

Abstract
Rho family GTPases are intracellular signaling proteins regulating multiple pathways involved in cell actomyosin organization, adhesion, and proliferation. Our knowledge of their cellular functions comes mostly from previous biochemical studies that used mutant overexpression approaches in various clonal cell lines. Recent progress in understanding Rho GTPase functions in blood cell development and regulation by gene targeting of individual Rho GTPases in mice has allowed a genetic understanding of their physiologic roles in hematopoietic progenitors and mature lineages. In particular, mouse gene–targeting studies have provided convincing evidence that individual members of the Rho GTPase family are essential regulators of cell type–specific functions and stimuli-specific pathways in regulating hematopoietic stem cell interaction with bone marrow niche, erythropoiesis, and red blood cell actin dynamics, phagocyte migration and killing, and T- and B-cell maturation. In addition, deregulation of Rho GTPase family members has been associated with multiple human hematologic diseases such as neutrophil dysfunction, leukemia, and Fanconi anemia, raising the possibility that Rho GTPases and downstream signaling pathways are of therapeutic value. In this review we discuss recent genetic studies of Rho GTPases in hematopoiesis and several blood lineages and the implications of Rho GTPase signaling in hematologic malignancies, immune pathology. and anemia.
Introduction
Mammalian Rho GTPases are a family of small GTP-binding proteins containing 22 members that are involved in many important cellular functions, including gene transcription, survival, adhesion, and cytoskeleton reorganization.1-3 They are closely related to Ras and share considerable structure–function similarities with Ras and other Ras-related small GTPases. As shown in Figure 1, similar to Ras, most Rho GTPases switch between the inactive GDP-bound form and the active GTP-bound form by a mechanism that is strictly regulated by the upstream guanine nucleotide exchange factors (GEFs). Once activated, the GTP-bound forms are capable of interacting with multiple downstream effector proteins in a spatially and temporally controlled manner. Conversely, GTPase-activating proteins (GAPs) stimulate the hydrolysis of bound GTP to GDP, switching Rho GTPases back to the inactive state. Rho GDP-dissociation inhibitors can sequester inactive GDP-bound Rho proteins in the cytosol and prevent their activation and intracellular trafficking. Further adding to the complexity of this tightly regulated mechanism, multiple Rho GEFs, Rho GAPs and Rho GDIs can activate or inactivate the same Rho GTPase, and each Rho GTPase may activate multiple downstream effectors, initiating a network of signals that affects cell functions.4,5

Intracellular signaling model of Rho GTPases in receptor-initiated pathways. Most Rho GTPases cycle between the GDP-bound, inactive state and the GTP-bound, active state. The GTP binding and GTP hydrolysis cycle is tightly regulated by Rho guanine nucleotide exchange factors (GEFs), Rho GTPase-activating proteins (GAPs), and Rho GDIs (GDP dissociation inhibitors), which also control their intracellular localization patterns. On activation by cytokine, chemokine, growth factor, or adhesion molecules, the GTP-bound Rho GTPases can transiently interact with a large panel of effector proteins to transduce signals that affect cell cycle, survival, transcription, adhesion, and cytoskeleton machineries.
Intracellular signaling model of Rho GTPases in receptor-initiated pathways. Most Rho GTPases cycle between the GDP-bound, inactive state and the GTP-bound, active state. The GTP binding and GTP hydrolysis cycle is tightly regulated by Rho guanine nucleotide exchange factors (GEFs), Rho GTPase-activating proteins (GAPs), and Rho GDIs (GDP dissociation inhibitors), which also control their intracellular localization patterns. On activation by cytokine, chemokine, growth factor, or adhesion molecules, the GTP-bound Rho GTPases can transiently interact with a large panel of effector proteins to transduce signals that affect cell cycle, survival, transcription, adhesion, and cytoskeleton machineries.
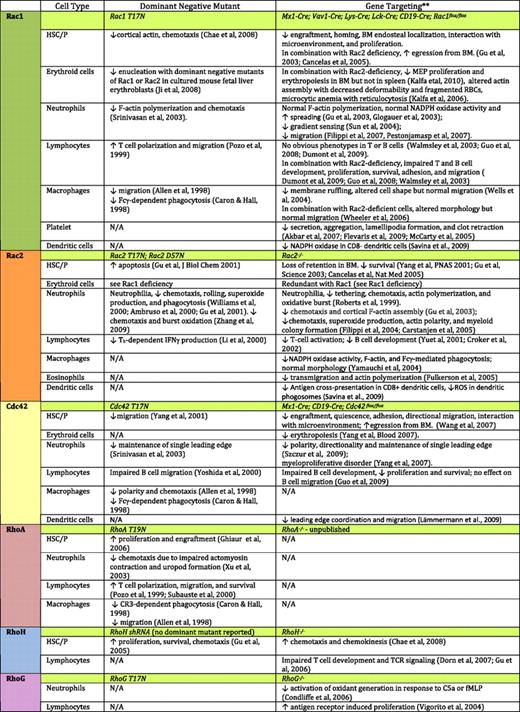
The role of Rho GTPase signaling in blood cell development and function was first implicated through studies of Rho regulators and effectors.5-7 GEFs such as Vav1, GAPs such as Bcr, and effectors such as WASP, are among the best documented ones. Although the use of dominant negative or constitutively active Rho GTPase mutant overexpression has led to some important observations in several blood lineages (Table 1), the inferred functions of Rho GTPases in blood development remained unclear because of the nonspecific and dose-dependent nature of this approach. Recent mouse gene–targeting studies have extended these studies to members of the Rho GTPases themselves,3,5,6 further showing many unique and redundant functions that each Rho GTPase plays in different blood lineages (Table 1). In this review we focus on our current understanding of Rho GTPases, mainly the Rac, Cdc42, Rho, and RhoH subfamilies that have been most extensively studied, in hematopoietic stem/progenitor cell engraftment, erythropoiesis and myelopoiesis, lymphopoiesis, and phagocyte regulation, with an emphasis on their contributions to both physiologic and pathologic conditions.
Effects of dominant negative mutant expression or gene targeting of Rho GTPases on hematopoietic cell functions. Please see the following additional references: Akbar et al127 ; Flevaris et al128 ; McCarty et al129 ; Savina et al130 ; Zhang et al131 ; Fulkerson et al132 ; Yoshida et al133 ; Lämmermann et al134 ; Xu et al135 ; Subauste et al136 . NA indicates data not available. **Only Rho GTPase gene-targeted mouse models are included.
Rho GTPases in hematopoietic stem and progenitor cell regulation
Hematopoietic stem and progenitor cell (HSC/P) fate is finely regulated to initiate the production of billions of blood cells every day. The endosteal space of adult bone marrow (BM) contains putative “niches” where a specialized microenvironment supports and nurtures HSC/Ps.8-10 There has been a tremendous advancement in knowledge about the molecular pathways involved in HSC/P regulation over the past few years, including that of trafficking to and from the BM niche.11 The cell functions involved in HSC/P homing, engraftment, and mobilization include cytoskeleton rearrangements, migration, transcription activation, survival, and cell-cycle progression. These events are regulated by multiple Rho GTPases, particularly Rac1, Rac2, Cdc42, RhoA, and RhoH (Figure 2).

Role of Rho GTPases in HSC/P homing, engraftment, and mobilization. Rac1, Rac2, Cdc42, and Rho control different hematopoietic stem and progenitor cell (HSC/P) functions. Signals required for HSC self-renewal are mediated by Rho, Cdc42, and Rac1. Although Rac1 appears to be required for proliferation, Rac2 controls survival. Cdc42 is necessary for cell-cycle entry of quiescent (G0) HSC/Ps in the bone marrow (BM) microenvironment, proliferation, and aging of HSC/Ps. Rac1 and Cdc42 control homing and interaction with the BM niche, whereas combined Rac1 and Rac2 activities and Cdc42 are necessary for the retention of HSC/Ps in the BM.
Role of Rho GTPases in HSC/P homing, engraftment, and mobilization. Rac1, Rac2, Cdc42, and Rho control different hematopoietic stem and progenitor cell (HSC/P) functions. Signals required for HSC self-renewal are mediated by Rho, Cdc42, and Rac1. Although Rac1 appears to be required for proliferation, Rac2 controls survival. Cdc42 is necessary for cell-cycle entry of quiescent (G0) HSC/Ps in the bone marrow (BM) microenvironment, proliferation, and aging of HSC/Ps. Rac1 and Cdc42 control homing and interaction with the BM niche, whereas combined Rac1 and Rac2 activities and Cdc42 are necessary for the retention of HSC/Ps in the BM.
HSC/P homing, trafficking, and interaction with the BM microenvironment
HSC/P homing and migration activities require Rac GTPases (Table 1). Rac1 and Rac2 integrate signals from β1-integrins and c-kit in HSC/Ps.12,13 CXCL12-induced chemoattraction is mediated by both Rac1 and Rac2 and is at least partly mediated by the GEF Tiam-1.14 c-Kit signals to Rac1 and Cdc42 at least in part through the GEF Vav1.15 Initial studies in Rac2−/− mice have shown that Rac2 is important for HSC/P adhesion and migration, and Rac2−/− mice showed increased numbers of circulating HSC/Ps and increased migration,16 suggesting an important role for Rac2 in retaining HSC/Ps in the BM niche. Further, Rac2 can affect Rac1 and Cdc42 activities, and such a cross talk between Rac2 and other Rho GTPases is involved in regulating HSC/P migration.16,17
Although Rac1 and Rac2 are highly homologous, they can regulate distinct HSC/P functions. Rac1 is required for engraftment of HSC/Ps into the BM,12 attributable specifically to its role in homing. Rac1 deficiency caused defective homing and localization of HSC/Ps in the BM endosteal space after transplantation of Rac1−/− HSC/Ps into wild-type recipients, whereas Rac2 deficiency decreased the retention of HSC/Ps in the BM.16,18 Deletion of both Rac1 and Rac2 led to a mobilization of HSC/Ps from BM to peripheral blood because of decreased adhesion and retention in the BM.12,18 These data strongly suggest that at the molecular level HSC/P homing and retention in the BM are not mirror image processes of each other, and both activities are subject to Rac1 and Rac2 regulation. In this aspect, Rac1, but not Rac2, appears to control fetal HSC/P trafficking. In the absence of Rac1, the number of circulating HSC/Ps in the blood of E10.5 embryos is severely diminished; consequently, embryonic hematopoiesis is significantly impaired in Rac1-deficient embryos as manifested by the absence of intraaortic clusters and near complete absence of fetal liver hematopoiesis.19 These deficiencies correlate with a decreased migration in response to CXCL12 and impaired interaction with the microenvironment-derived stromal cells.19
Cdc42 also uniquely regulates HSC/P trafficking and residence in the BM niche. Gene-targeted mice deficient in Cdc42 GAP, a negative regulator of Cdc42, exhibited disorganized F-actin structure, reduced adhesion and directional migration, and defective engraftment in HSC/P population.20 Cdc42−/− mice showed increased numbers of circulating HSC/Ps and hyperactive cell-cycle profiles, as well as profound defects in adhesion, migration, and homing activities (Table 1).21 Interestingly, aged HSC/Ps contain increased Cdc42 activity, and adhesion of HSC/Ps to the BM microenvironment is reduced in aged mice similar to that in Cdc42GAP−/− mice. Granulocyte colony-stimulating factor–induced HSC/P mobilization is also increased in aged mice, and this effect may be a consequence of the increased Cdc42 activity.22
RhoH constitutes a distinct subtype of Rho GTPases that is GTPase deficient and hematopoietic specific; it has been shown to modulate Rac1 signaling and function in primary HSC/Ps.23-25 RhoH gene targeting led to increased HSC/P chemotaxis and chemokinesis toward the chemoattractant CXCL12. This migratory response is due to increased Rac1 activity and translocation of Rac1 protein to the cell membrane, where it colocalizes with cortical F-actin and lipid rafts. Conversely, overexpression of RhoH in HSC/Ps blocks the membrane translocation of Rac1 and impairs cortical F-actin assembly and chemotaxis in response to CXCL12 stimulation (Table 1). It is possible that RhoH plays an opposite role from Rac1 and Rac2 in regulating HSC/P BM residency.25
Proliferation, survival, and self-renewal of HSC/Ps
Rac and Cdc42 GTPases are also required for proliferation and survival of HSC/Ps. Deletion of Cdc42 in the BM resulted in a loss of quiescence of long-term repopulating–HSCs and a transient accumulation and mobilization of short-term repopulating–HSCs, effects that are associated with transcription changes of β1-integrin, p21Cip, and c-Myc.21 Gain of function of Cdc42, in the Cdc42GAP knockout mice, caused elevated c-Jun N-terminal kinase (JNK) activity and increased apoptosis in the HSC/P compartment.20 However, Rac1-deficient HSC/Ps showed defective proliferative signaling by the c-Kit receptor tyrosine kinase, whereas loss of Rac2 activity led to a proapoptotic phenotype.12 Double-deficient Rac1−/−;Rac2−/− HSC/Ps showed a combination of reduced proliferation and increased apoptosis. It remains to be seen if any of the previously implicated Rac1 effectors, including PAK, POR1, IQGAP, and WAVE, and downstream transcription regulators such as p42/p44 extracellular signal-regulated kinases, p38, JNK, Akt, and/or β-catenin12,16,26-28 are directly involved in mediating Rac1/Rac2 proliferative signals in maintaining the healthy state of HSC/Ps. Interestingly, Rac1 and a GAP termed McgRacGAP are also required for nuclear localization of Stat5a through direct interaction,29 suggesting a role for Rac in regulation of transcription factor translocation to the nucleus and control of gene expression.
The effect of RhoA GTPase on hematopoiesis has been less studied compared with Rac1, Rac2, and Cdc42. RhoA activation can induce stress fiber formation and cell shape changes, decrease expression of Cdk inhibitors, and promote S phase cell-cycle progression in fibroblasts.1-3 However, the effect of RhoA on cell cycle and proliferation may be both cell type and agonist specific. Thus far, the role of RhoA in HSC/P functions has only been indirectly studied (Table 1). Inhibition of RhoA activity through expression of the dominant-negative mutant RhoAN19 by retrovirus-mediated gene transfer of murine BM cells was associated with a significant enhancement of HSC/P engraftment in vivo.30 More recently, HSC/Ps from p190-B-RhoGAP–deficient mice, which exhibit elevated RhoA activity, were found to have increased HSC self-renewal activity in serial transplantation experiments.31 This activity was independent of cell-cycle control; rather, p190-B seemed to affect HSC fate decision during cell division. These studies suggest that HSC maintenance probably requires a tightly regulated RhoA activity. Further analysis of RhoA function awaits the development of a RhoA knockout model.
Rho GTPases in myelopoiesis and erythropoiesis
The production of mature blood cells of the different lineages requires the differentiation of HSC/Ps through a successive series of increasingly lineage-restricted intermediate progenitors. The earliest step of commitment occurs at the level of the common lymphoid progenitor (CLP), and the common myeloid progenitor (CMP). CLPs differentiate into T, B, and natural killer cells,32 whereas CMPs give rise to the granulocyte-macrophage progenitor (GMP) and the megakaryocyte-erythroid progenitor (MEP).33 This differentiation program is regulated by an intertwining transcription factor network, tightly controlled by both dosage and timing.34-36
The role of Rho GTPases in regulating specific steps of myelopoiesis and erythropoiesis has just begun to be appreciated. The involvement of Rac in myelopoiesis has been examined in vitro with the use of isolated HSC/Ps from conditional knockout mice, and it appears that Rac1 and Rac2 GTPases regulate unique aspects of hematopoietic development. Rac1−/−;Rac2−/− as well as Rac1−/− HSC/Ps displayed reduced proliferation in response to stem cell factor, and this is associated with reduced cyclin D1 and decreased p42/p44 extracellular signal-regulated kinase phosphorylation.12 Increased apoptosis was noted in Rac1−/−;Rac2−/− and Rac2−/− progenitors, which was associated with reduced AKT activation after stem cell factor stimulation.12 How such observations relate to in vivo myeloid cell development remains unclear, because Rac2−/− and Rac1−/−;Rac2−/− mice show neutrophilia and defective mast cell functions.12,27,37
In erythropoiesis, Rac GTPases were shown to be essential for enucleation using constitutively active or dominant-negative mutants of Rac1 and Rac2 in cultured mouse fetal liver erythroblasts, in which either excessive activation or inhibition of Rac GTPases inhibits enucleation.38 Pharmacologic inhibition of Rac GTPases by NSC23766, a specific Rac inhibitor,39 was also shown to inhibit enucleation by disruption of the interaction of Rac with the diaphanous related-formin mDia2, leading to a disappearance of contractile actin ring in enucleating erythroblasts.38 However, enucleation is not decreased in Rac1−/−;Rac2−/− erythroblasts, in which up-regulated Rac3 may compensate for Rac1 and Rac2 deficiency. It is possible that the redundant activities of Rac1/Rac2/Rac3 GTPases collectively regulate the enucleation process.
Combined Rac1 and Rac2 deficiency in mice caused hemolytic anemia as evidenced by concurrent reticulocytosis, fragmented red blood cells (RBCs), and decreased deformability under shear stress, indicating an erythrocyte cytoskeleton defect. Indeed, Rac1−/−;Rac2−/− RBCs showed disturbed actin distribution in the cytoskeleton, with gaps and aggregates of F-actin and irregular clumping of band 3.40 Adducin, the capping protein of the actin oligomers at the rapidly exchanging barbed ends, is hyperphosphorylated on serine-724, a target of protein kinase C,41 thus facilitating uncapping with a potential promotion of actin polymerization as well as actin dissociation from spectrin40 (Figure 3). Although this pathology may be related to abnormal assembly of the cytoskeleton during erythropoiesis, a dynamic regulation of the actin oligomers by Rac GTPases during the life of the mature RBCs in the blood stream is an attractive possibility in explaining the remarkable deformability shown by normal erythrocytes under shear stress. The reticulocytosis that provides partial compensation of hemolytic anemia observed in Rac1−/−;Rac2−/− mice is produced by increased erythroid progenitors and precursors in the spleen, whereas MEPs along with erythroid colony-forming unit (CFU-E) activity are significantly decreased in the BM.42 These results indicate that Rac1 and Rac2 are essential for homeostatic erythropoiesis in the BM but are dispensable for stress erythropoiesis in the spleen.

In erythrocytes that lack Rac1 and Rac2 GTPases, there is increased phosphorylation of adducin by protein kinase C, leading to decreased F-actin capping at the barbed ends, dissociation of spectrin from actin, and increased fragility of the RBC cytoskeleton.
In erythrocytes that lack Rac1 and Rac2 GTPases, there is increased phosphorylation of adducin by protein kinase C, leading to decreased F-actin capping at the barbed ends, dissociation of spectrin from actin, and increased fragility of the RBC cytoskeleton.
Cdc42 has emerged as an essential regulator of the balance between myelopoiesis and erythropoiesis. Mice with deletion of Cdc42GAP (and therefore increased Cdc42 activity) displayed a reduction of BM cellularity with decreased granulocyte, erythroid, macrophage, megakaryocyte CFU; erythroid burst-forming unit, and CFU-E activities and anemia.20 This decrease in erythroid and myeloid progenitors correlated with increased apoptosis associated with increased JNK activity. Conditional deletion of Cdc42 in mice altered the frequency and number of primitive committed progenitors, leading to an increase in the GMP population and a decrease in the MEP population and significantly reduced erythroid burst-forming unit and CFU-E activities in Cdc42−/− BM cells.43 Hematopoietic-specific Cdc42−/− mice develop a fatal myeloproliferative disorder manifested by significant leukocytosis with neutrophilia in the peripheral blood along with myeloid hyperproliferation in the BM and spleen and myeloid cell infiltration of other organs, including lungs and liver.43 In parallel, severe anemia is noted in these mice, caused by a block in the early stages of erythropoiesis and associated with both proliferation and survival of Gr1+/Mac1+ cells. Because survival and proliferation in the erythroblast stages appeared normal, it is likely that Cdc42 deletion affects the erythroid differentiation program at a stage before proerythroblasts, such as the MEP level. These effects in both myeloid and erythroid lineages are probably caused by a biased gene transcription program in the Cdc42-deficient progenitors, including up-regulation of promyeloid genes (PU.1, C/EBP1α, and Gfi-1) in CMP, GMP, and MEP populations and down-regulation of proerythroid transcription factor GATA-2 in short-term repopulating-HSC, CMP, and MEP populations.43 Addressing how Cdc42 elicits signaling events to affect the key transcription factor network is an important subject for future studies.
Rho GTPases in lymphopoiesis
The role of Rho GTPases in lymphocyte biology was first appreciated by overexpressing dominant-negative and/or active forms of Rho GTPases in clonal cell lines (Table 1). With the use of this approach, Rho GTPases were suggested to mediate T-cell polarity, cytotoxic responses, spreading, migration, cytokine secretion, and T-cell receptor (TCR) and B-cell receptor (BCR) signaling.44-48 Analysis of Rac1 transgenic mice has shown that Rac1 regulates T-cell development by promoting the transition from CD4−CD8− double-negative stage III (DN3) to DN4 and by diverting CD4+CD8+ double-positive thymocytes from positive to negative selection.49,50 Examination of Rac2 and Cdc42 transgenic mice showed a role for Rac2 and Cdc42 in T-cell survival,51,52 whereas studies of a RhoA transgenic model showed that RhoA promotes positive selection of double-positive thymocytes and enhanced TCR signaling with increased proliferation on TCR cross-linking.53-55
Rho GTPases in T-cell development and activation
Gene targeting of either Rac1 or Rac2 found no apparent abnormalities in T-cell development.56-58 However, when both Rac1 and Rac2 are removed, T-cell development is severely impaired at the CLP stage and at checkpoints of β selection and positive selection. The developmental block is associated with defects in thymocyte proliferation, survival, adhesion, and migration. Further, simultaneous loss of Rac1 and Rac2 suppresses TCR-mediated interleukin-2 production and Akt activation and leads to hyperactivation of Notch signaling.57,58 Thus, Rac1 and Rac2 play a redundant but essential role in T-cell development. Rac2−/− mice show attenuated T helper type 1 differentiation of peripheral T cells associated with impaired production of interferon-γ.59 Moreover, Rac2 deficiency causes a disruption of actin polymerization and TCR clustering. It also produces a defect in calcium influx and Erk and p38 activation.56 Nonetheless, these defects are minor. Whether the defects are attenuated because of a compensatory effect of Rac1 remains to be elucidated.
In a case of cross talk among Rho GTPases, overexpression of RhoH inhibits Rac1- and RhoA-mediated activation of nuclear factor-κB, as well as p38 mitogen-activated protein kinase signaling, in Jurkat T cells.60 Thus, RhoH may function as an antagonist for the classic Rho GTPases. Physiologic functions of RhoH in T cells have recently been investigated in RhoH knockout mice. These mice display a defective T-cell maturation during transition from DN3 to DN4 and during positive selection.61,62 RhoH deficiency leads to defective TCR signaling manifested by decreased activation of CD3ζ, LAT, PLCγ, Vav1, and Erk, and by reduced calcium influx.61,62 Interestingly, RhoH was found to interact directly with tyrosine kinase Zap70. In the absence of RhoH, translocation of Zap70 to the immunologic synapse is impaired.62 RhoH is clearly involved in the early signaling steps of T cells, and this may be associated with its potential role in lymphocyte malignancies (see “Rho GTPases in hematologic abnormalities”).
Although the Rho family member RhoG shares high homology with Rac GTPases,63 it has been shown to uniquely regulate gene transcription in both T and B cells and to mediate cytoskeletal reorganization in T cells in response to fibronectin binding.64 RhoG knockout mice did not show a developmental defect in T cells; however, depletion of RhoG resulted in a moderate increase in T-cell proliferation in response to antigen receptor engagement.65
Rho GTPases in B-cell development and activation
Our understanding of Rho GTPases in B cells is limited in scope. Rac2-deficient mice show a reduction in B1a and marginal zone B cells associated with impaired proliferation and calcium influx.66 Furthermore, Rac2 deficiency leads to a decrease in immunoglobulin M–secreting plasma cells and hence an attenuated humoral immune response. In contrast, Rac1 deficiency has minimal effects on B cells.67 However, simultaneous ablation of Rac1 and Rac2 in B cells causes a more severe developmental block than the one caused by Rac2 deficiency alone.67 The B-cell developmental defects in Rac1/Rac2 double knockout mice are likely due to a survival defect resulting from impaired activation of Akt and a reduced expression of antiapoptotic Bcl-xL and blockade of B cell–activating factor receptor.67 These findings are reminiscent of the overlapping and critical role of Rac1 and Rac2 in T-cell development.57,58
A recent characterization of mice bearing B cell–specific deletion of Cdc42 showed that, similar to Rac1 and Rac2 simultaneous loss, Cdc42 deficiency leads to reductions in all splenic B-cell subpopulations and B1a cells, possibly because of defects in proliferation and B cell–activating factor receptor–mediated survival.68 In contrast to the Rac2 knockout effect, Cdc42 deficiency has no effect on B-cell migration.68 Therefore, although both Cdc42 and Rac1/Rac2 are important in late B-cell development in the spleen by mediating B-cell survival and proliferation, Rac1/Rac2, but not Cdc42, are required for B-cell migration.
Combining the results of overexpression and gene-targeting studies, the functional requirement of individual members of the Rho family in the critical steps of lymphopoiesis is becoming clear (Figure 4).

Scheme of Rho GTPase involvements in lymphocyte development. Rac1 and Rac2 are important for common lymphoid progenitor (CLP) differentiation from hematopoietic stem cells (HSCs) in the BM. Rac1 and Rac2, as well as RhoH, regulate T-cell development in the thymus by affecting β-selection and positive selection. Rac2 is also required for Th1 differentiation in peripheral lymphoid tissues. Rac1, Rac2, and Cdc42 are critical for multiple stages of B-cell development in the spleen, whereas Rac2, Cdc42, and RhoG also regulate antibody production. DN indicates CD4−CD8− double-negative thymocytes; DP, CD4+CD8+ double-positive thymocytes; SP, CD4+ or CD8+ single-positive T cells; Th, helper T cells; Tc, cytotoxic T cells; NFB, newly formed B cells; MZB, marginal zone B cells; and FOB, follicular B cells.
Scheme of Rho GTPase involvements in lymphocyte development. Rac1 and Rac2 are important for common lymphoid progenitor (CLP) differentiation from hematopoietic stem cells (HSCs) in the BM. Rac1 and Rac2, as well as RhoH, regulate T-cell development in the thymus by affecting β-selection and positive selection. Rac2 is also required for Th1 differentiation in peripheral lymphoid tissues. Rac1, Rac2, and Cdc42 are critical for multiple stages of B-cell development in the spleen, whereas Rac2, Cdc42, and RhoG also regulate antibody production. DN indicates CD4−CD8− double-negative thymocytes; DP, CD4+CD8+ double-positive thymocytes; SP, CD4+ or CD8+ single-positive T cells; Th, helper T cells; Tc, cytotoxic T cells; NFB, newly formed B cells; MZB, marginal zone B cells; and FOB, follicular B cells.
Rho GTPases in phagocyte regulation
Neutrophils provide the first line of defense by eradicating invading microbial pathogens. This is achieved through a series of highly coordinated responses of migration, ingestion, and ultimately killing of invading microbes. By regulating cytoskeleton rearrangements, Rho GTPases have been implicated as central regulators of specialized phagocyte functions69,70 (Table 1; Figure 5).

Phagocyte functions in response to bacterial infections. After sensing chemokines released by invading microorganism, leukocytes rapidly move toward the site of infection. This process involves initial changes in adhesive properties of the cells to the vessel endothelium followed by an extravasation step out of the blood vessel and directed migration into tissue. Once at the site of infection, cells phagocytize microbes and kill them by degranulation and reactive oxygen species (ROS) release.
Phagocyte functions in response to bacterial infections. After sensing chemokines released by invading microorganism, leukocytes rapidly move toward the site of infection. This process involves initial changes in adhesive properties of the cells to the vessel endothelium followed by an extravasation step out of the blood vessel and directed migration into tissue. Once at the site of infection, cells phagocytize microbes and kill them by degranulation and reactive oxygen species (ROS) release.
Migration
Directional neutrophil migration in response to a gradient of chemoattractant allows neutrophils to rapidly arrive at sites of infection. This process requires the establishment of a front–rear asymmetry, with a front enriched in filamentous actin and a rear made of actomyosin filaments. In general, Rac is the crucial regulator of actin polymerization, RhoA promotes myosin cytoskeleton organization at the rear, whereas Cdc42 is essential in organizing cell polarity, although it is not required for motility per se.69-72
Genetic models have shown that actin polymerization is specifically dependent on Rac2 rather than on Rac1 or Cdc42. Whereas Rac2−/− neutrophils exhibit a dramatic defect in polymerized actin, Rac1−/− neutrophils and neutrophils with deregulated Cdc42 exhibit normal F-actin polymerization.12,37,73-75 Instead, Rac1 has been shown to play significant roles in neutrophil shape and tail retraction.12,71 Recent work that used primary neutrophils from Cdc42GAP−/− and Cdc42−/− mice further confirm the involvement of Cdc42 in neutrophil polarity.76,77 Mechanistic studies suggest an unexpected mechanism in which CD11b may serve as a secondary signal of Cdc42-mediated polarity to determine the site of actomyosin assembly. Whereas the phenotypes of RhoA genetic deletion in phagocytes remain unknown, RhoA distributes at the rear of primary neutrophils and may regulate actomyosin contraction.73,78
Closely related to directional movement, gradient sensing, which is necessary to orient the cells toward high chemokine concentrations, also appears to depend on Rho GTPases but not on phosphatidylinositol 3-kinase, as initially proposed.79 Neutrophils deficient in the PAK-associated Rac GEF, PIXα, which exhibits low Cdc42 activity, fail to orient toward high chemokine (ie, C5a) concentration.80 Interestingly, this is also the case for Rac1−/− neutrophils, at least under defined experimental conditions.75
The mechanisms enabling macrophage migration are different from those used by neutrophils. For example, Rac1 and Rac2 appear to be largely dispensable for macrophage migration.81 It is not clear how macrophages migrate without Rac1 and Rac2. RhoB may contribute to this activity, because the speed of RhoB−/− macrophage migration is significantly impaired compared with wild-type macrophage migration.82
Phagocytosis
The uptake of bacteria by phagocytosis is the first step of bacterial killing. This process involves either integrin (CD11b or complement receptor 3 [cr3])–mediated or Fcγ receptor–mediated internalization. Because phagocytosis requires actin rearrangement beneath the phagocytic cup, Rho GTPases are implicated in this process.83,84 The prevailing view was that Rac and Cdc42, but not RhoA, activities were necessary for Fcγ receptor–mediated internalization. In contrast, RhoA seemed to be the main regulator of CR3-mediated uptake, also by controlling actin remodeling, although through a distinct structure than the one regulated by Rac and Cdc42.85 Recent work that used Rac1/Rac2-null macrophages have challenged this view.86 The genetic deletion of Rac1 and Rac2 prevents both CR3-mediated and Fcγ receptor–mediated phagocytosis (through defective actin rearrangement). In contrast, studies that used C3 transferase to inhibit RhoA in primary macrophages suggest that RhoA may also affect both FcγR and CR3-mediated phagocytosis, but independently of actin polymerization.86 Because other studies that used Rac2-null macrophages have suggested that Rac2 controls Fcγ receptor–mediated, but not CR3-mediated, phagocytosis,87 it will be important to further analyze the specific roles Rho GTPases play in phagocytosis in neutrophils or macrophages or both.
Bacteria killing
Once bacteria are internalized, the microbicidal function of phagocytes is largely ascribed to their ability to release reactive oxygen species (ROS) and hydrogen peroxide on activation of the reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex. This function has been attributed to Rac activity.69,70,88 Under cell-free conditions, both Rac1 and Rac2 can stimulate NADPH oxidase activity. However, genetic deletion of Rac2, but not Rac1, in neutrophils results in severe defects in ROS production, although in an agonist-dependent fashion.12,37,76,89,90 Importantly, ectopic expression of Rac1 in Rac2-null neutrophils cannot complement for the lack of Rac2 in superoxide production.89,91 Interestingly, Rac2-null macrophages exhibited defects in NADPH oxidase activation in a similar agonist-dependent fashion.87 RhoG has also been implicated in ROS production. RhoG−/− neutrophils exhibited a marked impairment in ROS production; however, this effect may be through Rac2 protein.92 The roles of other Rho GTPases in NADPH oxidase regulation remain to be analyzed in gene-targeted models. Cdc42 might be an important counter-regulator of Rac2-induced NAPH activation, because inactivation of Cdc42 by the introduction of the Cdc42/Rac interactive binding domain of WASP caused a dramatic increased in ROS production.93
Release of neutrophil granule content can also contribute to microbicidal activity. Both Rac1 and Rac2 are probably important during this process. Rac2−/− neutrophils failed to release primary granule content in response to chemoattractant stimulation, and Rac1 has been shown to play a role in myeloperoxidase release.94,95
The use of mouse infectious or inflammatory models has shown the importance of Rho GTPases in innate immunity. Rac2−/− mice show an increased mortality to invasive aspergillosis.37 Mice with Rac1−/− neutrophils are protected against neutrophilic lung inflammation.73 These analyses of Rho GTPase functions in primary cells derived from genetic models show a more complex picture than the original paradigm (Table 1).
Rho GTPases in hematologic abnormalities
Given their critical role in gene transcription, cell survival, adhesion, and cytoskeleton organization, it is no surprise that several Rho GTPases and their signaling pathways have been implicated in hematologic pathogenesis.
Hematologic abnormalities associated with decreased Rho GTPase activity
The first clearly defined mutation of a Rho GTPase family protein associated with a hematologic disease was described nearly a decade ago with the observation that an infant with severe immune defects harbored a missense mutation on 1 allele of the Rac2 gene, resulting in an asparagine residue in place of aspartic acid in the guanine nucleotide binding site.96,97 The patient had recurrent bacterial infections and a severe neutrophilia, phenotypic characteristics closely mimicking that of Rac2−/− mice.37 Biochemical and cell biology experiments showed that the Rac2 D57N mutant behaved as a dominant negative toward both Rac1 and Rac2, effectively ablating Rac activity in blood cells, causing growth and survival defects as well as engraftment and migration deficiencies.12,18,98
Recently, an association between decreased activity of Cdc42 and the hematologic disease Fanconi anemia (FA) has been described.99 FA is an inherited BM failure syndrome caused by mutations of a family of FA genes. Mouse model studies of FA have concluded that the defect is at the level of the HSC/P, but the mechanisms responsible for the HSC/P failure remain elusive.100-102 It was shown that aberrant down-regulation of Cdc42 activity by the loss of the Fanc-A gene could explain the defective homing, adhesion, and migration phenotype associated with patient-derived BM cells. On expression of an active form of Cdc42, the adhesion defect could be corrected.99 These studies implicate Cdc42 in the functional deficiency of FA and highlight the importance of this Rho GTPase in a hematopoietic stem/progenitor deficiency syndrome.
The effector of Cdc42, WASP, has been known to be involved in the rare immune deficiency disease, Wiskott-Aldrich syndrome.103 Mutations in the WASP gene result in loss of function of the WASp protein, and the defect seems to be at the level of the HSCs and is associated with defective migration.104 Clinically, this syndrome presents as an immune disorder with variable susceptibility to opportunistic infections. This is consistent with the idea that loss of Cdc42 effector function in hematopoietic cells can be causal for aberrant development and function of the immune system with associated immune deficiency and susceptibility to infection.
Hematologic abnormalities associated with increased GTPase activity
In contrast to the phenotypes associated with loss of Rho GTPase activity, the disease spectrum that is associated with increased Rho GTPase activity is often linked to enhanced proliferation and survival in diverse cancers of the blood. In non-Hodgkin lymphoma and multiple myeloma, transcriptional up-regulation of RhoH, a constitutively GTP-bound family member that is not regulated by the classic GEF/GAP molecular switch, is effected by recurrent chromosomal translocations t(3;4) and t(4;14) as well as through somatic hypermutation.105,106 The precise result of these genetic changes as they relate to the tumor phenotype is not clear, but studies from genetic models and biochemical analyses suggest that deregulated RhoH signaling may have profound effects on the survival, adhesion, and migration properties of cells.107,108 Interestingly, RhoH expression was recently found to be elevated in B cell chronic lymphocytic leukemia109 and suppressed in B hairy cell leukemia.110 In these diseases, RhoH depletion or RhoH overexpression, respectively, were effective in reversing the transforming phenotype, suggesting that RhoH may be distinctly involved in the initiation or maintenance of these B cell lymphoproliferative disorders.
The role of Rac GTPase signaling in myeloid-associated disease is well documented, especially in the context of the p210-BCR-ABL fusion protein and chronic myeloid leukemia. This particular subject has been the topic of a recent review.111 Rac1 and Rac2 appear to function downstream of BCR-ABL and are essential for the transforming ability of this oncogene (Figure 6). Rac3 has also been shown to play a role downstream of p190 BCR-ABL in the generation of lymphoblastic leukemia.112 Although the precise mechanism of activation and the interplay of Rac family members in the initiation, maintenance, or dissemination of BCR-ABL disease is not known, the importance of Rac GTPases in chronic myeloid leukemia is clearly documented in mice with the use of a genetic approach and confirmed in human cells with the use of a Rac inhibitor.113

A biochemical model of Rac GTPase involvement in hematopoietic cell transformation. Rac1 has unique as well as redundant roles with Rac2 and Rac3 as signal transducers in multiple oncogene or tumor suppressor gene–mediated leukemogenesis or lymphomagenesis, in addition to their physiologic roles in mediating receptor-stimulated signaling. They are required for p210 BCR-ABL–induced HSC transformation, and MLL-AF9–induced HSC/GMP transformation. Targeting of Rac GTPases has been suggested as a potential therapeutic means in the blood malignancies.
A biochemical model of Rac GTPase involvement in hematopoietic cell transformation. Rac1 has unique as well as redundant roles with Rac2 and Rac3 as signal transducers in multiple oncogene or tumor suppressor gene–mediated leukemogenesis or lymphomagenesis, in addition to their physiologic roles in mediating receptor-stimulated signaling. They are required for p210 BCR-ABL–induced HSC transformation, and MLL-AF9–induced HSC/GMP transformation. Targeting of Rac GTPases has been suggested as a potential therapeutic means in the blood malignancies.
Rho GTPase activation in acute myeloid leukemia (AML) has recently received significant scrutiny. It has been known for almost a decade that specific translocations in AML target signaling molecules involved in Rho GTPase activation. For example, a translocation involving the mixed lineage leukemia (MLL) gene on chromosome 11 was found to target a Rho GEF, LARG.114 The MLL-LARG fusion protein was shown to activate RhoA and play a mechanistic role in the development of the leukemia, implicating the constitutive Rho GTPase activity in the pathogenesis of AML.115 MLL has also been found in a translocation with GRAF, a GAP that is involved in “turning off” RhoA activity.116 The MLL-GRAF translocation inactivates the GAP activity of GRAF, resulting in constitutive activation of RhoA. A follow-up study analyzing GRAF expression in AML samples determined that 38% of samples showed CpG methylation of the GRAF promoter, implicating loss of expression of GRAF in AML development.117 In addition to RhoA activation in AML, loss of RhoA activity may also be associated with T-cell malignancy. T cell–specific expression of the bacterial toxin C3 transferase, a Rho inhibitor, promoted aggressive thymic lymphoblastic lymphoma in mice.118 Whether these differences are due to specific cell lineage requirements or to the different approaches used to generate the models remains to be experimentally determined.
It has also been reported that the Rac and Cdc42 GTPases are up-regulated in murine AML cells using a retroviral model of MLL-AF9 leukemia.119 These observations were subsequently confirmed in another model of this type of leukemia, using primary human rather than murine cells to model AML.120 Inhibition of Rac activity using either a pharmacologic or a knockdown approach was effective in inducing apoptosis in the human MLL-AF9 leukemia cells and showed a significant delay of the progression of AML induced by a human leukemia cell line.120,121 In addition, primary AML patient samples have been shown to be highly sensitive to Rac GTPase inhibition in vitro, potentially through a mechanism requiring stroma cell interaction.122
In addition to these blood malignancies, Rho GTPase activities have been suggested to play an important role in the hematologic pathologies associated with certain viral infections, including HIV and γ herpesviruses. The HIV protein v-Nef binds to Vav1 and activates the Rac signaling pathway.123 Down-regulation of Rho activity using statins or a geranylgeranyl transferase inhibitor, but not a farnesyl transferase inhibitor, specifically blocked entry of HIV-1–pseudotyped viruses, implicating Rho as a potential target for antiretroviral therapy.124 In the γ herpesvirus MHV-68, the viral protein M2 is necessary for activating the Vav1/Rac signaling cascade and promoting viral latency in B lymphocytes, a process critical in the life cycle of this virus and essential for effective colonization of the host.125,126
Although much remains to be studied about the specific mechanistic contributions of Rho GTPase signaling that are important in blood malignancies, the principle of pharmacologic targeting of Rho GTPase pathways, such as Rac signaling, presents an exciting new opportunity for future therapy (Figure 6). To this end, the development of lead inhibitors of this subfamily of Rho GTPases has shown promise, suggesting pharmacologic targeting of aberrant Rac activities may be a valid approach in reversing the malignant phenotypes of leukemia and lymphoma. As a demonstration of this principle, based on the crystal structure of Rac1 and the structure–function data of Rac1 interaction with GEFs, a small molecule inhibitor, NSC23766, was identified that showed specificity for a subset of GEF binding to Rac GTPases and blocked Rac activation by several GEFs.39 Although this lead inhibitor could in part mimic the Rac1 and Rac2 knockout phenotype of HSC/P mobilization from the BM,18 it displayed potent efficacy in suppressing the elevated Rac1 and Rac2 activities in BCR-ABL or MLL-AF9 oncogene–induced myeloproliferative disorder or AML-like syndrome and significantly slowing the disease progressions.113,120,121 These studies strongly imply that pharmacologic agents can be developed to tackle various pathologic conditions associated with malregulated activation states of individual Rho GTPases.
Conclusions and perspectives
The mouse gene–targeting approach has been invaluable in understanding Rho GTPase signaling functions in mammalian hematopoiesis and has led to our current understanding of some of the unique versus overlapping roles of individual Rho GTPases in different blood cells (Table 1). It is becoming clear that Rho GTPases Rac1, Rac2, Cdc42, RhoA, RhoH, and RhoG, as well as several regulators and effectors, are involved in multiple steps of hematopoiesis, including stem cell interaction with the BM niche, erythropoiesis, myelopoiesis, and lymphopoiesis, and it is likely that considerable cross talk may exist between the family members in defined functions (Figure 7). An emerging theme is that, although individual Rho GTPases may regulate unique aspects of cell proliferation, survival, or differentiation in defined blood cell types, they collectively play an essential role in regulating cytoskeleton organization, adhesion, and migration for most blood cell types.

A summary of the known involvement of Rho GTPases in the regulation of hematopoiesis based on mouse gene–targeting studies. Cdc42, Rac1, Rac2, RhoH, RhoG, and RhoA are among the best understood Rho GTPase family members for which mouse gene–targeting models have been characterized in various blood lineages. LTR indicates long-term repopulating; and STR, short-term repopulating. Other abbreviations of various progenitors are described in the text.
A summary of the known involvement of Rho GTPases in the regulation of hematopoiesis based on mouse gene–targeting studies. Cdc42, Rac1, Rac2, RhoH, RhoG, and RhoA are among the best understood Rho GTPase family members for which mouse gene–targeting models have been characterized in various blood lineages. LTR indicates long-term repopulating; and STR, short-term repopulating. Other abbreviations of various progenitors are described in the text.
Because Rho GTPases are capable of mediating signal transduction in multiple pathways, a continuing effort is to define blood cell type– and pathway-specific signaling functions using cell type–restricted knockout models. Inducible knockout or knockin mouse models under the control of endogenous promoters, as well as cross-breeding among various Rho GTPase or Rho GTPase regulator knockouts and other signaling component-gene–targeted mice, will help clarify the relationship between the Rho GTPases and other signaling networks. The small interfering RNA technology will be complementary to the mouse gene–targeting studies in analyzing Rho GTPase functions in human blood cells and may be especially suitable in determining the contributions of overexpressed or elevated levels of individual Rho GTPases in human blood malignancies. It can be expected that the mouse genetic studies will produce useful animal models to implicate specific Rho GTPase–mediated signaling pathways in multiple human blood abnormalities, including neutropenia, anemia, leukemia, lymphoma, and immune deficiency. With our growing understanding of the molecular mechanisms of Rho GTPase signaling important in various physiologic processes and disease states, the signal transduction pathways involving Rho GTPases and the cascades of protein–protein interactions as well as enzymatic activities can be pursued as drug targets for future therapeutic benefits.
Acknowledgments
We thank Dr David A. Williams for a critical reading of the manuscript, and Mr Xuan Zhou for a draft of the figures.
This work was supported by grants from the National Institutes of Health.
National Institutes of Health
Authorship
Contribution: J.C.M., J.A.C., M.-D.F., T.A.K., F.G., and Y.Z. surveyed the literature, generated the figures, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yi Zheng, Division of Experimental Hematology and Cancer Biology, Children's Hospital Medical Center, 3333 Burnet Ave, Cincinnati, OH 45229; e-mail: yi.zheng@cchmc.org.
