

Background: Studying gene expression and clinical outcome data from 136 clinical trials for patients with cancer (~21,000 patients with 26 cancer types), we found CD25 as one of the strongest predictors of poor clinical outcome in patients with B-cell malignancies, but not in other cancer types. This was unexpected because CD25 is known as one of three chains of the IL2 receptor on T-cells and NK-cells. Interleukin-2 (IL2) functions as essential T-cell growth factor. IL2 signals through b- and g-, but not a-chains (CD25) of its heterotrimeric receptor. CD25-deficiency causes lymphoproliferation and autoimmunity, however, its mechanistic role is unclear.
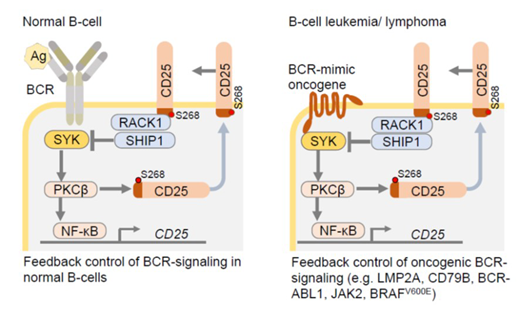
Results: Our experiments based on genetic mouse models and engineered patient-derived B-cell leukemia and lymphoma xenografts revealed that CD25 expressed on B-cells is not an IL2 receptor chain, but in fact binds downstream signaling molecules of the B-cell receptor (BCR). Through these interactions, CD25 mediates negative feedback to BCR signaling in response to antigen-encounter in normal B-cells. Defects in CD25-/-B-cells were not replicated in mice that express CD25 but lack expression of the IL2 cytokine. These findings demonstrate IL2-independent functions of CD25 in B-cells and B-cell derived leukemia and lymphoma. To comprehensively study the interactome of the short cytoplasmic tail of CD25, we performed proximity-dependent biotin identification (BioID). This analysis revealed that the CD25 tail exerts negative feedback control through recruitment of the PKCβ-scaffold RACK1 and the inhibitory phosphatase SHIP1 (see schematic, left). Interestingly, the cytoplasmic tail of CD25 harbors a PKCβ-substrate motif and mutation of a central serine residue (S268) to A268 compromised interactions with PKCβ, its scaffold RACK1 and SHIP1, demonstrating that feedback control was dependent on PKCβ-mediated phosphorylation of CD25-S268.
A genetic observation in a family with monogenic autoimmunity confirmed the functional importance of the cytoplasmic CD25-tail motif: a mutation immediately preceding S268 compromised CD25-surface translocation, which was restored by homology-directed repair of the S268. In vitro kinase assay with 62 candidate kinases against recombinant cytoplasmic tail of CD25-S268 or -A268 identified PKCβ as top-ranking kinase hit for CD25-S268 but not CD25-A268. Our genetic studies revealed that PKCβ is required for cell-membrane translocation of CD25, but also transcriptional expression of CD25 via NF-κB activation. Therefore, PKCβ act as critical effector molecule downstream of CD25 to mediate B-cell selection during normal B-cell development and calibrate oncogenic BCR signaling in B-cell tumors.
In B-cell malignancies, BCR-dependent survival and proliferation signals are often substituted by oncogenic BCR-mimics (e.g. BCR-ABL1, JAK2, BRAFV600E, LMP2A, CD79B mutations; see schematic, right). Accordingly, we identified CD25 surface-expression as biomarker of oncogenic BCR-signaling and predictor of poor clinical outcomes. CD25-/-B-cell leukemia failed to initiate fatal disease in transplant recipients. Owing to imbalances of oncogenic BCR-signaling and p53-checkpoint activation, CD25-/- B-cell leukemia failed to initiate fatal disease in transplant recipients. In patient-derived xenograft models of drug-resistant B-cell malignancies, treatment with a CD25-specific antibody drug-conjugate (ADCT-301) extended survival of transplant recipients or eradicated disease. These findings identified CD25 as previously unrecognized feedback regulator of oncogenic BCR-signaling and provide a rationale for therapeutic targeting of CD25 in refractory B-cell malignancies.
Zammarchi:ADC Therapeutics: Employment. Van Berkel:ADC Therapeutics: Research Funding. Melnick:Constellation: Consultancy; Janssen: Research Funding; Epizyme: Consultancy. Luger:Celgene: Research Funding; Cyslacel: Research Funding; Pfizer: Honoraria; Seattle Genetics: Research Funding; Agios: Honoraria; Ariad: Research Funding; Biosight: Research Funding; Kura: Research Funding; Onconova: Research Funding; Genetech: Research Funding; Jazz: Honoraria; Daichi Sankyo: Honoraria. Meffre:AbbVie: Consultancy, Other: Grant. Weinstock:Celgene: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal