dSXChfrUffwIKJRJUOblkgwkY4quWAhET6GTIt5xRQ9Lc6RIQAAIznoFDT9L7wsv3PoAV6KVKyvsqaIy60c9BSeqA2A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Prothrombotic activity of Low Fuc High Gal IgG1 immune complexes is inhibited by Syk, Btk, or P2Y12 inhibition. (A) Volume of thrombi formed in perfusion chambers on VWF plus immune complexes containing spike protein plus either WT IgG or IgG modified to have low fuc high gal after treatment with vehicle (dimethyl sulfoxide [DMSO]), 1 μM R406, 1 μM ibrutinib (Ibr), or 1 μM cangrelor (Cang). (B) Representative images (acquired at 20× magnification) of thrombi stained with DiOC6. Values are mean ± SEM. Significant differences were tested by 2-way ANOVA with the Tukey multiple comparisons test. *P < .05; **P < .01.
Prothrombotic activity of Low Fuc High Gal IgG1 immune complexes is inhibited by Syk, Btk, or P2Y12 inhibition. (A) Volume of thrombi formed in perfusion chambers on VWF plus immune complexes containing spike protein plus either WT IgG or IgG modified to have low fuc high gal after treatment with vehicle (dimethyl sulfoxide [DMSO]), 1 μM R406, 1 μM ibrutinib (Ibr), or 1 μM cangrelor (Cang). (B) Representative images (acquired at 20× magnification) of thrombi stained with DiOC6. Values are mean ± SEM. Significant differences were tested by 2-way ANOVA with the Tukey multiple comparisons test. *P < .05; **P < .01.
Immune complexes presented in suspension do not potently enhance platelet activation
Platelet aggregability is enhanced in patients with severe COVID-19,22 and we hypothesized that this might be a result of the presence of immune complexes containing anti-spike IgG with aberrant glycosylation of the IgG Fc domain. To test this hypothesis, we preincubated recombinant SARS-COV-2 spike protein with the same 4 COVA1-18 IgG variants used in previous experiments to enable formation of immune complexes in suspension. We then treated washed human platelets with the immune complexes and stimulated them with a range of collagen (type I) concentrations to induce aggregation (Figure 4A). We found that none of the immune complexes enhanced the potency (50% effective concentration [EC50]) of collagen-evoked aggregation (Figure 4B) or caused aggregation on their own. We also assessed the ability of the immune complexes to potentiate activation of integrin αIIbβ3 by measuring fibrinogen binding stimulated with ADP, TRAP-6, or CRP-XL by flow cytometry (Figure 4Ci-iii). We observed no significant difference between integrin activation stimulated by agonists in the presence of spike protein alone or immune complexes containing both spike and IgG. These data suggest that the manner of presentation of immune complexes may be an important part of the mechanism by which they activate platelets. Clustering of FcγRIIA induces intracellular signaling in platelets,20 and it is possible that immune complexes presented in suspension, rather than immobilized on a surface, are below the concentration threshold required to cause clustering of the receptor within our experimental system. Plasma samples from a subset of patients with COVID-19 who were positive for anti-spike IgG trigger platelet activation in suspension,23 and it is therefore possible that immune complexes of sufficient size and concentration are able to activate platelets.
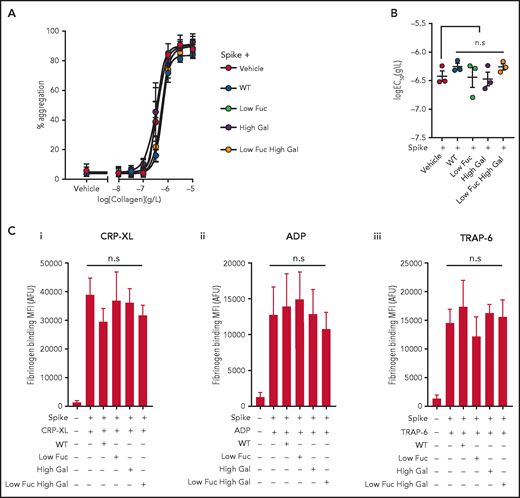
Aggregation and integrin αIIbβ3 activation are unaffected by COVID19 immune complexes. (Ai) Concentration response curves plotting platelet aggregation after stimulation with a range of type I collagen concentrations (from 10 μg/mL to 10 ng/mL) in the presence of immune complexes containing spike plus WT IgG or IgG with modified glycosylation and (Bi) plots of logEC50 for collagen in the presence of the different treatments, the bars represent the mean ± SEM. (C) Fibrinogen binding to platelets measured by flow cytometry after stimulation with (Ci) 10 μM ADP, (Cii) 1 μg/mL CRP-XL, and (Ciii) 1 μM TRAP-6 in the presence of spike only or immune complexes containing WT IgG or IgG with modified glycosylation. Significant differences were tested by 2-way ANOVA with the Tukey multiple comparisons test. AFU, active fluorescence units; MFI, mean fluorescent intensity.
Aggregation and integrin αIIbβ3 activation are unaffected by COVID19 immune complexes. (Ai) Concentration response curves plotting platelet aggregation after stimulation with a range of type I collagen concentrations (from 10 μg/mL to 10 ng/mL) in the presence of immune complexes containing spike plus WT IgG or IgG with modified glycosylation and (Bi) plots of logEC50 for collagen in the presence of the different treatments, the bars represent the mean ± SEM. (C) Fibrinogen binding to platelets measured by flow cytometry after stimulation with (Ci) 10 μM ADP, (Cii) 1 μg/mL CRP-XL, and (Ciii) 1 μM TRAP-6 in the presence of spike only or immune complexes containing WT IgG or IgG with modified glycosylation. Significant differences were tested by 2-way ANOVA with the Tukey multiple comparisons test. AFU, active fluorescence units; MFI, mean fluorescent intensity.
Discussion
There is a growing body of evidence that multiple complications arise that increase rates of thrombosis in severely ill patients who have COVID-19. These include damage to vascular endothelial cells after direct infection with SARS-CoV-2, which results in disruption of barrier function, exposure of subendothelial collagen, and release of prothrombotic plasma proteins, including VWF, from activated endothelial cells.17 The prothrombotic environment is exacerbated by a cytokine storm that may be driven by activation of macrophages by immune complexes containing afucosylated anti-spike IgG.9 Higher levels of hypofucosylated, hypergalactosylated IgG has been identified in the plasma of severely ill patients who have COVID-19 compared with patients who have a mild COVID-19 infection, and it is correlated with disease severity.9,15 In this study, we showed that immobilized immune complexes containing recombinant anti-spike IgG with low fucosylation and high galactosylation activate platelets to enhance thrombus formation on VWF, which is also increased in severely ill patients who have COVID-19.16-18
Vascular endothelial cells can be infected by SARS-CoV-2,24 and we hypothesize that subsequent expression of spike protein and formation of large immune complexes on the cell surface might combine with secreted VWF to form a highly prothrombotic surface (Figure 5). A similar mechanism for thrombosis induced by viral infection in the pulmonary circulation has been identified in severe H1N1 infection, whereby immune complexes present in the lungs activate platelets via FcγRIIA.25 The role of aberrant IgG glycosylation in stimulating this response was not investigated, but it has been suggested that afucosylated IgG may be common to immune responses against all enveloped viruses.9 Another study that investigated platelet activation mediated by plasma samples from severely ill patients who have COVID-19 also reported that platelet activation was dependent on FcγRIIA.23 The plasma samples were positive for anti-spike antibodies, but the glycosylation status was not measured. Another report identified a link between afucosylated IgG and FcγR-dependent activation of macrophages in severe COVID-19 illness, in which high antibody titers combined with altered glycosylation resulted in excessive secretion of cytokines.15 Direct binding of spike protein to platelet ACE2 has been reported as a potential mechanism for platelet hyperreactivity in severe COVID-19 infection26; however, expression of ACE2 in platelets is controversial,27 and we did not find evidence for direct platelet activation by the spike protein.

Aberrant glycosylation of anti-spike IgG in immune complexes act in concert with VWF to enhance platelet thrombus formation. SARS-CoV-2 infects vascular endothelial cells, and combined with other inflammatory signals, results in endothelial activation and release of prothrombotic factors including VWF. After the onset of adaptive immunity, anti-spike IgG accumulates in the circulation and binds to SARS-CoV-2. In critically ill patients with COVID-19, anti-spike IgG has abnormally low levels of fucosylation and high levels of galactosylation. Immune complexes containing this aberrant glycosylation pattern activate platelet FcγRIIA, which stimulates intracellular signals that synergize with the adhesive ligand VWF to promote platelet activation and thrombus formation. Schematic was created with BioRender.com.
Aberrant glycosylation of anti-spike IgG in immune complexes act in concert with VWF to enhance platelet thrombus formation. SARS-CoV-2 infects vascular endothelial cells, and combined with other inflammatory signals, results in endothelial activation and release of prothrombotic factors including VWF. After the onset of adaptive immunity, anti-spike IgG accumulates in the circulation and binds to SARS-CoV-2. In critically ill patients with COVID-19, anti-spike IgG has abnormally low levels of fucosylation and high levels of galactosylation. Immune complexes containing this aberrant glycosylation pattern activate platelet FcγRIIA, which stimulates intracellular signals that synergize with the adhesive ligand VWF to promote platelet activation and thrombus formation. Schematic was created with BioRender.com.
The role of platelets in COVID-19 is still emerging, but platelet-rich thrombi have been identified in both large arteries and microthrombi.4 The platelets of severely ill patients who have COVID-19 express markers of activation,28 and platelets from healthy donors exposed to plasma from these patients evoke activation.29 Platelets contain many inflammatory mediators within granules that might contribute toward the flood of cytokines present in critically ill patients who have COVID-19. Large numbers of platelet-monocyte and platelet-granulocyte aggregates have been identified in the blood of patients who have COVID-19 along with development of a pro-inflammatory phenotype in which expression of cytokines is increased.30 There is still scant information regarding the efficacy of anti-platelet drugs in patients who have COVID-19, but 1 study has suggested that patients receiving anti-platelet therapy with aspirin before being admitted to the hospital for COVID-19 seem to be partially protected and have better outcomes, whereas a separate study found that treating patient in the hospital with aspirin reduced mortality.31,32 Other non–anti-platelet drugs in trials for COVID-19 therapy target proteins also expressed in platelets and could therefore inhibit the contribution of platelets to thromboinflammation.
The Btk inhibitor acalabrutinib has been evaluated in clinical trials on the basis of its potential to block macrophage activation33; however, the potential of Btk inhibitors to reduce the contribution of platelets to thrombosis in COVID-19 infection34 and more generally in thomboinflammation35 has also been noted. We found that the Btk inhibitor ibrutinib reversed the enhancement of thrombus formation on VWF caused by IgG with Low Fuc High Gal, supporting the hypothesis that this strategy might have dual benefits on macrophage and platelet activation. The Syk inhibitor fostamatinib was identified as a potential COVID-19 therapeutic in a high-content screen of drugs that might protect against acute lung injury.36 The active metabolite of fostamatinib, R406, inhibits release of neutrophil extracellular traps37 and macrophage activation induced by plasma from patients who have COVID-19.15 R406 is also known to have inhibitory effects on signaling downstream of platelet GPVI and CLEC-2 receptors,38 although maximal collagen-evoked aggregation is unaffected by oral administration of R40639 and is currently in clinical trials for COVID-19 therapy in the United States (NCT04579393) and the United Kingdom (NCT04581954). We found that R406 reversed the potentiation of thrombus formation on VWF. This suggests that potential COVID-19 therapies such as fostamatinib targeting Syk or acalabrutinib targeting Btk may be effective not only in limiting the inflammatory response, but also in reducing platelet-mediated thrombosis.
Acknowledgments
This work was supported by Imperial College National Institute for Health Research bioresources and grants from the British Heart Foundation (RG/15/2/31224 and RG/20/7/34866), ZonMw (10430 01 201 0008) and by Amsterdam Infection and Immunity COVID-19 grant 24184.
Authorship
Contribution: A.P.B. designed the study, performed research, analyzed data, and helped write the manuscript; W.H., S.J., S.L., S.d.T., G.V., J.N., M.W., and M.v.G. contributed vital new reagents; J.L.M., T.S., and N.K. performed research; and N.C., I.J., J.d.D., and J.M.G. designed the study and helped write the manuscript.
Conflict-of-interest disclosure: J.M.G. has served as a consultant for Astra Zeneca and has received research funding from Celgene/Bristol Myers Squibb and Arena Pharmaceuticals. S.d.T. and M.v.G. have filed a patent application concerning the SARS-CoV-2 mAbs described in the article. N.C. has received honoraria and research funding from Rigel, Grifols, and Novartis. The remaining authors declare no competing financial interests.
Correspondence: Jonathan M. Gibbins, University of Reading, Health and Life Sciences Bldg, Whiteknights, Reading RG6 6EX, United Kingdom; e-mail: j.m.gibbins@reading.ac.uk.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.


