

Key Points
Extracellular ATP activates NLRP3 inflammasomes, resulting in MDSC dysfunction.
Preventing MDSC inflammasome activation and conserving IL-1β secretion decreases GVHD lethality.

Abstract
Myeloid-derived suppressor cells (MDSCs) can subdue inflammation. In mice with acute graft-versus-host disease (GVHD), donor MDSC infusion enhances survival that is only partial and transient because of MDSC inflammasome activation early posttransfer, resulting in differentiation and loss of suppressor function. Here we demonstrate that conditioning regimen–induced adenosine triphosphate (ATP) release is a primary driver of MDSC dysfunction through ATP receptor (P2x7R) engagement and NLR pyrin family domain 3 (NLRP3) inflammasome activation. P2x7R or NLRP3 knockout (KO) donor MDSCs provided significantly higher survival than wild-type (WT) MDSCs. Although in vivo pharmacologic targeting of NLRP3 or P2x7R promoted recipient survival, indicating in vivo biologic effects, no synergistic survival advantage was seen when combined with MDSCs. Because activated inflammasomes release mature interleukin-1β (IL-1β), we expected that IL-1β KO donor MDSCs would be superior in subverting GVHD, but such MDSCs proved inferior relative to WT. IL-1β release and IL-1 receptor expression was required for optimal MDSC function, and exogenous IL-1β added to suppression assays that included MDSCs increased suppressor potency. These data indicate that prolonged systemic NLRP3 inflammasome inhibition and decreased IL-1β could diminish survival in GVHD. However, loss of inflammasome activation and IL-1β release restricted to MDSCs rather than systemic inhibition allowed non-MDSC IL-1β signaling, improving survival. Extracellular ATP catalysis with peritransplant apyrase administered into the peritoneum, the ATP release site, synergized with WT MDSCs, as did regulatory T-cell infusion, which we showed reduced but did not eliminate MDSC inflammasome activation, as assessed with a novel inflammasome reporter strain. These findings will inform future clinical using MDSCs to decrease alloresponses in inflammatory environments.
Introduction
Graft-versus-host disease (GVHD) remains a major source of transplantation complications, with morbidity rates up to 15%, limiting the efficacy of allogeneic hematopoietic stem cell transplantation.1 GVHD prophylaxis consists primarily of globally immune suppressive drugs that largely target T cells. In the earliest phase of GVHD, T cells are primed by innate immune mediators, including myeloid cells, that drive their activation and expansion.2-4 Myeloid lineage cells, maintained in a relatively quiescent state, act as sentinels; upon activation, phenotype and motility changes occur to shape the T-cell response. To balance inflammation, regulatory myeloid lineage cell populations, such as myeloid-derived suppressor cells (MDSCs), are present. MDSCs, comprising a heterogeneous population of early myeloid progenitors defined by their functional ability to suppress innate and adaptive immune activation, have characteristics of immature granulocytes, macrophages, or dendritic cells. MDSCs increase in number under conditions of distress (eg, chronic inflammation, tumor burden) to limit pathology5-9 .
MDSC elimination or forced differentiation into mature myeloid cells has been used to subvert tumor-associated immune suppression.10-12 Conversely, MDSC expansion or infusion has been explored to buffer inflammation for therapeutic benefit. We and others have shown short-term bone marrow (BM) cultures with well-defined cytokine cocktails (eg, granulocyte-macrophage colony-stimulating factor (GM-CSF) plus granulocyte colony-stimulating factor (G-CSF) produce immature myeloid cells with suppressor function and the capacity to affect GVHD survival and clinical outcomes.13-16 MDSCs are remarkably malleable and employ multiple suppressor mechanisms dependent upon environmental signals. Antigen-independent suppression can occur via upregulation of coinhibitory ligands, soluble factor production, and essential amino acid depletion. Arginase-1 (Arg1) or nitric oxide (Nos2) production metabolizes extracellular L-arginine, which is essential for activated T-cell expansion.13,17,18 Interleukin-13 (IL-13) activation supports an Ly6C+CD11b+Arg1+ (MDSC-IL13) population, promoting metabolic stress and T-cell dysfunction.13 Other reported mechanisms include catabolic disruption through cysteine, tryptophan depletion, or induction of regulatory T cells (Tregs).19,20
Under inflammatory conditions, MDSC-IL13 efficacy is limited by cell-intrinsic inflammasome activation, release of inflammatory mediators, and myeloid differentiation.14 The inflammasome is an intracellular multiprotein complex that forms in response to pathogen- or danger-associated molecular patterns, consisting of caspase-1 and adaptor protein apoptosis-associated speck-like protein containing CARD (ASC), and is required for maturation of proinflammatory IL-1β, IL-18, and pyroptosis-inducer gasdermin-D.21-23 An initiating signal (signal 1), such as bacterial lipopolysaccharide (LPS)–triggered TLR4, promotes NFκB activity and caspase-1 activation. Canonical inflammasome activation requires a secondary stimulus to engage unique adaptor proteins tailored to sense distinct danger signals. Absent in melanoma 2 (AIM2) inflammasome activation by damaged or foreign cytosolic DNA, NLR family CARD domain containing 4 (NLRC4) activation via bacterial flagellin, and NLR pyrin family domain 3 (NLRP3 or cryopyrin) activation by stress or danger signals (alum, urate, or ATP) all lead to cleavage of inactive pro–caspase-1 into an active form, leading to pro–IL-1β processing and secretion.21,23-25 Here we sought to interrogate inflammatory pathways linked to myeloid cell maturation and define mediators of MDSC inflammasome-associated loss of function to identify targets for enhancing MDSC potency.
Methods
Experimental animals
Female 8- to 12-week-old BALB/cAnNCr (H2d, catalog #555) and C57BL/6NCr (B6, H2b, #556) mice were purchased from the National Cancer Institute colony at Charles River; B6.129S7-Il1r1tm1Imx/J (IL-1 receptor [IL-1R] knockout [KO], #003245) and B6.129P2-P2rx7tm1Gab/J (P2x7R KO, #005576) mice were purchased from The Jackson Laboratory. Myosin regulatory light chain interacting protein (IDOL)–transgenic mice were bred and maintained in house. Bones from MyD88 KO MyD88/TRIF double KO (dKO) donors were provided by Samithamby Jeyaseelan Jey (Louisiana State University); caspase-1/11 dKO, caspase-11 KO, IL-1β KO, AIM2 KO, NLRC4 KO, and NLRP3 KO bones were provided by J.P.-Y.T. Unless otherwise noted, all KO and transgenic mice were maintained on a C57Bl/6 (B6) background. Mice were housed in a specific pathogen-free facility in microisolator cages under approved protocols by the University of Minnesota Institutional Animal Care and Use Committee.
MDSC-IL13, inflammasome activation and detection
Murine MDSC-IL13s were generated by culturing donor BM with 100 ng/mL human granulocyte CSF (Neupogen; Amgen), and 0.4 to 2.5 ng/mL of recombinant murine granulocyte-macrophage CSF (415-ML), with recombinant murine IL-13 (413-ML; R&D Systems) added on day 3 for arginase-1 induction.13 Trypsin/EDTA and light scraping on day 4 recovered >92% CD11b+ cells, with expected enrichment of Ly6Chi monocytic MDSCs, as seen previously.13 Where indicated, on day 4, MDSC-IL13s were treated for 3 hours with LPS (0.2 µg/mL; LPS-EK Ultrapure; Invivogen) and then for 1 hour with ATP (2 mM; A7699; Sigma) before harvest. Where indicated, flagellin (0.12 μg/mL; FLA-ST Ultrapure; Invivogen) in profect-P1 was delivered to engage NLRC4. In AIM2 inflammasome activation, Lipofectamine 2000 was used to deliver poly(deoxyadenylic-thymidylic) acid sodium salt (1.6 μg/mL; P0883; Sigma). The FAM-FLICA Caspase-1 assay kit (#98; ImmunoChemistry Technologies) for detection of active caspase-1 was used per manufacturer instructions; active caspase-1 was detected by flow cytometry.
GVHD and reagents
BALB/c recipients were lethally irradiated (700 cGy of total-body irradiation by X-ray) on day −1 and given B6 donor BM (1e7) and purified CD25-depleted T cells (2e6; Stemcell Technologies EasySep negative cell isolation system) on day 0 to induce acute GVHD (aGVHD). Some cohorts were coinfused with B6 MDSC-IL13s (6e6) and/or Tregs (2e6) on day 0. MDSC-IL13s from KO donors were confirmed to have similar Ly6Chi cell surface phenotypes, suppressive capacities, and growth characteristics after culture. Clinical GVHD and daily survival monitoring were as described previously.26 MCC950 (20-50 mg/kg; #5.38120; Sigma),27 3,5,7-trihydroxy-4′-methoxy-8-(3-hydroxy-3-methylbutyl)-flavone icariin derivative (ICTA; 30 mg/kg; provided by Dr Sheng Wei, Moffitt Cancer Center),28 and A-438079 (16 mg/kg; A9736; Sigma)29,30 were administered intraperitoneally on day −1 and then daily or every other day for 1 to 4 weeks; apyrase (4 units; A6237; Sigma) was administered daily from day 0 to 4.
Flow cytometry, BLI, and suppression assay
Flow cytometric data were acquired on a BD LSRFortessa and analyzed using FlowJo software. MDSC lineage (granulocytic vs monocytic) was identified using the following antibodies: CD11b eFlour450, M1/70; Ly6C PerCP-Cy5.5, AFS98; Ly6G APC, 1A8; MHC class 2 APC-eFluor 780 M5/114.15.2; CD115 Alexa Fluor 488, AFS98 (eBioscience); and CD11c PE, N418 (Becton Dickenson), as previously reported.13 Bioluminescent imaging (BLI) was performed using a Xenogen IVIS 200 (Caliper Life Sciences) after injection with 3 mg of firefly luciferin substrate (122799; PerkinElmer) administered intraperitoneally 5 minutes before imaging. Whole-body images were captured, and the region of interest was used to quantify radiance. After imaging, animals were euthanized, and organs were transferred to plates containing 0.5 mg/mL of luciferin in phosphate-buffered saline for explant imaging. For T-cell suppression, titrated MDSCs were diluted in complete RPMI media containing 150 µM of L-arginine; responding T cells (1e6 per mL) stained with 3.5 µM of carboxyfluorescein succinimidyl ester (CFSE; C34554, Thermo Fisher), T cell–depleted splenocytes (0.5e6 per mL), and anti-CD3ε (0.25 µg/mL; 14-0031-86; Invitrogen) were added to 96-well flat-bottom plates and incubated for 3 days before analysis of total CD8 T-cell proliferation. Indicated inhibitors were added 1 hour before ATP addition: glyburide (100 µM; sc-200982a; Santa Cruz Biotechnology), MCC950 (10 µM; 5.38120; Sigma), ICTA (200 µg/mL; as in “GVHD and reagents”), β-hydroxybutyrate (BHB; 200 µM; provided by Dr Vishwa Dixit), A-438079 (25 µM; A9736; Sigma), and ruxolitinib (1 µM; S1378; Selleck Chemicals).
ELISA and arginase assay
Culture supernatant murine IL-1β was quantified 1 hour after ATP addition with the IL-1β/IL-1F2 DuoSet enzyme-linked immunosorbent assay (ELISA) kit (DY401; R&D Systems), and cell-associated arginase-1 activity was determined using the QuantiChrom Arginase assay kit (DARG-100; Bioassay Systems) according to manufacturer instructions.
Statistics
Kaplan-Meier survival curves were analyzed by log-rank test and in vitro data by Student t test or 2-way analysis of variance as indicated (GraphPad Prism Software). Bar graphs represent mean values and 1 standard deviation from the mean; statistical significance was defined as P ≤ .05.
Results
TLR-triggered MDSC loss of function
In GVHD mice, MDSC efficacy is rapidly compromised in part because of inflammasome activation.14 To uncover druggable targets, extrinsic inflammasome-activating factors were investigated. Conditioning-associated tissue damage promotes bacteria-derived pathogen-associated molecular pattern release, including LPS from mucosal barrier disruption that promotes GVHD.31,32 The TLR family of receptors specialize in sensing environmental dangers (eg, TLR4-LPS), promoting immune activation via 2 intracellular signaling adaptor proteins, MyD88 and TRIF.33-36 In vitro LPS stimulation of MDSCs augmented caspase-1 activity, measured by FLICA, a cell-permeable fluorescent probe that binds active caspase-1 (Figure 1A). TLR4 downstream MyD88 signaling promotes pro–IL-1β transcription, and TRIF signaling increases caspase-1 activity.37-39 LPS-stimulated MyD88 KO and wild-type (WT) MDSC-IL13s had comparable FLICA MFI (Figure 1A). In contrast, MyD88/TRIF dKO MDSC-IL13s did not respond to LPS or LPS plus ATP, as evidenced by a reduced FLICA MFI compared with MyD88 KO or WT MDSC-IL13s. Supernatants from cultured MDSCs treated with LPS plus ATP were assayed by ELISA for caspase-1–mediated IL-1β secretion. Reflective of FLICA MFI, LPS plus ATP–stimulated MyD88/TRIF dKO MDSC-IL13s had a reduction in IL-1β production relative to WT (P = .0006) or MyD88 KO MDSCs (P = .002; Figure 1B), consistent with resistance to LPS/ATP inflammasome induction. The lower IL-1β concentration in MyD88 KO MDSC-IL13 supernatants was likely a result of defective MyD88 pro–IL-1β production; constitutive low-level IL-1β expression could explain the modest IL-1β levels detected.37,39 Although LPS-triggered TLR4 can induce myeloid maturation, LPS was insufficient to compromise MDSC-IL13 suppressive function, whereas LPS plus ATP caused almost complete loss in WT MDSC and MyD88 KO but not MyD88/TRIF dKO MDSC-IL13 suppression (Figure 1C). To determine whether TLR signaling facilitated myeloid maturation and inflammasome assembly, leading to limited MDSC-IL13 function in GVHD suppression, MyD88 KO, MyD88/TRIF dKO, and WT MDSC-IL13s were compared. MyD88 KO MDSC-IL13s were unable to promote survival over WT MDSCs (Figure 1D). Recipients of MyD88/TRIF dKO MDSCs had improved survival compared with those receiving MDSC-IL13s, which did not reach statistical significance (Figure 1E; P = .056; mean survival time, 35 vs 60 days). These findings show a role for TLR but suggest that inhibiting TLR signaling alone is not sufficient to rescue MDSC-IL13s from loss of suppression in a highly inflammatory setting. Future studies using TRIF KO MDSC-IL13s will be needed to determine whether TRIF single KO vs WT MDSC-IL13s have a demonstrable survival benefit.
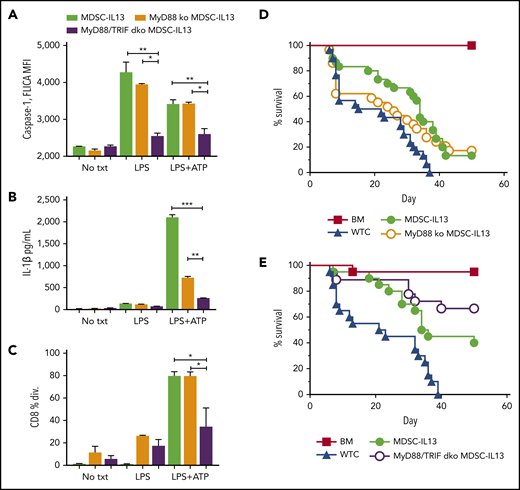
TLR4 signaling in MDSCs. (A) MDSC-IL13s treated as indicated for inflammasome induction with LPS (0.2 μg/mL) for 3 hours followed by ATP (2 mM) for 1 hour were harvested and aliquoted for FLICA analysis, shown as mean fluorescence intensity (MFI), gated on CD11b+Ly6C+ fraction; 2 replicates per condition. (B) Supernatants from cultures were assayed for IL-1β production by ELISA; 2 replicates per condition. (C) Harvested MDSC-IL13s were washed, counted, and plated in a CFSE suppression assay at a 1:1 ratio with naïve B6-purified CD25-depleted whole T cells stimulated with anti-CD3ε. Data represent fraction of CD8+ T cells having undergone ≥1 division; 3 replicates per condition. (D) Kaplan-Meier survival curve for lethally irradiated Balb/c recipients given 1e7 B6 BM cells plus 2e6 purified CD25-depleted whole T cells (WTCs) and 6e6 MDSC-IL13s (M13) or MyD88 KO MDSC-IL13s, as indicated. WTCs vs M13, P = .0001; WTCs vs MyD88 KO, P = .0460; M13 vs MyD88 KO, P = .5988. Data represent 3 independent pooled experiments with n = 30 animals per group. (E) Kaplan-Meier survival curve, as in panel D, giving 6e6 WT MDSC-IL13s (M13) or 6e6 MyD88/TRIF dKO MDSC-IL13s. WTCs vs M13, P = .0012; WTCs vs MyD/TRIF dKO, P < .0001; M13 vs MyD88/TRIF dKO, P = .0561. Data represent 2 independent pooled experiments with n = 20 per group. *P < .05, **P < .01, ***P < .001. txt, treatment.
TLR4 signaling in MDSCs. (A) MDSC-IL13s treated as indicated for inflammasome induction with LPS (0.2 μg/mL) for 3 hours followed by ATP (2 mM) for 1 hour were harvested and aliquoted for FLICA analysis, shown as mean fluorescence intensity (MFI), gated on CD11b+Ly6C+ fraction; 2 replicates per condition. (B) Supernatants from cultures were assayed for IL-1β production by ELISA; 2 replicates per condition. (C) Harvested MDSC-IL13s were washed, counted, and plated in a CFSE suppression assay at a 1:1 ratio with naïve B6-purified CD25-depleted whole T cells stimulated with anti-CD3ε. Data represent fraction of CD8+ T cells having undergone ≥1 division; 3 replicates per condition. (D) Kaplan-Meier survival curve for lethally irradiated Balb/c recipients given 1e7 B6 BM cells plus 2e6 purified CD25-depleted whole T cells (WTCs) and 6e6 MDSC-IL13s (M13) or MyD88 KO MDSC-IL13s, as indicated. WTCs vs M13, P = .0001; WTCs vs MyD88 KO, P = .0460; M13 vs MyD88 KO, P = .5988. Data represent 3 independent pooled experiments with n = 30 animals per group. (E) Kaplan-Meier survival curve, as in panel D, giving 6e6 WT MDSC-IL13s (M13) or 6e6 MyD88/TRIF dKO MDSC-IL13s. WTCs vs M13, P = .0012; WTCs vs MyD/TRIF dKO, P < .0001; M13 vs MyD88/TRIF dKO, P = .0561. Data represent 2 independent pooled experiments with n = 20 per group. *P < .05, **P < .01, ***P < .001. txt, treatment.
Inflammasome activation causes MDSC-IL13 loss of function
We next sought to determine which downstream inflammasome components and inciting signaling events were associated with loss of function. Activation requires a combination of induction signals (ATP/double-stranded DNA), intrinsic molecular scaffolds to certain stimuli (NLRP3/AIM2/NLRC4), shared adapters and effector enzymes (ASC, caspase-1), and/or cytokines (IL-1β/IL-18) produced from conditioning regimen and GVHD injury.40-42
To interrogate pathway contributions to loss of function in GVHD mice, MDSC-IL13s were generated from AIM2, NLRP3, or NLRC4 KO donors, and inflammasomes were activated with relevant activation stimuli. When triggered by poly(dT), a mimic for viral DNA, WT but not AIM2 KO MDSC-IL13s produced copious amounts of IL-1β (Figure 2A). Flagellin-activated WT but not NLRC4 KO MDSC-IL13s secreted IL-1β, whereas NLRP3 KO MDSC-IL13s failed to respond to ATP, as expected (Figure 2A).
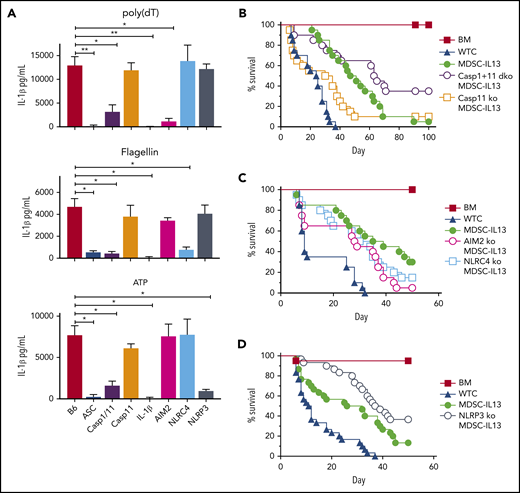
MDSC-IL13s responsive to inflammasome induction. MDSC-IL13s were generated from WT (B6), ASC KO, caspase-1/-11 dKO, caspase-11 KO, IL-1β KO, AIM2 KO, NLRC4 KO, or NLRP3 KO donors and then challenged with LPS (0.2 µg/ml) for 3 hours followed by poly(dT) (0.8 µg/mL), flagellin (0.12 µg/mL), or ATP (2 mM), as indicated. (A) One hour after full inflammasome induction, culture supernatants were analyzed for IL-1β by ELISA. Data represent 3 replicate cultures and 2 independent experiments. (B) Kaplan-Meier survival curve for Balb/c-recipient GVHD mice given BM, BM plus 2e6 CD25-depleted whole T cells (WTCs), and BM plus WTCs with MDSC-IL13s (M13) or caspase-1/-11 dKO MDSC-IL13s. WTCS vs M13, P < .0001; WTCS vs caspase-1/-11 dKO, P < .0001; M13 vs caspase-1/-11 dKO, P = .0389. Data represent combination of 2 independent experiments, n = 20 per group. (C) Same as in panel B, using AIM2 KO or NLRC4 KO MDSC-IL13s. WTCs vs all M13 groups, P < .003; M13 vs AIM2 KO or NLRC4 KO P = not significant. Data represent 2 independent pooled experiments, n = 20 per group. (D) Same as in panel B, using NLRP3 KO MDSC-IL13s. WTCs vs M13, P = .0001; WTCs vs NLRP3 KO, P < .0001; M13 vs NLRP3 KO, P = .0250. Data are representative of 3 independent pooled experiments, n = 30 per group. *P < .05, **P < .01.
MDSC-IL13s responsive to inflammasome induction. MDSC-IL13s were generated from WT (B6), ASC KO, caspase-1/-11 dKO, caspase-11 KO, IL-1β KO, AIM2 KO, NLRC4 KO, or NLRP3 KO donors and then challenged with LPS (0.2 µg/ml) for 3 hours followed by poly(dT) (0.8 µg/mL), flagellin (0.12 µg/mL), or ATP (2 mM), as indicated. (A) One hour after full inflammasome induction, culture supernatants were analyzed for IL-1β by ELISA. Data represent 3 replicate cultures and 2 independent experiments. (B) Kaplan-Meier survival curve for Balb/c-recipient GVHD mice given BM, BM plus 2e6 CD25-depleted whole T cells (WTCs), and BM plus WTCs with MDSC-IL13s (M13) or caspase-1/-11 dKO MDSC-IL13s. WTCS vs M13, P < .0001; WTCS vs caspase-1/-11 dKO, P < .0001; M13 vs caspase-1/-11 dKO, P = .0389. Data represent combination of 2 independent experiments, n = 20 per group. (C) Same as in panel B, using AIM2 KO or NLRC4 KO MDSC-IL13s. WTCs vs all M13 groups, P < .003; M13 vs AIM2 KO or NLRC4 KO P = not significant. Data represent 2 independent pooled experiments, n = 20 per group. (D) Same as in panel B, using NLRP3 KO MDSC-IL13s. WTCs vs M13, P = .0001; WTCs vs NLRP3 KO, P < .0001; M13 vs NLRP3 KO, P = .0250. Data are representative of 3 independent pooled experiments, n = 30 per group. *P < .05, **P < .01.
ASC, a shared and critical adaptor for inflammasome assembly, is essential for GVHD-associated MDSC-IL13 loss of function.14,25,43 Under all tested conditions, ASC KO MDSC-IL13s failed to support full inflammasome activation (Figure 2A). Caspase-1 is a core component of the canonical inflammasome that undergoes autocatalytic cleavage when active and is required to process pro–IL-1β, whereas caspase-11 is involved in noncanonical inflammasome activation.44,45 We compared caspase-1/-11 dKO MDSCs with caspase-11 KO MDSCs, because recent reports have found the strain of origin for caspase-1 KO mice to be deficient for both caspase-1 and -11.44,45 In vitro activation with LPS and poly(dT), flagellin, or ATP stimulated WT and caspase-11 KO MDSC-IL13s to produce similar IL-1β levels (Figure 2A). In contrast, caspase-1/-11 dKO MDSC-IL13s were resistant to IL-1β production. When KO MDSCs were applied therapeutically, caspase-11 KO MDSCs failed to enhance survival relative to MDSC-IL13s, whereas caspase-1/-11 dKO MDSCs provided a significant (P = .0389) survival advantage, supporting canonical inflammasome activation as key to limiting MDSC-IL13 suppressive activity in vivo (Figure 2B).
Although AIM2 and NLRC4 KO MDSC-IL13s both improved aGVHD, neither provided additional protection compared with WT MDSC-IL13s, indicating AIM2- and NLRC4-independent loss of function (Figure 2C). In contrast, NRLP3 KO MDSC-IL13s significantly increased survival (P = .025 vs WT MDSCs; Figure 2D). These data point to NLRP3 inflammasome activation as a negative regulator of MDSC-IL13 suppressor function in vivo.
Targeting NLRP3 inflammasome activation diminishes MDSC-IL13 loss of function
NLRP3 polymorphisms are associated with autoinflammatory diseases referred to as cryopyrin-associated periodic syndromes.21 We tested small molecules MCC950 (diarylsuflonylurea-containing compound) and ICTA (derivative of icariin, the major herba epimedii active ingredient), which specifically inhibit NLRP3 activation27,28,31,46,47 ; glyburide (glibenclamide), a sulfonylurea drug that preserves pancreatic β-cell function in type 2 diabetes48,49 ; and BHB, a natural ketone metabolite elevated during starvation, for efficacy in suppressing NLRP3 inflammasome activation of LPS plus ATP–treated MDSC-IL13s.50 Glyburide and MCC950 consistently demonstrated IL-1β inhibition (Figure 3A), retained suppressor function (Figure 3B) and arginase-1 activity (Figure 3C). ICTA showed more variable inflammasome suppression despite varying doses and formulations that promoted solubility, and BHB-nLG (nanolipogel format to improve bioavailability) also failed to equal MCC950 or glyburide in preventing reduced suppressor capacity and arginase activity (Figure 3). Notably, BHB-nLGs at high 5- to 10-mM concentrations lowered but did not eliminate IL-1 p17 by western blot, suggesting suboptimal potency (data not shown).
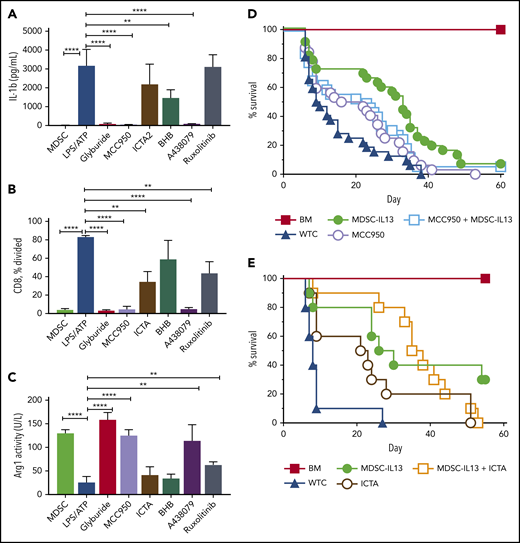
NLRP3 inflammasome inhibition protects function of MDSC-IL13s. Cultured WT MDSC-IL13s were treated as indicated with reagents to inhibit inflammasome induction or myeloid maturation 1 hour before LPS plus ATP inflammasome induction: glyburide (100 µM), MCC950 (10 µM), ICTA2 (200 µg/ml), BHB (200 µM), A-438079 (25 µM), and ruxolitinib (1 µM). One hour after ATP stimulation, cultures were assayed as indicated. (A) Culture supernatants were assessed for IL-1β. (B) Harvested MDSCs were counted and plated for CFSE suppression assay at a 1:1 ratio with responding CD25-depleted whole T cells (WTCs); data represent frequency of CD8+ T cells undergoing ≥1 division. (C) MDSCs were washed, counted, and assayed for cell-associated arginase-1 activity. (A-C) In vitro data represent 3 replicates per condition and 2 independent experiments. (D) Kaplan-Meier survival curve for B6 > Balb/c GVHD animals given MDSC-IL13s (M13) and/or the NLRP3-specific inhibitor MCC950 (MCC) (50 mg/kg intraperitoneally) every other day starting at day −1 for 3 weeks. WTCs vs M13, P < .001; WTCs vs MCC, P = .0055; WTCs vs M13 plus MCC, P < .0001; M13 vs MCC, P = .0152; M13 vs M13 plus MCC, P = .4291; MCC vs MCC plus M13, P = .1226. Data represent combination of 3 independent experiments, n = 30 per group. (E) Kaplan-Meier survival curve for B6 > Balb/c GVHD animals given MDSC-IL13 (M13) and/or the NLRP3-specific inhibitor ICTA. WTCs vs M13, P = .0028; WTCs vs ICTA2, P = .0128; WTCs vs M13 plus ICTA, P = .0001; M13 or ICTA vs M13 plus ICTA, P = not significant. Data represent a single experiment, n = 10 per group. **P < .01, ****P < .0001.
NLRP3 inflammasome inhibition protects function of MDSC-IL13s. Cultured WT MDSC-IL13s were treated as indicated with reagents to inhibit inflammasome induction or myeloid maturation 1 hour before LPS plus ATP inflammasome induction: glyburide (100 µM), MCC950 (10 µM), ICTA2 (200 µg/ml), BHB (200 µM), A-438079 (25 µM), and ruxolitinib (1 µM). One hour after ATP stimulation, cultures were assayed as indicated. (A) Culture supernatants were assessed for IL-1β. (B) Harvested MDSCs were counted and plated for CFSE suppression assay at a 1:1 ratio with responding CD25-depleted whole T cells (WTCs); data represent frequency of CD8+ T cells undergoing ≥1 division. (C) MDSCs were washed, counted, and assayed for cell-associated arginase-1 activity. (A-C) In vitro data represent 3 replicates per condition and 2 independent experiments. (D) Kaplan-Meier survival curve for B6 > Balb/c GVHD animals given MDSC-IL13s (M13) and/or the NLRP3-specific inhibitor MCC950 (MCC) (50 mg/kg intraperitoneally) every other day starting at day −1 for 3 weeks. WTCs vs M13, P < .001; WTCs vs MCC, P = .0055; WTCs vs M13 plus MCC, P < .0001; M13 vs MCC, P = .0152; M13 vs M13 plus MCC, P = .4291; MCC vs MCC plus M13, P = .1226. Data represent combination of 3 independent experiments, n = 30 per group. (E) Kaplan-Meier survival curve for B6 > Balb/c GVHD animals given MDSC-IL13 (M13) and/or the NLRP3-specific inhibitor ICTA. WTCs vs M13, P = .0028; WTCs vs ICTA2, P = .0128; WTCs vs M13 plus ICTA, P = .0001; M13 or ICTA vs M13 plus ICTA, P = not significant. Data represent a single experiment, n = 10 per group. **P < .01, ****P < .0001.
These findings indicate that glyburide and the selective inflammasome inhibitor MCC950 maintain MDSC-IL13 in vitro suppressor function and arginase-1 activity. Glyburide was not well tolerated in GVHD mice (data not shown), possibly complicated by increased stress from hypoglycemia. MCC950 has shown efficacy in experimental autoimmune encephalomyelitis, multiple sclerosis, and cryopyrin-associated periodic syndrome models.27 Although MCC950 was well tolerated and improved aGVHD survival overall, despite dose escalations and varied treatment regimens, we were unable to demonstrate the expected synergistic effect of combining MCC950 and MDSC-IL13s (Figure 3D). The same trend was observed for ICTA treatment with MDSC-IL13s, showing modestly improved survival when administered alone but no advantage when combined with MDSC-IL13s (Figure 3E). In contrast with the previously published results for ASC KO14 or from caspase-1/-11 KO and NLRP3 KO MDSC-IL13s, pharmacologically inhibiting NLRP3 (MCC950) was not additive to MDSC-IL13s in promoting survival, although targeting inflammasomes globally improved outcomes in GVHD modestly. As shown in Figure 2A, AIM2 KO MDSCs produced very little IL-1β in response to poly(dT), a mimic for viral DNA. Because AIM2 is a downstream target of the gain-of-function Jak2V617F mutation in polycythemia vera,51 ruxolitinib, a potent JAK1/2 inhibitor that inhibits inflammation and interferon-γ–driven MHC class 2 upregulation on myeloid cells, was also tested.3,52 Consistent with the results of AIM2 KO MDSC-IL13s as being comparable to WT MDSC-IL13s for aGVHD protection, ruxolitinib was largely ineffective (IL-1β) or at best modestly effective (arginase-1; suppression) in preventing inflammasome activation in vitro and therefore was not further pursued.
Extracellular ATP regulates MDSC-IL13 inflammasome-associated loss of function
Tissue damage associated with aGVHD can lead to necrotic cell death and bacterial product translocation, resulting in innate immune activation via MyD88/TRIF-mediated TLR signaling.46 Cell death increases extracellular ATP, stimulates innate and adaptive immunity,29,53,54 and is implicated in GVHD pathogenesis.31,55,56 Extracellular ATP is a leading indicator of system stress, rendering purinergic receptors (eg, P2x7R/P2y2R) especially attuned to sensing noninfectious damage55 ; GvHD upregulates P2x7R.56 Compared with nonirradiated controls, radiation-induced peritoneal extracellular ATP peaked at 3 to 4 days after irradiation (Figure 4A). The ATP diphosphohydrolase apyrase, administered 2.5 hours prelavage, abrogated high extracellular ATP levels. Further supporting ATP as the primary driver of inflammasome-associated loss of function, survival was improved, with MDSCs rendered nonresponsive to ATP via P2x7 receptor deficiency, demonstrating enhanced survival relative to WT MDSCs (Figure 4B). In vitro, the highly selective P2x7R inhibitor A-438079 also reduced inflammasome activation (Figure 3A), preserved suppressive capacity (Figure 3B), and maintained arginase-1 levels (Figure 3C). However, similar to NLRP3 pharmacologic targeting, A-438079 promoted survival of GVHD mice but was not additive with WT MDSC-IL13s (Figure 4C).
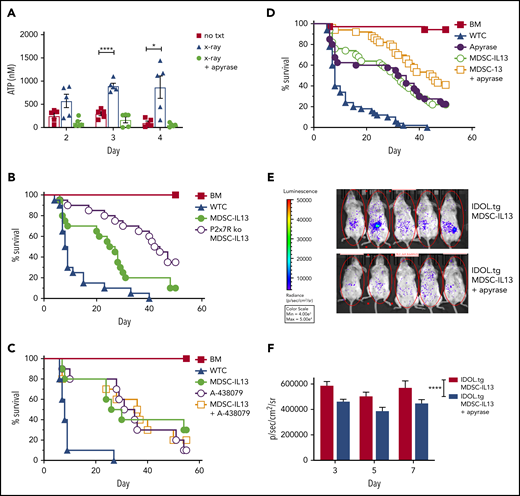
Extracellular ATP associated with MDSC loss of function. Balb/c mice were lethally irradiated then euthanized on the day indicated, injected intraperitoneally with 1 mL of cold RPMI. Animals marked X-ray plus apyrase received a single intraperitoneal injection of 10 units of apyrase 2.5 hours before euthanization. (A) Fluid was recovered from the peritoneal space to prechilled tubes and assayed for extracellular ATP. Data are representative of 2 independent experiments, n = 5 per group. (B) Kaplan-Meier survival curve of Balb/c-recipient GVHD animals receiving MDSC-IL13s (M13) or P2x7R KO MDSC-IL13s. Whole T cells (WTCs) vs M13, P = .0052; WTCs vs P2x7R KO, P < .0001; M13 vs P2x7R KO, P = .0158. Data are representative of 2 independent pooled experiments, n = 20 per group. (C) Kaplan-Meier survival of Balb/c GVHD mice treated with A-438079 (80 mg/kg intraperitoneally daily from day 0 to +4) with or without MDSC-IL13s as indicated. WTCs vs M13, P = .0028; WTCs vs A438079, P = .0002; WTCs vs A438079 plus M13, P = .0012; M13 vs A438079 or A438079 plus M13, P = not significant. Data shown represent a single experiment, n = 10 per group. (D) Kaplan-Meier survival curve of GVHD mice treated with apyrase (4 U intraperitoneally daily from day 0 to 4) or MDSC-IL13 plus apyrase, as indicated. WTCs vs M13, apyrase, or M13 plus apyrase, P < .0001; M13 vs apyrase, P = not significant; M13 vs apyrase plus M13, P = .0063; apyrase vs M13 plus apyrase, P = .0164. Data represent 5 combined independent experiments, n = 50 per group. (E) Animals were treated as in panel D using IDOL-transgenic donors and then on day 3 posttransfer assayed for whole-body bioluminescent imaging 5 minutes after D-luciferin injection. (F) Summary data are from a single experiment, n = 12 to 14 mice per group, and are representative of 2 independent experiments. *P < .05, ****P < .0001. txt, treatment.
Extracellular ATP associated with MDSC loss of function. Balb/c mice were lethally irradiated then euthanized on the day indicated, injected intraperitoneally with 1 mL of cold RPMI. Animals marked X-ray plus apyrase received a single intraperitoneal injection of 10 units of apyrase 2.5 hours before euthanization. (A) Fluid was recovered from the peritoneal space to prechilled tubes and assayed for extracellular ATP. Data are representative of 2 independent experiments, n = 5 per group. (B) Kaplan-Meier survival curve of Balb/c-recipient GVHD animals receiving MDSC-IL13s (M13) or P2x7R KO MDSC-IL13s. Whole T cells (WTCs) vs M13, P = .0052; WTCs vs P2x7R KO, P < .0001; M13 vs P2x7R KO, P = .0158. Data are representative of 2 independent pooled experiments, n = 20 per group. (C) Kaplan-Meier survival of Balb/c GVHD mice treated with A-438079 (80 mg/kg intraperitoneally daily from day 0 to +4) with or without MDSC-IL13s as indicated. WTCs vs M13, P = .0028; WTCs vs A438079, P = .0002; WTCs vs A438079 plus M13, P = .0012; M13 vs A438079 or A438079 plus M13, P = not significant. Data shown represent a single experiment, n = 10 per group. (D) Kaplan-Meier survival curve of GVHD mice treated with apyrase (4 U intraperitoneally daily from day 0 to 4) or MDSC-IL13 plus apyrase, as indicated. WTCs vs M13, apyrase, or M13 plus apyrase, P < .0001; M13 vs apyrase, P = not significant; M13 vs apyrase plus M13, P = .0063; apyrase vs M13 plus apyrase, P = .0164. Data represent 5 combined independent experiments, n = 50 per group. (E) Animals were treated as in panel D using IDOL-transgenic donors and then on day 3 posttransfer assayed for whole-body bioluminescent imaging 5 minutes after D-luciferin injection. (F) Summary data are from a single experiment, n = 12 to 14 mice per group, and are representative of 2 independent experiments. *P < .05, ****P < .0001. txt, treatment.
Together, these data (Figures 3 and 4) point to MDSC-intrinsic targeting rather than systemic inflammasome targeting to augment MDSC-IL13 survival effects. Because gastrointestinal GVHD is associated with higher morbidity and mortality, we sought to target extracellular ATP directly in the peritoneal space. Mice given MDSC-IL13s with apyrase intraperitoneally on days 0 to 4 had significantly higher survival than those receiving either individual treatment (Figure 4D). To confirm apyrase intraperitoneal injections limited MDSC-IL13 inflammasome conversion locally, MDSC-IL13s were generated from a novel reporter-transgenic donor strain, IDOL-tg, which has IL-1β promoter–driven luciferase expression in which IL-1β transcription and caspase-1 enzymatic processing are both required to yield a luminescent signal, whereas under noninflammasome conditions, IDOL proteasomal degradation prevents luminescence.57 Under GVHD conditions, IDOL-MDSC-IL13s exhibited a robust signal, which was reduced by apyrase (Figure 4E). On days 3, 5, and 7, MDSC-IL13 inflammasome activation was diminished by apyrase treatment (Figure 4E-F). Together, reduced peritoneal extracellular ATP and MDSC-IL13 inflammasome activation suggest early posttransplantation ATP targeting better maintains suppressor function in vivo.
Optimal in vitro and in vivo MDSC-IL13 suppression supported by IL-1R expression after inflammasome activation
IL-1β is at the center of local and systemic inflammation, and as a hallmark of inflammasome activation produced by myeloid cells, IL-1β is tightly controlled.21,40,58 IL-1β, linked to GVHD, is highly enriched in murine GVHD organs56 and, in some studies, correlates with GVHD severity in patients.59 IL-1R antagonists (IL-1RA) neutralize IL-1β and IL-1α to reduce murine GVHD.60,61 Furthermore, IL-1RA and soluble IL-1R clinical efficacy was seen in steroid-resistant GVHD.62,63 However, in the peri–BM transplantation period, IL-1RA (days −4 to +10) was ineffective, consistent with IL-1β operating later post–BM transplantation.64,65 To determine whether donor MDSC-IL13 inflammasome-generated IL-1β directly contributes to loss-of-suppressor function, IL-1β KO donors were used to generate MDSC-IL13s. Activated inflammasome-mediated MDSC-IL13 maturation would be unchanged (ie, ariginase-1 loss) as core inflammasome components remained intact, thereby isolating effects to MDSC-secreted IL-1β. Interestingly, although IL-1β KO MDSC-IL13s improved survival compared with no MDSCs (Figure 5A; P = .0434), we found that IL-1β KO MDSC-IL13s were inferior to WT MDSC-IL13s (P = .0027). This suggests MDSC IL-1β secretion is not directly driving inflammasome-mediated graft loss as expected.
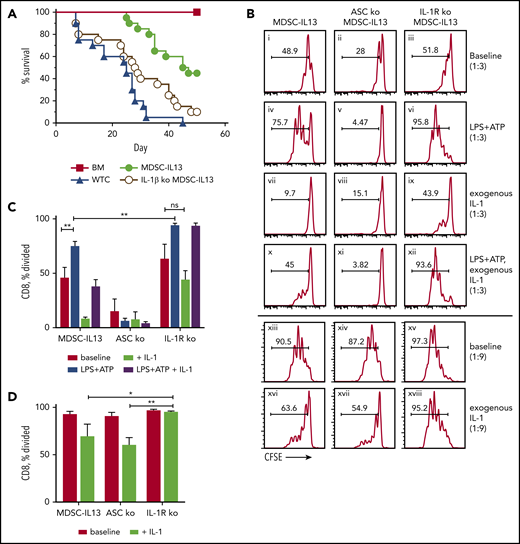
IL-1β plays a protective role by suppressing T cells. (A) Kaplan-Meier survival curve of Balb/c-recipient GVHD mice receiving MDSC-IL13s (M13) from WT or IL-1β–deficient donors. Whole T cells (WTCs) vs M13, P < .0001; WTCs vs IL-1β KO, P = .0434; M13 vs IL-1β KO, P = .0027. Data represent 2 independent pooled experiments, n = 20 per group. (B) Cultured MDSC-IL13s from WT or IL-1R KO donors were plated in a suppression assay at a 1:3 or 1:9 ratio as indicated with CFSE-labeled naïve responder IL-1R KO T cells and T cell–depleted stimulators (0.5×) from IL-1R KO donors with 0.25 µg/mL of anti-CD3ε. Histograms represent loss of CFSE fluorescence from cell division 3 days after coculture and are coded for clarity with text. Summary data for the 1:3 (C) and 1:9 ratios, representing 3 independent cultures per group, representative of 3 independent experiments. *P < .05, **P < .01. ns, not significant.
IL-1β plays a protective role by suppressing T cells. (A) Kaplan-Meier survival curve of Balb/c-recipient GVHD mice receiving MDSC-IL13s (M13) from WT or IL-1β–deficient donors. Whole T cells (WTCs) vs M13, P < .0001; WTCs vs IL-1β KO, P = .0434; M13 vs IL-1β KO, P = .0027. Data represent 2 independent pooled experiments, n = 20 per group. (B) Cultured MDSC-IL13s from WT or IL-1R KO donors were plated in a suppression assay at a 1:3 or 1:9 ratio as indicated with CFSE-labeled naïve responder IL-1R KO T cells and T cell–depleted stimulators (0.5×) from IL-1R KO donors with 0.25 µg/mL of anti-CD3ε. Histograms represent loss of CFSE fluorescence from cell division 3 days after coculture and are coded for clarity with text. Summary data for the 1:3 (C) and 1:9 ratios, representing 3 independent cultures per group, representative of 3 independent experiments. *P < .05, **P < .01. ns, not significant.
To discern IL-1β alloresponse effects, CFSE-labeled responder T cells, T cell–depleted splenocyte stimulators, and anti-CD3ε were admixed with or without exogenous IL-1β and MDSC-IL13s. To isolate the effects of exogenous IL-1β to MDSC-IL13s, IL-1R KO responder T cells and stimulators were used. At 1:3 MDSC/T cell ratios, MDSC-IL13 suppression began showing reduced efficacy in fully inhibiting proliferation (Figure 5B). Pretreating MDSC-IL13s with LPS plus ATP further reduced suppression in an ASC-dependent manner (Figure 5Bi vs 5Biv; Figure 5Biv vs 5Bv). Interestingly, activating NLRP3 inflammasomes in IL-1R KO MDSCs showed still further reduced suppression vs WT (Figure 5Biv vs 5Bvi), suggesting inflammasome activation–induced IL-1β production promotes suppression even with reduced arginase-1–mediated suppression resulting from MDSC-IL13 maturation.14 Conversely, inflammasome-activated MDSC-IL13 cocultures required IL-1R expression for increased suppression by exogenous IL-1β (Figure 5Bi vs 5Bvii; Figure 5Biv vs 5Bx; Figure 5Bx vs 5Bxii); even without inflammasome activation, at MDSC/T cell ratios of 1:9, IL-1β supplementation increased suppression (Figure 5Bxiii vs 5Bxvi) in an IL-1R–dependent manner (Figure 5Bxvi vs 5Bxviii). These findings are summarized for the 1:3 (Figure 5C) and 1:9 ratios (Figure 5D) and may help explain why global inflammasome inhibition combined with MDSC-IL13s is not additive, because reduced systemic IL-1β production may limit T-cell suppression in GVHD.
Suppression of inflammasome-mediated MDSC conversion by Tregs
Our GVHD model deliberately segregated MDSC from Treg function by depleting Tregs from the donor T-cell graft. It has been reported that MDSCs support Treg expansion through CD40-CD40L triggering and release of Treg-supporting cytokines.66 Because Tregs can reduce inflammation, and MDSC-IL13s and Tregs can suppress alloresponses via different mechanisms, we reasoned that adding Tregs to MDSC-IL13 therapy might be additive or synergistic by supporting MDSC function as well. Although each cell type prolonged survival compared with T cells alone, a marked increase in overall survival was seen with cotransfer of MDSCs and Tregs (P < .0065; MDSC-IL13s or Tregs vs both; Figure 6A). To determine if MDSC-IL13 and Treg coinfusion inhibited inflammasome-associated conversion of MDSCs, MDSC-IL13 inflammasome activation was assessed using IDOL-transgenic MDSC-IL13s. Indeed, Tregs inhibited MDSC-IL13 inflammasome activation, as evidenced by reduced BLI signal (Figure 6B); the effect was transient, peaking day 5, when MDSC conversion was maximal (Figure 6C; IDOL vs IDOL plus Tregs; P = .0022). Whole-organ explants from IDOL-MDSC-IL13 GVHD mice indicated that at day 5, the large and small intestine are primary inflammasome activation sites and are protected when Tregs are present (Figure 6D-E). These findings support Tregs acting to protect MDSC function in a mutually beneficial manner, resulting in a synergistic survival advantage.
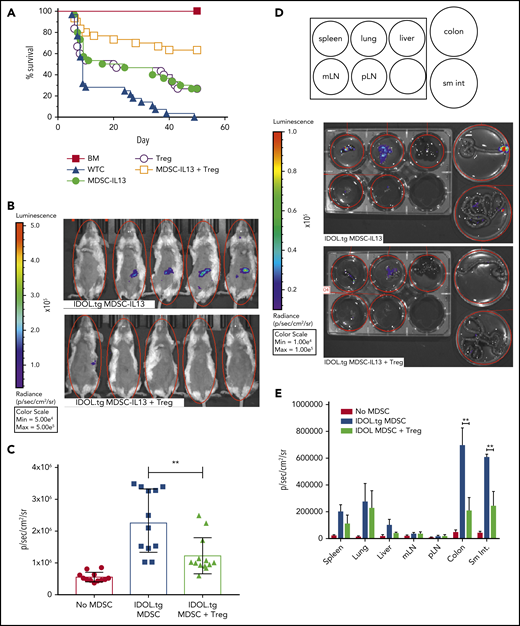
Regulatory T cells promote enhanced survival by protecting MDSC-IL13 function. (A) Kaplan-Meier survival curve of Balb/c GVHD mice given MDSC-IL13s (6e6) and/or freshly isolated regulatory T cells (2e6). Whole T cells (WTCs) vs M13, P = .0022; WTCs vs Tregs, P = .0083; WTCs vs M13 plus Tregs, P < .0001; M13 vs Tregs, P = not significant; M13 vs M13 plus Tregs, P = .0064; Tregs vs M13 plus Tregs, P = .0047. Data are a combination of 3 independent experiments, n = 30 per group. (B) GVHD animals were treated as in panel A with MDSC-IL13s from IDOL-transgenic donors with or without Tregs. On day 5, animals were shaved and given 3 mg of D-luciferin intraperitoneally; BLI was performed after 5 minutes. (C) Summary BLI data, day 5, indicating the radiance values for the region of interest shown in Figure 6B. No MDSCs vs IDOL MDSCs, P < .0001; no MDSCs vs IDOL plus Tregs, P = .0006; IDOL vs IDOL plus Tregs, P = .0022. Data are from n ≥ 12 per group, representative of 2 independent experiments. (D) After whole-body imaging, animals were euthanized and organs explanted to dishes containing D-luciferin in solution for imaging as indicated. (E) Day 5 summary data of organ explants, n = 5 per group. **P < .01. mLN, mesenteric lymph node; pLN, peripheral LN; sm int, small intestine.
Regulatory T cells promote enhanced survival by protecting MDSC-IL13 function. (A) Kaplan-Meier survival curve of Balb/c GVHD mice given MDSC-IL13s (6e6) and/or freshly isolated regulatory T cells (2e6). Whole T cells (WTCs) vs M13, P = .0022; WTCs vs Tregs, P = .0083; WTCs vs M13 plus Tregs, P < .0001; M13 vs Tregs, P = not significant; M13 vs M13 plus Tregs, P = .0064; Tregs vs M13 plus Tregs, P = .0047. Data are a combination of 3 independent experiments, n = 30 per group. (B) GVHD animals were treated as in panel A with MDSC-IL13s from IDOL-transgenic donors with or without Tregs. On day 5, animals were shaved and given 3 mg of D-luciferin intraperitoneally; BLI was performed after 5 minutes. (C) Summary BLI data, day 5, indicating the radiance values for the region of interest shown in Figure 6B. No MDSCs vs IDOL MDSCs, P < .0001; no MDSCs vs IDOL plus Tregs, P = .0006; IDOL vs IDOL plus Tregs, P = .0022. Data are from n ≥ 12 per group, representative of 2 independent experiments. (D) After whole-body imaging, animals were euthanized and organs explanted to dishes containing D-luciferin in solution for imaging as indicated. (E) Day 5 summary data of organ explants, n = 5 per group. **P < .01. mLN, mesenteric lymph node; pLN, peripheral LN; sm int, small intestine.
Discussion
Although MDSC therapy is effective at prolonging survival in a robust aGVHD model, the functionally effective window is relatively narrow because of the harsh postconditioning environment that results in MDSC differentiation away from immature suppressive myeloid lineage cells.14 Here we identify radiation conditioning–induced peritoneal extracellular ATP as a primary driver of inflammasome activation. Acute GVHD injury increases LPS67-69 and ATP,46,56 and we show here that prohibiting ATP binding to its receptor (P2x7R) or targeting downstream NLRP3-associated inflammasome activation on transferred MDSCs better maintains functional activity and hence recipient survival. Of the other well-characterized inflammasome pathways tested, MDSC-IL13s from AIM2 or NLRC4 KO donors ameliorated in vitro MDSC-IL13 loss of function upon exposure to their unique inciting stimuli (poly[dT] and flagellin, respectively) but did not improve in vivo survival. These data suggest there are insufficient stimuli (DNA/flagellin) under these in vivo conditions in specific pathogen-free mice to trigger AIM2 or NLRC4 inflammasome activation in MDSC-IL13s.
To begin to define mechanisms leading to MDSC dysfunction, we focused initially on MDSC-intrinsic pathways. NLRP3 inflammasome activation by LPS plus ATP required MyD88 and TRIF for full caspase-1 activation, IL-1β secretion, and MDSC-IL13 loss of function. The dual requirement for deleting MyD88/TRIF to abrogate in vitro NLRP3 inflammasome activation is consistent with the known signaling of TLR4 and MyD88 upregulation by P2x7R signaling.70 Consistent with these data, MDSC-IL13s deficient in ASC, caspase-1, NLRP3, or P2x7R each improved survival relative to WT MDSC-IL13s. Although pharmacologic inhibitors targeting these pathways provided varying efficacy, our finding that ATP/NLRP3 inflammasome disruption resulted in maintained MDSC function despite inflammasome-activating conditions in vitro was largely validated. Despite drug-dose escalations and varied schedules, combining these inhibitors with MDSC-IL13 transfer fundamentally failed to phenocopy the additive effects seen with MDSC-IL13s generated from inflammasome KO donors.
IL-1 cytokine family literature is rich and varied, with pro- and anti-inflammatory properties, decoy receptors, and differential expression patterns all suggesting a tightly regulated and highly conserved system for governing immune regulation.71 Findings supporting IL-1RA as a treatment approach for chronic conditions suggest IL-1R signaling is proinflammatory31 ; however, confounding data exist in the acute setting, where recovery from tissue damage and immune pathology, especially at the intestinal barrier, a primary GVHD organ, depends on IL-1 responsiveness.72-74 IL-1R signaling shares the intracellular adaptor MyD88 with TLRs,71 which could enlighten the underlying causes of limited MyD88 KO MDSC-IL13 efficacy. Despite an inability to signal via IL-1R, the trend toward survival extension with MyD88/TRIF dKO MDSC-IL13s may be due to better maintained suppressive function seen with dual adapter protein deficiency.
Extracellular ATP, a known NLRP3 inflammasome trigger, was increased in the peritoneal cavity from conditioning regimen injury and GVHD56 and was susceptible to catabolic hydrolysis with the ATP diphosphohydrolase apyrase. The resulting reduced MDSC-associated inflammasome activation as measured by BLI of IDOL-transgenic MDSC-IL13s and improved survival conferred by MDSC-IL13s support the notion that danger signals such as ATP and LPS initiate the early GVHD events that mature MDSC-IL13s,14 circumventing suppressor function and only transiently inhibiting GVHD lethality. Peri–BM transplantation apyrase administration in the peritoneal cavity focused catabolic activity to the peak time of ATP production in a critical GVHD organ, the gut. Our presumption that IL-1β contributed directly to MDSC-associated loss of function and reduced survival proved incorrect, because IL-1β KO MDSC-IL13s showed consistently reduced in vitro suppression and in vivo decreased survival relative to WT MDSC-IL13s in GVHD mice. Indeed, IL-1β production by MDSC-IL13s and IL-1β binding to IL-1R actually promoted suppressor function, as assayed in vitro and in vivo, consistent with data shown in Figure 5. As such, the failure of drug-based NLRP3 inflammasome inhibition to add to the in vivo MDSC-IL13 protective effect seems to be a function of prolonged, systemic IL-1β inhibition in concert with MDSC dysfunction rather than incomplete penetrance, because survival benefits were seen with the inhibitors alone, as similarly reported elsewhere.56 Findings are suggestive of a more complex dynamic between MDSC inflammasome activation and IL-1β secretion in tempering MDSC-IL13 therapeutic benefits. Although beyond the scope of this study, it will be of interest to elucidate the divergent roles of inflammasome activation and IL-1β signaling. Intriguingly, Tregs cotransferred with MDSC-IL13s cooperated to improve survival over either therapy alone. Using IDOL-MDSC imaging, we demonstrated reduced inflammasome activation when Tregs were present. Alternatively, blunting caspase-1 cleavage of intracellular pro–IL-18, particularly in the absence of IL-1β, could mitigate the known IL-18 benefits in augmenting murine survival in CD4 T cell–dominated GVHD systems,75-77 but this will require further study.
In summary, the transient MDSC-IL13 improvement in GVHD recipient survival is caused, at least in part, by extracellular ATP triggering of P2x7R in conjunction with NLRP3 inflammasome activation. IL-1R signaling of MDSC-IL13s and IL-1β production by activated inflammasomes reduces GVHD lethality. Collectively, these results suggest that MDSC-IL13s genetically modified to selectively delete P2x7R (or other purinergic receptors), NLRP3, or the shared ASC inflammasome component or local control of ATP levels in the peritoneal cavity warrant further consideration for human studies in light of the known susceptibility of human MDSCs to loss-of-suppression function with inflammasome activation.14
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Vishwa Deep Dixit (Yale University) for providing the β-hydroxybutyrate nanolipogel compound, Sheng Wei (Moffitt) for providing ICTA, Samithamby Jeyaseelan Jey (Louisiana State University) for providing bones from MyD88 KO and MyD88/TRIF dKO mice, and colleagues of B.R.B.’s laboratory for technical assistance.
This study was supported by National Institutes of Health Research Program Grants from the National Heart, Lung, and Blood Institue (R01 HL56067), National Institue of Allergy and Infectious Diseases (R37 AI34495), and National Heart, Lung, and Blood Institue (R01 HL11879).
Authorship
Contribution: B.H.K. designed and performed experiments, analyzed data, and wrote the manuscript; A.S., C.M.-H., M.L., G.T., L.M., M.Z., J.D., W.K., and J.P. performed and/or consulted on experiments; K.H., S.C.J., J.S.M., M.A.C., C.J.F., T.I., J.P.-Y.T., J.S.S., W.J.M., G.R.H., V.B., and D.H.M. provided material and consulted on experimental design and manuscript preparation; P.J.M. and R.Z. designed experiments, consulted on data, and edited the manuscript; and B.R.B. designed experiments, discussed data, and edited the manuscript.
Conflict-of-interest disclosure: B.R.B. receives remuneration as an advisor to Kadmon Pharmaceuticals, Inc., Five Prime Therapeutics, Inc., Regeneron Pharmaceuticals, Magenta Therapeutics, and BlueRock Therapeutics and research support from Fate Therapeutics, RXi Pharmaceuticals, Alpine Immune Sciences, Inc., AbbVie, Inc., the Leukemia and Lymphoma Society, the Children’s Cancer Research Fund, and the KidsFirst Fund and is a cofounder of Tmunity Therapuetics, Inc.; C.J.F. is an employee of Novartis Pharma AG; J.S.M. receives remuneration as a consultant to and receives research funds from Fate Therapeutics and GT Biopharma and serves on the scientific advisory boards of OnkImmune, Dr Reddy’s Laboratories, Moderna, Nektar, and CytoSen; and B.H.K., R.Z., P.J.M., V.B., and B.R.B. disclose patent licensing to Fate Therapeutics, Inc. The remaining authors declare no competing financial interests.
Correspondence: Bruce R. Blazar, MMC 109, 420 Delaware St SE, University of Minnesota, Minneapolis, MN 55455; e-mail: blaza001@umn.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal