

Key Points
The DNA glycosylase MBD4 acts as a safeguard against damage from 5mC deamination.
Germ line MBD4 deficiency stimulates clonal hematopoiesis and guides the development of leukemia via recurrent mutations in DNMT3A.

Abstract
The tendency of 5-methylcytosine (5mC) to undergo spontaneous deamination has had a major role in shaping the human genome, and this methylation damage remains the primary source of somatic mutations that accumulate with age. How 5mC deamination contributes to cancer risk in different tissues remains unclear. Genomic profiling of 3 early-onset acute myeloid leukemias (AMLs) identified germ line loss of MBD4 as an initiator of 5mC-dependent hypermutation. MBD4-deficient AMLs display a 33-fold higher mutation burden than AML generally, with >95% being C>T in the context of a CG dinucleotide. This distinctive signature was also observed in sporadic cancers that acquired biallelic mutations in MBD4 and in Mbd4 knockout mice. Sequential sampling of germ line cases demonstrated repeated expansion of blood cell progenitors with pathogenic mutations in DNMT3A, a key driver gene for both clonal hematopoiesis and AML. Our findings reveal genetic and epigenetic factors that shape the mutagenic influence of 5mC. Within blood cells, this links methylation damage to the driver landscape of clonal hematopoiesis and reveals a conserved path to leukemia. Germ line MBD4 deficiency enhances cancer susceptibility and predisposes to AML.
Introduction
Cells are exposed to a variety of stresses that damage DNA. Most damage arises from endogenous sources, including exposure to reactive molecules and replication errors.1 Although the vast majority of these events are repaired, some are propagated and introduce mutations. This decay in genomic integrity has major implications for our health, particularly for modulating cancer incidence as we age. Fanconi anemia provides an illustration of this within the hematopoietic system. The specific DNA repair defects that underpin this family of diseases set the stage for a high risk of development of myelodysplasia and acute myeloid leukemia (AML) at an early age.2
DNA methylation on cytosine residues provides a major mutagenic stimulus, as 5-methylcytosine (5mC) has a tendency to undergo spontaneous deamination to thymine.3 Therefore, it is not surprising that CG>TG mutations are a prominent feature of age-related DNA damage, as detected in human cancers,4 normal stem cells,5 and de novo mutations passed through the germ line.6 This form of damage is so ubiquitous that it has been proposed as a molecular clock to track aging.4 CG>TG mutations make an important contribution to the somatic mutation landscape of cancer,7 and it is important to delineate how the repair pathways that restrict methylation damage modify cancer risk.
Methylation damage is repaired by the base excision repair (BER) pathway. After deamination of 5mC, removal of the mispaired thymine is accomplished by 1 of 2 DNA glycosylases, methyl-binding domain 4 (MBD4)8 or thymine DNA glycosylase (TDG).9 Inactivation of Mbd4 in mice confirmed a functional role in repair of methylation damage,10,11 but whether it protects against cancer remains unclear. In this report, we characterize familial cases with germ line inactivation of MBD4 and demonstrate its crucial role in safeguarding against methylation damage and vulnerability to the development of AML and some solid cancers.
Methods
Patient characteristics and sample collection
Patients provided informed consent in accordance with the Declaration of Helsinki for participation in research and for collection of samples over the course of their treatment. This research project was approved by our respective human research ethics committees (HRECs) (Erasmus Medical Center [EMC] Medical Review Ethics Committee project MEC 2015-155, Walter and Eliza Hall Institute of Medical Research [WEHI] HREC project 13/01, Melbourne Health HREC project 2012.274). EMC-AML-1, WEHI-AML-1, and WEHI-AML-2 were diagnosed with AML and treated with combination chemotherapy as per the protocols at their respective institutions.
EMC-AML-1 was 33 years old when diagnosed with acute monocytic leukemia (AML, World Health Organization [WHO] International Classification of Diseases [ICD] 9891/3). The AML had trisomy 11 on karyotyping and was negative for NPM1, FLT3, and CEBPA mutations. His medical history included colonic polyps requiring a hemicolectomy 2 years prior to his AML diagnosis. His AML was refractory to induction chemotherapy (standard dose cytarabine and daunorubicin). Repeat induction with intermediate dose cytarabine resulted in complete morphologic and cytogenetic remission. He then had an autologous hematopoietic stem cell transplant (HSCT) with BU-CY conditioning (busulfan and cyclophosphamide). He relapsed 2 years and 3 months postautologous HSCT. The AML at relapse had a normal karyotype and was negative for NPM1, FLT3, and CEBPA mutations. Salvage induction chemotherapy (high-dose cytarabine, mitoxantrone, and etoposide) resulted in complete morphologic remission. This was followed by an allogeneic HSCT from a matched unrelated donor with myeloablative and total body irradiation conditioning. He achieved complete morphologic remission with full donor chimerism. He developed extensive graft-versus-host disease with secondary graft failure responsive to steroids and Epstein-Barr virus reactivation requiring rituximab. He died 2 years postallogeneic HSCT with relapsed AML.
WEHI-AML-1 was 31 years old when diagnosed with AML with myelodysplasia-related changes (myelodysplastic syndrome–associated cytogenetic abnormality, monosomy 7, WHO ICD 9895/3). The AML was negative for NPM1, FLT3, and CEBPA mutations. She had induction chemotherapy (high-dose cytarabine, idarubicin, and etoposide) and achieved complete morphologic and cytogenetic remission. This was followed by 2 cycles of consolidation chemotherapy (standard-dose cytarabine, idarubicin, and etoposide). Early morphologic relapse was detected on bone marrow examination prior to allogeneic HSCT from her female sibling (WEHI-AML-2) with BU-CY conditioning. Bone marrow examination 5 weeks postallogeneic HSCT showed complete morphologic and cytogenetic remission, as well as full donor chimerism. Relapsed AML (of WEHI-AML-1 origin) occurred 11 weeks postallogeneic HSCT. Salvage therapy with FLAG chemotherapy regimen (fludarabine, cytarabine, and filgrastim) proved unsuccessful. WEHI-AML-1 died of relapsed AML <12 months after diagnosis.
WEHI-AML-2 was 30 years old when she donated peripheral blood stem cells to WEHI-AML-1. Her medical history included iron deficiency anemia secondary to menorrhagia and bleeding from descending colon and rectal polyps. Her full blood count was normal at the time of stem cell donation. Her routine full blood count 4 years later, at 34 years old, showed pancytopenia. A diagnosis of AML with myelodysplasia-related changes (myelodysplastic syndrome–associated cytogenetic abnormality, monosomy 7, WHO ICD 9895/3) was made on bone marrow examination. The AML was negative for NPM1, FLT3, and CEBPA mutations. She had induction chemotherapy (high-dose cytarabine and idarubicin) and achieved complete morphologic and cytogenetic remission. This was followed by 1 cycle of consolidation chemotherapy (standard-dose cytarabine, idarubicin, and etoposide). She then had an allogeneic HSCT using 2 partially HLA-matched umbilical cord blood units following FLU-CY-TBI conditioning (fludarabine; cyclophosphamide, and total body irradiation). She developed grade 1 graft-versus-host disease of the gut. She remains in complete morphologic and cytogenetic remission.
Samples from bone marrow and peripheral blood were collected over the course of their treatment (supplemental Table 1; available on the Blood Web site). WEHI-AML-2 was the donor for an allogeneic HSCT for WEHI-AML-1 and had peripheral blood taken for chimerism analysis at time of donation that was available for analysis.
Whole exome sequencing and whole genome sequencing
Whole exome sequencing on EMC-AML-1 was performed as previously described.12 For WEHI-AML-1 and WEHI-AML-2, 50 to 100 ng of DNA and the TruSeq Nano DNA Sample Preparation Kit (Illumina) were used to generate indexed DNA libraries. Whole genome sequencing was performed on a HiSeq X Ten (Illumina). Exome capture was performed with the Human All Exon v5_UTR Capture Library and the SureSelectXT2 Target Enrichment System (Agilent Technologies) before sequencing on a HiSeq2500 (Illumina). Alignment and variant calling are detailed in the supplemental Methods.
Assessment of MBD4 status and proportion of CG>TG mutations in TCGA
To assess the frequency of CG>TG mutations in The Cancer Genome Atlas (TCGA) samples, somatic single nucleotide variant (SNV) calls available through the National Cancer Institute Genomic Data Commons were filtered to restrict the analysis to variants with a variant allele frequency >20%, with minimum 20 reads coverage and that were recognized by at least 3 out of the 4 callers: SomaticSniper, VarScan2, MuTect2, and MuSE. This approach correlated well with results from our own analysis pipeline. Candidate germ line loss-of-function variants impacting MBD4 were sourced from Genomic Data Commons (September 2016) and analysis restricted to variants with a variant allele frequency >10%, found with a population frequency <1% in ExAC (non-TCGA cohort).13 The variant allele frequency and local copy number around MBD4 were assessed in the matched cancer sample to designate cases as either monoallelic or biallelic inactivation.
Reduced representation bisulfite sequencing (RRBS)
For WEHI-AML-1 and WEHI-AML-2, RRBS libraries were made from 75 to 100 ng of DNA using the Ovation RRBS Methyl-Seq System (NuGEN) with bisulfite conversion using the Epitect kit (Qiagen). The libraries were sequenced on a HiSeq2500. Enhanced RRBS data from EMC-AML-1 were available through the Database of Genotypes and Phenotypes (dbGaP) (phs001027), and RRBS data from a glioblastoma, GBM1063T, were available from Gene Expression Omnibus (GSE70175).14 RRBS sequencing reads were trimmed to remove adapters and low-quality sequence with Trim_Galore. Diversity adaptors were removed with a NuGEN python script (trimRRBSdiversityAdaptCustomers.py). Alignment to hg19 was performed with Bismark 0.13.0, and methylation status was assessed using bismark_methylation_extractor, ignoring 5 bases at the 5′ end of each read.15
Whole genome sequencing of Mbd4 wild-type and knockout mice
Mbd4 knockout mice (JAX stock #004989) were obtained from Jackson Laboratory.11 The mice were backcrossed an additional generation to C57BL/6, prior to intercrossing. All animal studies were approved by the WEHI Animal Ethics Committee (Project 2014.010). Mouse bone marrow cells were collected in Dulbecco modified Eagle medium (Thermo Fisher Scientific) containing 10% HyClone bovine calf serum, iron supplemented (Thermo Fisher Scientific). Ten thousand cells were cultured in 1 mL Dulbecco modified Eagle medium with 20% bovine calf serum, 0.3% agar (BD), 100 ng/mL murine stem cell factor, 10 ng/mL murine interleukin-3 (IL-3), and 2 IU erythropoietin.16 Cultures were incubated for 11 days at 37°C in a fully humidified atmosphere with 10% CO2. Individual colonies were isolated, and DNA was extracted using QIAamp DNA Micro Kit (Qiagen). DNA from individual colonies was amplified using the TruePrime WGA Kit (SYGNIS), and the amplified DNA was purified using QIAamp DNA Mini Kit (Qiagen). Mouse bone marrow DNA was extracted using DNeasy Blood & Tissue Kit (Qiagen). DNA was measured using the Agilent 2200 Tapestation Genomic DNA ScreenTape Assay (Agilent Technologies). Whole genome sequencing was performed on a NovaSeq (Illumina). DNA sequencing data were aligned to the mouse genome (mm10) using bwa-mem. Alignment, variant calling, and calculation of relative mutation rate were performed using the same approach outlined for the human sequencing data. Welch's t test was used to compare between the groups of samples (n = 3 per group).
Genomic profiling of single-cell–derived colonies (SCDCs) from EMC-AML-1
EMC-AML-1’s autologous stem cell transplant and diagnosis peripheral blood samples were used to obtain single hematopoietic progenitor cell colonies. Briefly, cells were thawed and sequentially diluted in Iscove modified Dulbecco medium (Thermo Fisher) supplemented with 5% human serum albumin (Thermo Fisher) and 20 U/mL heparin (ie, initially 1:1; after 10 minutes, 1:10; and after 20 minutes, 1:20). The cell suspension was centrifuged at 4°C, and cells were resuspended in cold phosphate-buffered saline. Cells were plated at different densities (0.04 to 2 × 105 cells per mL) in MethoCult GF H84434 Methylcellulose medium with cytokines (Stemcell Technologies) for 14 days at 37°C and 5% CO2. DNA was isolated from individual colonies using the QiaAmp DNA Micro Kit (Qiagen) and quantified using Qubit DNA HS assay kit (Life Technologies). The Illumina TruSight Myeloid Sequencing Panel (Illumina) was applied to detect mutations in genes frequently mutated in myeloid malignancy.
MBD4 glycosylase activity assays
MBD4 glycosylase activity assays were performed as previously described with the following modifications.17 The glycosylase activity of MBD4 protein (0.5 μM) on double-stranded FAM-labeled 32bp-oligonucleotides (0.1μM) was assessed and monitored by denaturing gel electrophoresis. The resulting FAM-labeled single-stranded DNA was visualized using the 473-nm laser (Blue LD Laser) and 530DF20 emission filter on a Typhoon FLA9500 (GE Healthcare).
The following 32-bp oligonucleotides were obtained from Integrated DNA Technologies: (FAM)-5′-TCGGATGTTGTGGGTCAGXGCATGATAGTGTA-3′ (where X = C or T); 5′-TACACTATCATGCGCTGACCCACAACATCCGA-3′. The double-stranded FAM-labeled matched and mismatched oligonucleotides were prepared by hybridization whereby 100 µM of oligodinucleotides were mixed in 50 µL annealing buffer containing 10 mM tris(hydroxymethyl)aminomethane HCl, 1 mM EDTA, and 50 mM NaCl (pH 8.0), then incubated at 95°C for 2 minutes, followed by a steady temperature reduction over 45 minutes to 25°C. The double-stranded duplexes were cooled and stored at 4°C.
Results
Germ line loss of MBD4 predisposes to AML with a novel mutational signature
We identified 3 patients with AML, including 2 siblings, that were distinctive because of their high mutational burden (∼33-fold above what is typical for AML) and unique mutational signature, where >95% of mutations were CG>TG (Figure 1A-B; supplemental Figure 1A). This signature differs from the distribution of C>T mutations generally observed in AML and is more refined than the mutational signature ascribed to aging,4 suggesting a near complete dependence on 5mC deamination. Although CG>TG mutations are an integral feature of age-related DNA damage and AML is most commonly a disease of older age (median age of onset is >70 years), all 3 patients were younger than 35 years at diagnosis.
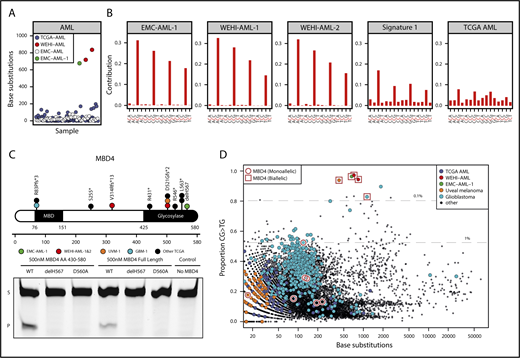
MBD4-deficient cancers exhibit a distinctive mutational signature. (A) Mutation burden in AML, presented as number of base substitutions per exome. Data sourced from dbGaP; cases are ordered on patient identifier (EMC: phs00102712 and TCGA: phs00017824 ). (B) Trimer context of C>T mutations in 3 MBD4-deficient AML cases. The center of origin is reflected in the sample label. For comparison, we show signature 1, the established signature associated with 5mC deamination, and all C>T mutations present in TCGA-AML. (C) Schematic representation of MBD4, highlighting germ line loss-of-function variants detected in the AML cases and cases within TCGA (at top). A glycosylase assay was performed to assess the activity of recombinant MBD4 (either AA430-580 or full length), wild-type (WT), delH567, or the catalytically inactive mutant D560A. Substrate (S) and product (P). Consistent results were obtained in 5 experiments for MBD4 AA430-580 and 3 experiments for full length. (D) The proportion of CG>TG mutations observed is set out against the total number of base substitutions detected for all TCGA samples. Samples with germ line MBD4 loss-of-function variants were designated either as heterozygous (monoallelic) or completely inactivated (biallelic) based on the genotype of the cancer (includes somatic mutations). Gray lines mark the top 1% and 0.1% of cases with the highest proportion of CG>TG mutations. A select set of tumor types are highlighted.
MBD4-deficient cancers exhibit a distinctive mutational signature. (A) Mutation burden in AML, presented as number of base substitutions per exome. Data sourced from dbGaP; cases are ordered on patient identifier (EMC: phs00102712 and TCGA: phs00017824 ). (B) Trimer context of C>T mutations in 3 MBD4-deficient AML cases. The center of origin is reflected in the sample label. For comparison, we show signature 1, the established signature associated with 5mC deamination, and all C>T mutations present in TCGA-AML. (C) Schematic representation of MBD4, highlighting germ line loss-of-function variants detected in the AML cases and cases within TCGA (at top). A glycosylase assay was performed to assess the activity of recombinant MBD4 (either AA430-580 or full length), wild-type (WT), delH567, or the catalytically inactive mutant D560A. Substrate (S) and product (P). Consistent results were obtained in 5 experiments for MBD4 AA430-580 and 3 experiments for full length. (D) The proportion of CG>TG mutations observed is set out against the total number of base substitutions detected for all TCGA samples. Samples with germ line MBD4 loss-of-function variants were designated either as heterozygous (monoallelic) or completely inactivated (biallelic) based on the genotype of the cancer (includes somatic mutations). Gray lines mark the top 1% and 0.1% of cases with the highest proportion of CG>TG mutations. A select set of tumor types are highlighted.
Sequencing germ line DNA from the 3 cases identified loss-of-function variants in the gene encoding the DNA glycosylase MBD4, which plays a key role in initiating repair after 5mC deamination8 (Figure 1C; supplemental Table 2). Case EMC-AML-1 carried a homozygous deletion of Histidine 567 (H567) in the glycosylase domain of MBD4. An in vitro glycosylase assay confirmed that loss of H567 results in a catalytically inactive MBD4 protein (Figure 1C). The siblings (WEHI-AML-1, WEHI-AML-2) were compound heterozygotes with a frameshift in exon 3 and a variant that disrupts the splice acceptor of exon 7 of MBD4 (Figure 1C; supplemental Figure 2A). Analysis of the MBD4 messenger RNA allowed for phasing of the variants to distinct alleles and confirmed aberrant splicing that excludes exon 7 and disrupts the glycosylase domain (supplemental Figure 2B). MBD4 has not previously been associated with hematological malignancy, but somatic mutations, predominantly frameshifts, have been detected in sporadic colon cancers with mismatch repair deficiency.18,19 Two patients (EMC-AML-1, WEHI-AML-2) also had colorectal polyps, a common manifestation of DNA repair defects, including those associated with loss of BER components MUTYH20 and NTHL1.21
Inactivation of MBD4 is associated with a methylation damage signature across different types of cancer
We mined large cancer databases to explore the link between MBD4 deficiency and the distinctive CG>TG signature. Analysis of TCGA, comprising 10 683 cancers (including 200 AMLs), identified 9 cases that carried germ line loss-of-function variants in MBD4 (Figure 1C; supplemental Figure 1A-B and supplemental Table 2). In 2 of these cases, a uveal melanoma (TCGA-UVM-1) and a glioblastoma multiforme (TCGA-GBM-1), splice site mutations were accompanied by loss of the wild-type MBD4 allele (supplemental Figure 3A). Analysis of RNA sequencing from both tumors confirmed aberrant splicing of MBD4, predicted to result in protein truncation and loss of function (supplemental Figure 3B). Both cases exhibited an elevated mutation rate and strong enrichment for CG>TG mutations, similar to the MBD4-deficient AMLs (Figure 1D; supplemental Figure 1A). This signature was also observed in a glioma cell line, SW1783, that carries a homozygous truncating variant in MBD4 at Leucine 563 (supplemental Figure 1A). Cancers that retained a wild-type allele did not display a prominent CG>TG signature (Figure 1D; supplemental Figure 1A). These results suggest both alleles of MBD4 must be inactivated to inhibit its repair activity, which is consistent with other BER-associated cancer syndromes.20,21
Genetic and epigenetic features that impact methylation damage
Whole genome sequencing and methylation profiling were performed to refine the mutational signature associated with MBD4 deficiency in AML. Overall, >15 000 substitution mutations were identified in each AML genome, of which >90% were CG>TG (supplemental Figure 1B). Insertions and deletions were uncommon, suggesting the mismatch repair pathway remains intact. The mutation rate was linked to 5mC abundance. Sparsely methylated regions, such as promoters and CG islands, were rarely mutated (Figure 2A). Correcting for 5mC abundance measured in normal CD34+ cells revealed a consistent mutation rate across different genomic features (Figure 2A). Direct assessment of the methylation status in MBD4-deficient cancers, or matched control tissue, confirmed that mutations occurred at methylated CG sites (supplemental Figure 4).

Damage introduced by 5mC deamination is influenced by genetic and epigenetic features. (A) Observed relative mutation rates (RMRs) at different genomic features in whole genome sequencing from WEHI-AML-1 and WEHI-AML-2, calculated per Mb of CG dinucleotides (CG corrected), or corrected for methylation status in normal CD34+ cells (5mC corrected). (B) Abundance and methylation status for NCG trimers from whole genome bisulfite sequencing derived from normal CD34+ cells.37 An RMR value was calculated for WEHI-AML-1 and WEHI-AML-2 for each NCG trimer, accounting for differences in abundance and 5mC status in normal CD34+ cells and scaled to account for total mutation load (see supplemental Methods). Individual values are plotted (n = 2), and bars show the mean. (C) RMR values were calculated from exome data for the 5 MBD4-deficient cancers. There was a significant enrichment of mutations in the ACG context compared with TCG (P = .0079, Mann-Whitney U test). (D) RMR values were calculated from whole genome sequencing data generated from Mbd4 knockout (Mbd4-KO) murine blood cell progenitors at 4 months of age. Values from individual colonies are plotted (n = 3), and the bar shows the mean. There was a significant enrichment of mutations in the ACG context compared with TCG (P = .019, Welch’s t test). (E) RMR values were calculated for NCGN tetramers in WEHI-AML-1 and WEHI-AML-2, then separated by replication timing (n = 2).
Damage introduced by 5mC deamination is influenced by genetic and epigenetic features. (A) Observed relative mutation rates (RMRs) at different genomic features in whole genome sequencing from WEHI-AML-1 and WEHI-AML-2, calculated per Mb of CG dinucleotides (CG corrected), or corrected for methylation status in normal CD34+ cells (5mC corrected). (B) Abundance and methylation status for NCG trimers from whole genome bisulfite sequencing derived from normal CD34+ cells.37 An RMR value was calculated for WEHI-AML-1 and WEHI-AML-2 for each NCG trimer, accounting for differences in abundance and 5mC status in normal CD34+ cells and scaled to account for total mutation load (see supplemental Methods). Individual values are plotted (n = 2), and bars show the mean. (C) RMR values were calculated from exome data for the 5 MBD4-deficient cancers. There was a significant enrichment of mutations in the ACG context compared with TCG (P = .0079, Mann-Whitney U test). (D) RMR values were calculated from whole genome sequencing data generated from Mbd4 knockout (Mbd4-KO) murine blood cell progenitors at 4 months of age. Values from individual colonies are plotted (n = 3), and the bar shows the mean. There was a significant enrichment of mutations in the ACG context compared with TCG (P = .019, Welch’s t test). (E) RMR values were calculated for NCGN tetramers in WEHI-AML-1 and WEHI-AML-2, then separated by replication timing (n = 2).
We next assessed the influence of genetic and epigenetic features on the mutation rate.22 When we examined the local sequence context, we observed that the proportion of mutations was higher in the context of the ACG triplet and lower in the context of TCG, with CCG and GCG being intermediate. The preference for ACG remained after correction for trimer abundance and methylation status (Figure 2B) and was found to be significant in the exome data from 5 MBD4-deficient cancers (P = .007937, Mann-Whitney U test) (Figure 2C). The same mutational signature, including the preference for the ACG trimer, was recapitulated in blood cell progenitors isolated from Mbd4 knockout mice, both at 4 months of age (Figure 2D) and in animals aged for over a year, which had a higher mutation burden (supplemental Figure 5). The ACA trimer was the most commonly mutated site outside of a CG context in the cancers, and this matches the most common site of non-CG methylation.23 Extending the analysis of sequence context to include 1 base on either side of the CG identified higher mutation rates in the context of a 3′ cytosine (NCGC). The relative mutation rate was not influenced by the transcriptional strand (supplemental Figure 6A) but was higher in late replicating regions (Figure 2E) and at lowly expressed genes (supplemental Figure 6B). The differences between tetramers and enrichment in late replicating regions were also evident in rare germ line CG>TG single nucleotide polymorphisms from the gnomAD database13 (supplemental Figure 6C). Collectively, these results suggest that although 5mC is the dominant factor contributing to the mutation rate, the local sequence context, replication timing, and expression status also contribute.
MBD4 deficiency drives a common path of clonal evolution to AML
The 3 cases of AML with germ line MBD4 deficiency exhibited common molecular features, including biallelic DNMT3A mutations and IDH1 or IDH2 hot spot mutations, all of which were CG>TG (Figure 3A-C). This is a relatively rare path to AML, affecting <3% of patients in TCGA-AML24 ; therefore, it is highly unlikely that these 3 individuals share this pattern of driver mutations by chance. Analysis of sequential bone marrow biopsies taken during treatment and single-cell genotyping allowed us to refine the order of somatic mutation acquisition in 2 cases (EMC-AML-1, WEHI-AML-1), with DNMT3A mutations preceding IDH mutations (Figure 3A-B; supplemental Figure 7). DNMT3A mutations present in the AML at diagnosis were also detected in nonleukemic bone marrow populations in both cases, indicating that these mutations are among the first acquired. Mutations in DNMT3A are known to alter the self-renewal capacity of hematopoietic stem cells (HSCs)25 and are associated with age-related clonal hematopoiesis (ARCH), also known as clonal hematopoiesis of indeterminate potential.26-29 For both cases, a marked expansion of clones carrying DNMT3A mutations occurred in the remission phase following treatment (Figure 3A-B). EMC-AML-1 experienced multiple clonal outgrowths, with 9 distinct DNMT3A mutations, and repeated selection of clones with biallelic DNMT3A mutations, which appears to be a key step in the development of leukemia in these patients. Broader testing of other AMLs with biallelic DNMT3A mutations demonstrated that 24 out of 30 (80%) have coincident mutations in IDH1 or IDH2, suggesting cooperation between these mutations that may explain this conserved path to leukemia.
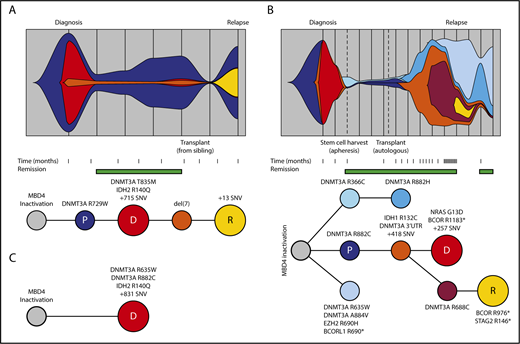
Germ line MBD4-deficient patients share a common path to AML. Clonal evolution and phylogenetic tree diagram highlighting the acquisition of key driver mutations and clonal dynamics in WEHI-AML-1 (A) and in EMC-AML-1 (B). (C) The phylogenetic tree diagram for key driver mutations in WEHI-AML-2. Variant allele frequencies were derived from whole exome sequencing data or deep sequencing for all cases. For EMC-AML-1 single-cell genotyping was used to resolve the clonal relationships. Clones are represented by different colors, and the vertical lines in the top panels indicate sampling points. The premalignant clone (P, in dark blue) and the AML clones evident at diagnosis (D, in red) and relapse (R, in yellow) are designated. Both WEHI-AML-1 and EMC-AML-1 experienced clonal hematopoiesis during remission. The transplant for WEHI-AML-1 was provided by WEHI-AML-2, which occurred 4 years prior to her own diagnosis of AML.
Germ line MBD4-deficient patients share a common path to AML. Clonal evolution and phylogenetic tree diagram highlighting the acquisition of key driver mutations and clonal dynamics in WEHI-AML-1 (A) and in EMC-AML-1 (B). (C) The phylogenetic tree diagram for key driver mutations in WEHI-AML-2. Variant allele frequencies were derived from whole exome sequencing data or deep sequencing for all cases. For EMC-AML-1 single-cell genotyping was used to resolve the clonal relationships. Clones are represented by different colors, and the vertical lines in the top panels indicate sampling points. The premalignant clone (P, in dark blue) and the AML clones evident at diagnosis (D, in red) and relapse (R, in yellow) are designated. Both WEHI-AML-1 and EMC-AML-1 experienced clonal hematopoiesis during remission. The transplant for WEHI-AML-1 was provided by WEHI-AML-2, which occurred 4 years prior to her own diagnosis of AML.
MBD4 deficiency stimulates clonal hematopoiesis through inactivation of DNMT3A
To determine the influence of this mutational process on the composition of MBD4-deficient bone marrow, we genotyped additional single cells, or SCDCs, isolated from EMC-AML-1 at multiple points during treatment. As expected, the leukemic clones were dominant at the time of diagnosis and relapse, but genotyping individual cells revealed that they continue to acquire CG>TG mutations (supplemental Figure 8). When HSCs collected at remission were examined, we found that 20 of 30 (67%) SCDCs carried mono- or biallelic CG>TG mutations in DNMT3A that were mostly distinct (Figure 4A). A further 2 (7%) SCDCs carried CG>TG mutations in TP53 (Figure 4B). Deep variant calling across all EMC-AML-1 samples uncovered additional CG>TG mutations in ARCH-associated genes: 28 in DNMT3A, 10 in TP53, 5 in ASXL1, and 7 in TET2 (Figure 4A-D). When these findings are extrapolated to the entire bone marrow compartment, it suggests a rich diversity of clones carrying mutations in ARCH-associated genes, predominantly in DNMT3A. Three observations support the notion that the mutations in DNMT3A are functionally important: first, their repeated expansion in the blood indicates a fitness advantage; second, there is clear enrichment of nonsynonymous mutations (assessed with dNdScv,30 q = 4.63e-05, Benjamini-Hochberg corrected); and third, the majority of mutations (65%) have been observed in ARCH26,28,31 (Figure 4A). Taken together, these results emphasize the importance of 5mC damage as a source of mutations that drive clonal expansion in the blood, representing a key contributor to ARCH.
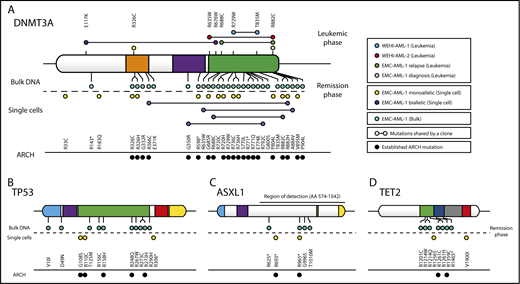
Recurrent C>T mutations in genes implicated in age-related clonal hematopoiesis (ARCH). (A) DNMT3A mutations were detected in MBD4-deficient patients at time of disease (leukemic phase) or remission (remission phase). EMC-AML-1 had additional DNMT3A mutations that were detected through sequencing of bulk DNA, SCDCs obtained from diagnostic bone marrow, and SCDCs from autologous stem cells collected during complete remission. The majority of the DNMT3A mutations had been detected in healthy individuals with ARCH. Additional point mutations were identified in remission material from EMC-AML-1, in TP53 (B), ASXL1 (C), and TET2 (D). A more detailed phylogenetic tree is provided in supplemental Figure 8.
Recurrent C>T mutations in genes implicated in age-related clonal hematopoiesis (ARCH). (A) DNMT3A mutations were detected in MBD4-deficient patients at time of disease (leukemic phase) or remission (remission phase). EMC-AML-1 had additional DNMT3A mutations that were detected through sequencing of bulk DNA, SCDCs obtained from diagnostic bone marrow, and SCDCs from autologous stem cells collected during complete remission. The majority of the DNMT3A mutations had been detected in healthy individuals with ARCH. Additional point mutations were identified in remission material from EMC-AML-1, in TP53 (B), ASXL1 (C), and TET2 (D). A more detailed phylogenetic tree is provided in supplemental Figure 8.
Discussion
Here we describe a new genetic predisposition to cancer, in which germ line MBD4 deficiency is associated with the development of early-onset AML, through the acquisition of pathogenic mutations in driver genes, most particularly DNMT3A. Although additional investigation is required to determine the frequency with which MBD4 deficiency contributes to familial cancer predisposition and to refine the disease spectrum and penetrance, our results highlight a crucial role for MBD4 in safeguarding against the damage wrought by 5mC deamination. Concomitantly, 2 other groups have also identified the link between MBD4 inactivation and methylation damage, through identification of sporadic solid cancers with a combination of germ line and somatic mutations (Rodrigues et al32 and Jan Korbel, manuscript submitted November 2017). Our study, in addition, reveals the impact of constitutive inactivation of MBD4 on the development of early-onset AML and reveals that blood cell progenitors are particularly sensitive to methylation damage.
As noted earlier, methylation damage accumulates as part of normal aging.4,5 Our current understanding of how methylation damage manifests largely depends on mutational profiles garnered from large collections of human cancers,4 but distilling a clear signature has been complicated by the diverse DNA damage processes and repair defects present in those cancers. MBD4-deficient cancers, particularly cases with constitutive loss, provide a unique opportunity to refine the mutational signature for methylation damage, and we have identified genetic and epigenetic factors that shape its influence. This distinctive damage signature was recapitulated in blood cells from Mbd4 knockout mice, indicating that the DNA repair pathway guarding against methylation damage is broadly conserved. The ubiquitous nature of methylation damage means even small fluctuations in mutation rate are relevant if we wish to understand its influence on genomic integrity. Our results demonstrate a profound link between methylation damage and the development of hematological malignancy, which is reshaping our understanding of how 5mC contributes to cancer risk over a lifetime.
One manifestation of methylation damage is clonal hematopoiesis, a phenomenon typically observed in people >70 years of age.26-29 The influence of methylation damage is reflected in the prevalence of C>T mutations in clonal hematopoiesis, which has been noted previously.28,33 Individuals with biallelic loss of MBD4 in the germ line confirm this link; they sustain high levels of damage from 5mC deamination throughout their lifetime and experience clonal expansions decades earlier, which eventually progress to AML. Repeated sampling and single-cell genotyping of blood cell progenitors revealed a rich diversity of mutations that overlap the driver landscape of clonal hematopoiesis, including mutations in DNMT3A particularly, but also in TP53, ASXL1, and TET2. The coexistence of this diverse array of mutant clones, and our ability to monitor their prevalence dynamically, offers new insight into the fitness landscape of clonal hematopoiesis. Future studies will need to explore the latency and degree of penetrance of mutations associated with clonal hematopoiesis and AML in Mbd4 knockout mice as they age, in order to fully investigate the human disease pathogenesis we have identified.
There are >40 million 5mC residues in the genome, yet the 3 individuals that lack MBD4 constitutively all developed the same type of cancer, AML, with a common set of driver mutations. A small set of genes have been defined that predispose to AML (reviewed by Godley and Shimamura34 ), including DNA repair genes, such as those in the Fanconi anemia pathway, but to our knowledge none exhibit such a conserved path to malignancy. Our results indicate this convergence results from the combination of a highly restricted mutational signature, which accesses a select set of driver genes, and the role of DNMT3A, which regulates HSC self-renewal capacity and protects against transformation.25,35,36 This interaction between mutational process, driver landscape, and stem cell biology may explain the tissue-restricted pattern of disease in this and other cancer predisposition syndromes and has broader implications for understanding how the aging process shapes cancer risk.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank S. He, A. Rijneveld, K. van Lom, and K. Gussinklo for providing clinical information; M. Wall for assistance with cytogenetics; N. Sprigg for assistance with sample collection; L. Di Rago for assistance with mouse agar colonies; E. Rombouts for assistance with single-cell sorting; I. Martincorena and F. Abascal for advice on the dNdScv model; S. van Rossum and J. Lebbink for assistance with recombinant protein isolation; the Australasian Leukaemia and Lymphoma Group for access to clinical samples; and S. Wilcox for technical assistance with sequencing. Additional sequencing was performed at the Australian Genome Research Facility (Melbourne, VIC, Australia) and the Kinghorn Centre for Clinical Genomics (Sydney, NSW, Australia). Sean Grimmond, Jason Wong, Oliver Sieber, Alicia Oshlack, and Stephen Nutt provided valuable feedback on the manuscript.
This work was supported by the Australian National Health and Medical Research Council (NHMRC) (program grant 1113577 [W.S.A. and A.W.R.] and project grant 1145912 [I.J.M.]), an Independent Research Institutes Infrastructure Support Scheme Grant (9000220), a Victorian State Government Operational Infrastructure Support Grant, The Netherlands Organisation for Scientific Research (NWO), and the Center for Translational Molecular Medicine (CTMM). M.A.S. is supported by a grant from CTMM (GR03O-102) and a Rubicon fellowship from NWO (019.153LW.038). E.C. is supported by a PhD scholarship from the Leukaemia Foundation of Australia. A.S.a.H. is supported by a PhD scholarship from the Ministry of Health, Sultanate of Oman. M.E.B. is supported by the Bellberry-Viertel fellowship. W.S.A. and A.W.R. are supported by fellowships from NHMRC (1058344 and 1079560, respectively). I.J.M. is supported by the Victorian Cancer Agency. The authors wish to acknowledge the generous philanthropic support of the Felton Bequest, Malcolm Broomhead, and BHP Billiton.
The results are based, in part, on data generated by the TCGA Research Network (http://cancergenome.nih.gov/) and the Epigenetic studies in Acute Myeloid Leukemia (phs001027), which was supported by National Institutes of Health, National Cancer Institute (K08CA169055) (F. E. Garrett-Bakelman), Starr Cancer Consortium I4-A442 (A. M. Melnick, R. Levine, and C. E. Mason), and LLS SCOR 7006-13 (A. M. Melnick). Sequencing data from WEHI-AML-1 and WEHI-AML-2 have been deposited at the European Genome Phenome Archive (EGA) (EGAS00001002581). The data are available for ethically approved research into hematological malignancy upon completion of a data transfer agreement. Sequencing data from EMC-AML-1 were sourced from the dbGaP under accession phs001027. Sequencing data from the Mbd4 knockout mice is available through the National Center for Biotechnology Information (NCBI) Short Read Archive (SRP126117). The code for reproducing figures is made available through GitHub (https://github.com/MathijsSanders/AML-RoaMeR).
Authorship
Contribution: M.A.S., E.C., A.W.R., P.J.M.V., and I.J.M. conceived and designed research; M.A.S., E.C., C.F., A.Z., S.E.M., A.S.a.H., A.B., B. Luiken, M.R., T.M., R.M.H., F.G.K., A.W.R., P.J.M.V., and I.J.M. developed methodology and performed research; M.A.S., E.C., C.F., A.Z., S.E.M., R.M.H., F.G.K., S.F., M.E.B., E.M.B., W.S.A., A.W.R., P.J.M.V., and I.J.M. analyzed data; and M.A.S., E.C., C.F., W.S.A., B. Löwenberg, A.W.R., P.J.M.V., and I.J.M. wrote the manuscript or contributed to revision of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ian J. Majewski, Cancer and Haematology Division, The Walter and Eliza Hall Institute, 1G Royal Parade, Parkville 3052, VIC, Australia; e-mail: majewski@wehi.edu.au.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal