

Key Points
Unsupervised consensus clustering put together patients with similar morphology or mutations into 5 morphologic and 8 genetic profiles.
Machine-learning techniques interrogated morphologic feature interdependencies and potential associations with mutations and survival.

Abstract
Morphologic interpretation is the standard in diagnosing myelodysplastic syndrome (MDS), but it has limitations, such as varying reliability in pathologic evaluation and lack of integration with genetic data. Somatic events shape morphologic features, but the complexity of morphologic and genetic changes makes clear associations challenging. This article interrogates novel clinical subtypes of MDS using a machine-learning technique devised to identify patterns of cooccurrence among morphologic features and genomic events. We sequenced 1079 MDS patients and analyzed bone marrow morphologic alterations and other clinical features. A total of 1929 somatic mutations were identified. Five distinct morphologic profiles with unique clinical characteristics were defined. Seventy-seven percent of higher-risk patients clustered in profile 1. All lower-risk (LR) patients clustered into the remaining 4 profiles: profile 2 was characterized by pancytopenia, profile 3 by monocytosis, profile 4 by elevated megakaryocytes, and profile 5 by erythroid dysplasia. These profiles could also separate patients with different prognoses. LR MDS patients were classified into 8 genetic signatures (eg, signature A had TET2 mutations, signature B had both TET2 and SRSF2 mutations, and signature G had SF3B1 mutations), demonstrating association with specific morphologic profiles. Six morphologic profiles/genetic signature associations were confirmed in a separate analysis of an independent cohort. Our study demonstrates that nonrandom or even pathognomonic relationships between morphology and genotype to define clinical features can be identified. This is the first comprehensive implementation of machine-learning algorithms to elucidate potential intrinsic interdependencies among genetic lesions, morphologies, and clinical prognostic in attributes of MDS.
Introduction
The pathogenesis of myelodysplastic syndromes (MDSs) is founded in progressive acquisition of genomic lesions (mutations, chromosomal defects)1,2 ; yet, since the introduction of aniline dyes by Paul Ehrlich, morphologic evaluation of blood and marrow cells has been the gold standard for diagnoses of hematologic neoplasia such as MDS.3 The spectra of morphologic abnormalities include continuums from dysplasia to myeloproliferative features, low to high blast counts, and changes in different blood cell lineages of varying degree. These continuums have been used across generations of morphologic disease classifications, which along with functional parameters and cytogenetics, form the basis for current prognostic schemes.4,5 Subjectivity is a downside of morphologic evaluations, with interpathologist reliability of assessment shown to be variable.6 Although morphologic abnormalities provide some clues as to the mechanisms of MDS evolution, somatic mutations and chromosomal defects are directly linked to the pathogenesis of this disease and are likely responsible for the pathognomonic morphologic changes.7 A few well-known genotype/morphology associations provide a general proof of principle for the usefulness of genotype/phenotype associations. They include those of the del(5q) syndrome, the link of SF3B1 mutations to ring sideroblasts, the presence of JAK2/SF3B1 mutations in refractory anemia with ring sideroblasts with thrombocytosis (RARS-T),8 and MYH9 mutations in May-Hegglin anomaly.9 Correction between the presence of ring sideroblasts and SF3B1 mutations has been well established and does not require further bioanalytic workup. Erythroid dysplasia may exist with and without ring sideroblasts. For the purpose of this study, erythroid dysplasia was evaluated on bone marrow smears according to Wright staining. In the last decade, systematic application of next-generation sequencing (NGS) has led to important discoveries of somatic mutation associations with MDSs. Combined with large spectra of recurrent chromosomal lesions in MDSs, the tremendous complexity of morphologic and genetic changes imposes challenges to studies endeavoring to establish correlations among them. Indeed, the extent to which diverse genetic and epigenetic alterations share phenotypes is unresolved; their successful integration may offer a new avenue to improve diagnosis and prognosis of MDSs. To that end, modern statistical approaches exploiting machine-learning and artificial intelligence bioinformatic tools, along with the availability of sufficiently large data sets, provide an opportunity for the most efficient, combined analysis of genomic and morphologic data. Such an integration could resolve many of the limitations of current diagnostic schemes, including subjectivity, labor intensity, incomplete reproducibility, and disconnect between genetic/functional and morphologic/phenotypic biomarkers. Our study applies these techniques to identify relationships between morphologic features and genomic changes with different clinical MDS phenotypes. The goal is to establish more precise descriptions of MDS patients with subtyping schemes that integrate multiple features of the disease.
Methods
Patients
A total of 1079 patients with MDS (n = 654), MDS/myeloproliferative neoplasms (MPNs) (n = 231) and secondary acute myeloid leukemia (AML; sAML) from MDS or MDS/MPN (n = 194) were screened and enrolled in this study (Table 1).1 Therapy-related MDSs were not included. Patients had fully annotated outcomes with follow-up and pathomorphologic evaluations. All samples were obtained after written informed consent, according to protocols approved by Cleveland Clinic’s Institutional Review Board (IRB-5024). Two hundred thirty-one patients were diagnosed with MDS/MPN, 155 with chronic myelomonocytic leukemia (CMML; 15%), 54 with MDS/MPN unclassifiable (MDS/MPN-U; 5%), and 22 with RARS-T (2%). sAML cases arose from MDS (n = 175) or MDS/MPN (n = 19). World Health Organization (WHO) classification was used to dichotomize morphologic features.2 MDS patients were separated based on Revised International Prognostic Scoring System (IPSS-R) scores of ≤3.5 vs >3.5 as lower-risk (LR) vs higher risk (HR) of transformation to sAML,10 MDS/MPN patients were grouped by WHO classification; MDS/MPN-U and RARS-T are LRs, CMML were HRs. All secondary AML patients derived from MDS or MDS/MPN belong to the HR group. Fifty-seven percent of the patients (620 of 1079) were LR, 43% (459 of 1079) were HR (supplemental Table 2, available on the Blood Web site). Germline DNA was obtained from buccal mucosa or CD3+ T cells in peripheral blood.11 MDS DNA was from bone marrow or peripheral blood. Bone marrow smears or biopsy specimens were evaluated to establish cytomorphologic diagnosis and assess the individual cytogenetics abnormalities used in the analysis (supplemental Table 3). Bone marrow smears were used for cytomorphologic assessment by a skilled hematopathologist. Fibrosis was assessed on bone marrow biopsy specimens.
Patient characteristics
| Total cohort, n = 1079 | |
|---|---|
| Median age, y | |
| ≥60 | 883 |
| <60 | 196 |
| Male:female (ratio) | 682:397 (1.7) |
| Cases with follow-up, mo | 7.6 |
| Subtypes* | |
| MDS | |
| 5q− | 23 |
| RCUD | 57 |
| RARS | 68 |
| RCMD | 214 |
| RCMD-RS | 14 |
| MDS-U | 41 |
| RAEB-1 | 117 |
| RAEB-2 | 120 |
| Secondary AML† | 194 |
| MDS/MPN | |
| MDS/MPN-U | 54 |
| CMML-1 | 127 |
| CMML-2 | 28 |
| RARS-T | 22 |
| Cytogenetics,‡n (%) | |
| Normal karyotype | 392 (46) |
| Aberrant karyotype | 455 (55) |
| Total cohort, n = 1079 | |
|---|---|
| Median age, y | |
| ≥60 | 883 |
| <60 | 196 |
| Male:female (ratio) | 682:397 (1.7) |
| Cases with follow-up, mo | 7.6 |
| Subtypes* | |
| MDS | |
| 5q− | 23 |
| RCUD | 57 |
| RARS | 68 |
| RCMD | 214 |
| RCMD-RS | 14 |
| MDS-U | 41 |
| RAEB-1 | 117 |
| RAEB-2 | 120 |
| Secondary AML† | 194 |
| MDS/MPN | |
| MDS/MPN-U | 54 |
| CMML-1 | 127 |
| CMML-2 | 28 |
| RARS-T | 22 |
| Cytogenetics,‡n (%) | |
| Normal karyotype | 392 (46) |
| Aberrant karyotype | 455 (55) |
5q−, myelodysplastic syndrome with isolated del(5q); AML, acute myeloid leukemia; CMML, chronic myelomonocytic leukemia; RAEB, refractory anemia with excess blasts; RCMD, refractory cytopenia with multilineage dysplasia; RCMD-RS, RCMD with ringed sideroblasts; RCUD, refractory cytopenia with unilineage dysplasia.
WHO classification 2008.1
AML from MDS (n = 175), sAML from MDS/MPN (n = 19).
Cytogenetics data from 847 patients are available.
Whole-exome sequencing
Whole-exome sequencing (WES) was performed as previously described.11,12 Paired disease and normal germline DNA was used. Whole-exome capture was accomplished by hybridizing sonicated genomic DNA to a bait complementary DNA (cDNA) library synthesized on magnetic beads (SureSelect Human All Exon 50 Mb or V4 kit; Agilent Technologies). Captured targets were sequenced using a HiSeq 2000 (Illumina) and the standard protocol for 100-bp paired-end reads. Reads were aligned to the human genome (hg19) by a Burrows-Wheeler aligner (http://bio-bwa.sourceforge.net/) using a genome analysis tool kit (GATK) version 4.0 pipeline that also extracted candidate variants/polymorphisms to reduce sequencing errors. Validations were performed by Sanger or polymerase chain reaction (PCR) amplicon targeted sequencing as previously described.12
Targeted sequencing
Targeted sequencing was performed using a TruSeq Custom Amplicon (Illumina) or a custom cDNA bait library (SureSelect; Agilent Technology) as previously described.12-14 Two panels had 33 genes in common (supplemental Table 4). Sequencing libraries were generated according to an Illumina paired-end library protocol. The enriched targets were sequenced using a Hiseq 2000 or Miseq (Illumina), at 862× coverage. Variants were annotated using Annovar15 and filtered by removing: (1) synonymous single-nucleotide variants; (2) variants only present in unidirectional reads; and (3) variants in repetitive genomic regions (supplemental Figure 4). Only variants with a minimum depth of 20 and 5 positive high-quality reads were called as mutants. A bioanalytic pipeline, devised in-house, as previously described,13 was applied to identify somatic mutations by comparison with sequenced controls and mutational databases such as dbSNP138,16 1000 Genomes17 or ESP 6500 database, and Exome Aggregation Consortium (ExAC).18 Mapping errors were removed by visual inspection with the Integral Genomics Viewer. Validation by Sanger sequencing or PCR amplicon sequencing was performed as previously described.13 Variant allelic frequencies were adjusted according to zygosity and copy number based on conventional metaphase karyotyping/single-nucleotide polymorphism array results.13 An overall accuracy of our platform for detection of somatic mutations was estimated to be 98.7% (74 of 75).19
Associations among mutations and morphology
Frequent mutations and morphologic changes were assessed for mutual correlation. Any combination of these variants was exhaustively tested in a pairwise manner using the Fisher exact test, and multiple testing was corrected with the Benjamini-Hochberg q value (assumed significant when q < 0.01 for coexistence). Significant correlations were plotted with transition colors (magenta for positive and green for negative correlations), together with circle diameters indicating the degree of significance.
Cluster and subtype analyses
Noting patterns of interdependence among morphologic features, unsupervised clustering was applied to define the intrinsic patterns of cooccurrence exhibited among 24 individual morphologic features and identify morphologic subtypes of MDS. Consensus clustering was used to identify and assign MDS patients to morphologic subtypes. Implementation used the Consensus Cluster Plus package in R20 with the partitioning around medoids algorithm and binary distance measures. Pairwise inter-patient dissimilarity, was computed from the consensus values by aggregating iterative clustering results from subsamples of MDS patients in the discovery cohort. The clustering process was performed for ranks from 2 to 15, with the optimal k determined by the proportion of ambiguous clustering (PAC) score.
Five discrete morphologic patterns were evident from unsupervised analyses. MDS patients classified as LR comprised 4 these profiles, which were further interrogated for orientation by patterns of genetic mutations. A machine-learning technique based on Bayesian partial exchangeability interrogated the extent to which patterns of mutation incidence and cooccurrence discriminate morphologic subtypes of MDS.21-23 The subtyping methodology was applied to LR MDS patients and targeted discrimination of the 4 morphologic profiles. The Bayesian model was used to define the extent to which patients i and j are pairwise exchangeable (or the extent to which the results can be averaged) when predicting the pathologically observed morphologic profile. The Bayesian framework facilitates an individualized predictive probability for each profile yielding a set of precision-recall and receiver operating characteristic curves. The minimum distance to perfect discrimination was identified for each curve. An optimal set of pairwise patient-exchangeability measures was selected to minimize the averaged distance. The resultant exchangeability relationships define an undirected, fully connected graph with respect to the patient sample space. The spin-glass algorithm was used to partition the individual patients into discrete subtypes.24,25 Implementation used the igraph package.26 A single model, selected and subsequently validated using the independent test set, yielded genetic signatures demonstrating morphologic orientation. The resultant mapping from mutations to MDS subtypes is described by a classification decision tree that was created using the Caret package in R27 after application of the random forest algorithm with subtype assignment as the response and genetic mutation as the independent variables.
HR patients comprised a single-morphologic subtype. Therefore, we did not assess patterns of cooccurrence among morphologic characteristics and mutations for patients classified clinically as HR. Instead, genetic subtypes of HR MDS were interrogated for their prognostic utility through association with survival. The random survival forest method was used to identify an optimal matrix of interpatient proximity measures with implementation in R using the randomForestSRC package.28 Discrete prognostic subtypes of HR MDS were defined from consensus clustering using the ConsensusClusterPlus package with the hierarchical clustering algorithm applied with complete linkage method and Pearson distance measure.
The decision tree provides this and thus disseminates the findings for practical use without the need for computation. Specifically, we have applied an open source tool supported by R to describe the relationship between subtypes and mutations using a simple set of decision thresholds. The tree is not a part of the Bayesian model. It rather describes the patterns identified by the model.
Validation
Model validation was conducted in an independent cohort of MDS patients. Patients in the validation cohort were assigned 1 clustering membership by the k-nearest neighbor algorithm (k = 5 here), by evaluating their relative similarity to each patient in the discovery cohort. The dissimilarity measure was computed with the same feature support as the discovery cohort with binary distance used to define the extent of dissimilarity between any 2 patients based on the presence of mutations.
Statistical analysis
Comparisons of proportions were performed by using 2-sided Fisher exact tests. Paired data were analyzed by the Wilcoxon signed-rank test. Continuous variables were compared using the Mann-Whitney U test. Kaplan-Meier methods were used for survival analysis. The log-rank test was used to compare survival curves. Analyses were performed with R (https://www.r-project.org), SPSS software (IBM) and Prism (GraphPad). Significance was determined at a 2-sided α level of 0.05, except for P values in multiple comparisons, which were adjusted according to the method described by Benjamini and Hochberg.29
Results
Spectrum of morphologic features
We analyzed 1079 patients with MDS or MDS/MPN overlap including LR and HR subtypes (Table 1). Bone marrow morphologic features were evaluated by an independent pathologist, blinded to mutational status, based on uniformly defined WHO criteria (supplemental Table 1).2 Other morphology-related clinical variables, such as extent and types of cytopenias, the presence of fibrosis, increased megakaryocytes, and monocytosis were also investigated (supplemental Table 2). In addition, all cases were separated into 2 risk groups according to IPSS-R (LR ≤3.5 and HR >3.5),10 each of which was randomly divided in a 3:1 ratio into discovery and validation groups (supplemental Figure 1). To address the challenge of revealing complexities of morphologic features and their combinations, we devised a step-wise simplified strategy; that is, >10% of a single-lineage cell which had at least 1 morphologic abnormality in the bone marrow were defined as having dysplasia. Patients were dichotomized into dysplasia positive vs negative in each lineage. Myeloid, erythroid, and megakaryocytic dysplasia occurred in 54%, 70%, and 72% of patients, respectively (supplemental Table 2). Ninety-four percent of patients had at least 1 dysplasia, 37% bilineage dysplasia, and 32% trilineage dysplasia (supplemental Figure 2). Focusing on 276 patients with single-lineage dysplasia, myeloid, erythroid, and megakaryocytic dysplasia were identified in 5%, 11%, and 10% of patients, respectively. In these patients, 46%, 62%, and 60% had neutropenia, anemia, and thrombocytopenia, respectively. Eighty-nine percent of patients had at least 1 cytopenia; 57% had multiple cytopenias. Proportions of patients with bone marrow fibrosis, elevated megakaryocytes, and monocytosis, were 19%, 31%, and 19%, respectively; 50% had at least 1 of these features.
These morphologic and clinical features were highly correlated, cooccurrence and mutual exclusivity were observed for several morphologic features (supplemental Figure 3): myeloid dysplasia cooccurred with dysplasias of other lineages, thrombocytopenia, and HR subtypes, and erythroid dysplasia was mutually exclusive of monocytosis. There are thus interactions between the molecular pathways that underlie different morphologic features.
Associations between mutations and morphologic features
In total, 33 genes were examined by NGS, focusing on mutations which were present in >10% cells which matched with the criteria for the morphologic features (supplemental Table 4); 1929 somatic mutations were identified in this manner after removing single-nucleotide polymorphisms/sequencing errors (supplemental Figure 4). The most frequently mutated genes were TET2 (20%), ASXL1 (17%), SF3B1 (13%), SRSF2 (11%), DNMT3A (11%), and RUNX1 (10%). Morphologic feature (present/absent) correlations with mutations (mutant/wild type) were quantified by odds ratios (supplemetnal Figures 5-8). There were 11 morphologic and clinical features that were associated with 33 mutated genes, so the number of possible associations was very large. We thus devised strategies that sequentially examined associations in an automated fashion. The goal was to identify causal and therefore recurrent genotype/phenotype relationships biologically and medically, as these would be more likely to be instructive. The utility of identifying an n × m relation (cluster of n features with m genes) was then substantiated by its impact on prognosis and relevance in risk of progression to AML (HR vs LR subtypes). Analyses of 11 × 33 = 363 univariate relations yielded 52 morphology/genotype associations (q < .1) (Figure 1A; Table 2). Examples include: myeloid dysplasia associating positively with STAG2, NRAS, SRSF2, TP53, and TET2 mutations and negatively with SF3B1 mutations (Figure 1B); erythroid and megakaryocyte dysplasia being enriched in SF3B1 and ASXL1 mutations, respectively; neutropenia being more frequent in patients with IDH1 mutations; anemia being positively associated with ETV6 mutations and negatively associated with TET2, mutations; thrombocytopenia being associated positively with TP53 mutations and negatively with JAK2, SF3B1, and BCORL1 mutations (Figure 1C); and fibrosis being associated more with JAK2 mutations and less with BCOR and BCORL1 mutations (Figure 1A).
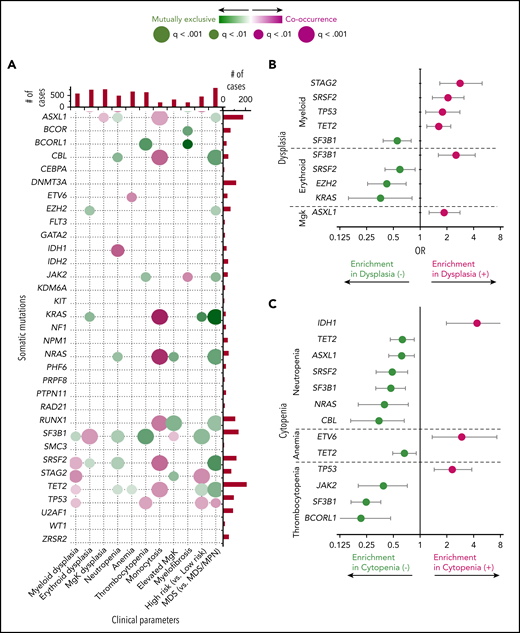
Associations between individual clinical features and somatic mutations. (A) Directions, magnitude, and strength of evidence of associations between pairs of selected morphologic features and genetic mutations are shown. Pairs shown have q values (P values corrected for multiple hypothesis testing) <0.1; larger dots represent stronger evidence. Color is used to depict the magnitude of pairwise odds ratios (OR): magenta represents cooccurring traits (positive association); green reflects mutually exclusive traits (negative association). (B-C) Estimated ORs and their confidence intervals for selected pairs of genetic mutations and dysplastic (B) or cytopenic (C) features. MgK, megakaryocyte.
Associations between individual clinical features and somatic mutations. (A) Directions, magnitude, and strength of evidence of associations between pairs of selected morphologic features and genetic mutations are shown. Pairs shown have q values (P values corrected for multiple hypothesis testing) <0.1; larger dots represent stronger evidence. Color is used to depict the magnitude of pairwise odds ratios (OR): magenta represents cooccurring traits (positive association); green reflects mutually exclusive traits (negative association). (B-C) Estimated ORs and their confidence intervals for selected pairs of genetic mutations and dysplastic (B) or cytopenic (C) features. MgK, megakaryocyte.
Significant associations between morphologic features and somatic mutations
| Clinical events | Mutated genes | OR | 95% CI | P | q | Cooccurrence | Clinical only | Mutation only | Intact | |
|---|---|---|---|---|---|---|---|---|---|---|
| Lower | Upper | |||||||||
| MDS (vs MDS/MPN) | SRSF2 | 0.20 | 0.14 | 0.30 | 5.57E-15 | 5.85E-13 | 55 | 774 | 65 | 185 |
| Thrombocytopenia | SF3B1 | 0.25 | 0.17 | 0.36 | 1.96E-13 | 1.85E-11 | 42 | 601 | 96 | 340 |
| MDS (vs MDS/MPN) | TET2 | 0.32 | 0.23 | 0.44 | 1.31E-11 | 1.03E-09 | 123 | 706 | 88 | 162 |
| Monocytosis | SRSF2 | 3.89 | 2.60 | 5.81 | 2.23E-10 | 1.41E-08 | 51 | 153 | 69 | 806 |
| Monocytosis | NRAS | 6.74 | 3.77 | 12.23 | 3.11E-10 | 1.84E-08 | 29 | 175 | 21 | 854 |
| MDS (vs MDS/MPN) | KRAS | 0.07 | 0.02 | 0.19 | 6.64E-09 | 3.31E-07 | 5 | 824 | 19 | 231 |
| Monocytosis | TET2 | 2.72 | 1.93 | 3.82 | 2.41E-08 | 1.09E-06 | 70 | 134 | 141 | 734 |
| Monocytosis | RUNX1 | 3.43 | 2.25 | 5.20 | 4.07E-08 | 1.68E-06 | 44 | 160 | 65 | 810 |
| High risk (vs low risk) | SF3B1 | 0.36 | 0.24 | 0.54 | 5.02E-07 | 1.64E-05 | 32 | 427 | 106 | 514 |
| Monocytosis | KRAS | 7.64 | 3.35 | 18.43 | 2.33E-06 | 6.88E-05 | 15 | 189 | 9 | 866 |
| MDS (vs MDS/MPN) | NRAS | 0.26 | 0.14 | 0.46 | 6.52E-06 | 0.000176 | 24 | 805 | 26 | 224 |
| Monocytosis | CBL | 4.11 | 2.25 | 7.45 | 7.49E-06 | 0.000197 | 22 | 182 | 25 | 850 |
| MDS (vs MDS/MPN) | CBL | 0.25 | 0.13 | 0.44 | 7.72E-06 | 0.000198 | 22 | 807 | 25 | 225 |
| Monocytosis | ASXL1 | 2.31 | 1.60 | 3.31 | 1.44E-05 | 0.000336 | 56 | 148 | 123 | 752 |
| MDS (vs MDS/MPN) | RUNX1 | 0.40 | 0.26 | 0.60 | 2.18E-05 | 0.000492 | 65 | 764 | 44 | 206 |
| Elevated MgK | RUNX1 | 0.33 | 0.18 | 0.56 | 2.56E-05 | 0.000564 | 15 | 316 | 94 | 654 |
| High risk (vs low risk) | STAG2 | 2.84 | 1.75 | 4.75 | 2.64E-05 | 0.000568 | 49 | 410 | 25 | 595 |
| Erythroid dysplasia | SF3B1 | 2.53 | 1.60 | 4.17 | 4.41E-05 | 0.000888 | 116 | 636 | 22 | 305 |
| Thrombocytopenia | BCORL1 | 0.22 | 0.09 | 0.47 | 7.27E-05 | 0.001323 | 8 | 635 | 24 | 412 |
| Neutropenia | SF3B1 | 0.47 | 0.32 | 0.68 | 7.91E-05 | 0.001412 | 42 | 454 | 96 | 487 |
| Myeloid dysplasia | STAG2 | 2.81 | 1.66 | 4.98 | 9.41E-05 | 0.00159 | 56 | 528 | 18 | 477 |
| Neutropenia | IDH1 | 4.37 | 1.97 | 11.03 | 0.000223 | 0.003351 | 25 | 471 | 7 | 576 |
| High risk (vs low risk) | TET2 | 0.55 | 0.40 | 0.75 | 0.000252 | 0.003722 | 66 | 393 | 145 | 475 |
| High risk (vs low risk) | TP53 | 2.22 | 1.45 | 3.42 | 0.000317 | 0.004611 | 58 | 401 | 38 | 582 |
| Myeloid dysplasia | SRSF2 | 2.05 | 1.37 | 3.11 | 0.000445 | 0.006184 | 83 | 501 | 37 | 458 |
| Neutropenia | SRSF2 | 0.49 | 0.32 | 0.72 | 0.000438 | 0.006184 | 37 | 459 | 83 | 500 |
| Thrombocytopenia | TP53 | 2.30 | 1.44 | 3.81 | 0.000467 | 0.006396 | 73 | 570 | 23 | 413 |
| Erythroid dysplasia | EZH2 | 0.42 | 0.26 | 0.70 | 0.000865 | 0.010233 | 34 | 718 | 33 | 294 |
| MDS (vs MDS/MPN) | ASXL1 | 0.55 | 0.39 | 0.78 | 0.000955 | 0.011153 | 120 | 709 | 59 | 191 |
| Myelofibrosis | JAK2 | 2.97 | 1.55 | 5.54 | 0.001163 | 0.013097 | 17 | 187 | 26 | 849 |
| Myeloid dysplasia | SF3B1 | 0.55 | 0.38 | 0.79 | 0.00134 | 0.014735 | 57 | 527 | 81 | 414 |
| Neutropenia | CBL | 0.34 | 0.17 | 0.66 | 0.001486 | 0.016163 | 11 | 485 | 36 | 547 |
| Elevated MgK | STAG2 | 0.37 | 0.18 | 0.69 | 0.001623 | 0.017445 | 11 | 320 | 63 | 685 |
| MgK dysplasia | ASXL1 | 1.86 | 1.26 | 2.81 | 0.001983 | 0.02084 | 145 | 627 | 34 | 273 |
| Myelofibrosis | BCORL1 | 0.06 | Infinity | 0.49 | 0.002015 | 0.020943 | 0 | 204 | 32 | 843 |
| Elevated MgK | NRAS | 0.30 | 0.11 | 0.65 | 0.002546 | 0.024574 | 6 | 325 | 44 | 704 |
| MDS (vs MDS/MPN) | JAK2 | 0.36 | 0.20 | 0.68 | 0.002488 | 0.024574 | 24 | 805 | 19 | 231 |
| Myeloid dysplasia | TET2 | 1.62 | 1.19 | 2.21 | 0.002619 | 0.025022 | 134 | 450 | 77 | 418 |
| High risk (vs low risk) | KRAS | 0.19 | 0.04 | 0.55 | 0.002666 | 0.025219 | 3 | 456 | 21 | 599 |
| Elevated MgK | SF3B1 | 1.77 | 1.23 | 2.55 | 0.002897 | 0.02687 | 58 | 273 | 80 | 668 |
| Myelofibrosis | BCOR | 0.26 | 0.08 | 0.63 | 0.003344 | 0.029642 | 4 | 200 | 63 | 812 |
| MDS (vs MDS/MPN) | TP53 | 2.48 | 1.36 | 5.00 | 0.003384 | 0.029642 | 85 | 744 | 11 | 239 |
| Neutropenia | TET2 | 0.63 | 0.46 | 0.86 | 0.003448 | 0.029922 | 78 | 418 | 133 | 450 |
| Neutropenia | NRAS | 0.40 | 0.20 | 0.74 | 0.003555 | 0.030576 | 13 | 483 | 37 | 546 |
| Thrombocytopenia | JAK2 | 0.39 | 0.20 | 0.72 | 0.003784 | 0.031771 | 16 | 627 | 27 | 409 |
| Neutropenia | ASXL1 | 0.62 | 0.44 | 0.86 | 0.00516 | 0.042569 | 65 | 431 | 114 | 469 |
| MDS (vs MDS/MPN) | EZH2 | 0.48 | 0.29 | 0.81 | 0.006814 | 0.05327 | 42 | 787 | 25 | 225 |
| Anemia | ETV6 | 2.94 | 1.37 | 7.30 | 0.007257 | 0.055363 | 33 | 640 | 7 | 399 |
| Anemia | TET2 | 0.67 | 0.49 | 0.90 | 0.009031 | 0.067803 | 115 | 558 | 96 | 310 |
| Myeloid dysplasia | TP53 | 1.78 | 1.15 | 2.80 | 0.010013 | 0.073428 | 64 | 520 | 32 | 463 |
| Erythroid dysplasia | SRSF2 | 0.59 | 0.40 | 0.88 | 0.011132 | 0.079778 | 71 | 681 | 49 | 278 |
| Erythroid dysplasia | KRAS | 0.36 | 0.16 | 0.81 | 0.01355 | 0.094253 | 11 | 741 | 13 | 314 |
| Clinical events | Mutated genes | OR | 95% CI | P | q | Cooccurrence | Clinical only | Mutation only | Intact | |
|---|---|---|---|---|---|---|---|---|---|---|
| Lower | Upper | |||||||||
| MDS (vs MDS/MPN) | SRSF2 | 0.20 | 0.14 | 0.30 | 5.57E-15 | 5.85E-13 | 55 | 774 | 65 | 185 |
| Thrombocytopenia | SF3B1 | 0.25 | 0.17 | 0.36 | 1.96E-13 | 1.85E-11 | 42 | 601 | 96 | 340 |
| MDS (vs MDS/MPN) | TET2 | 0.32 | 0.23 | 0.44 | 1.31E-11 | 1.03E-09 | 123 | 706 | 88 | 162 |
| Monocytosis | SRSF2 | 3.89 | 2.60 | 5.81 | 2.23E-10 | 1.41E-08 | 51 | 153 | 69 | 806 |
| Monocytosis | NRAS | 6.74 | 3.77 | 12.23 | 3.11E-10 | 1.84E-08 | 29 | 175 | 21 | 854 |
| MDS (vs MDS/MPN) | KRAS | 0.07 | 0.02 | 0.19 | 6.64E-09 | 3.31E-07 | 5 | 824 | 19 | 231 |
| Monocytosis | TET2 | 2.72 | 1.93 | 3.82 | 2.41E-08 | 1.09E-06 | 70 | 134 | 141 | 734 |
| Monocytosis | RUNX1 | 3.43 | 2.25 | 5.20 | 4.07E-08 | 1.68E-06 | 44 | 160 | 65 | 810 |
| High risk (vs low risk) | SF3B1 | 0.36 | 0.24 | 0.54 | 5.02E-07 | 1.64E-05 | 32 | 427 | 106 | 514 |
| Monocytosis | KRAS | 7.64 | 3.35 | 18.43 | 2.33E-06 | 6.88E-05 | 15 | 189 | 9 | 866 |
| MDS (vs MDS/MPN) | NRAS | 0.26 | 0.14 | 0.46 | 6.52E-06 | 0.000176 | 24 | 805 | 26 | 224 |
| Monocytosis | CBL | 4.11 | 2.25 | 7.45 | 7.49E-06 | 0.000197 | 22 | 182 | 25 | 850 |
| MDS (vs MDS/MPN) | CBL | 0.25 | 0.13 | 0.44 | 7.72E-06 | 0.000198 | 22 | 807 | 25 | 225 |
| Monocytosis | ASXL1 | 2.31 | 1.60 | 3.31 | 1.44E-05 | 0.000336 | 56 | 148 | 123 | 752 |
| MDS (vs MDS/MPN) | RUNX1 | 0.40 | 0.26 | 0.60 | 2.18E-05 | 0.000492 | 65 | 764 | 44 | 206 |
| Elevated MgK | RUNX1 | 0.33 | 0.18 | 0.56 | 2.56E-05 | 0.000564 | 15 | 316 | 94 | 654 |
| High risk (vs low risk) | STAG2 | 2.84 | 1.75 | 4.75 | 2.64E-05 | 0.000568 | 49 | 410 | 25 | 595 |
| Erythroid dysplasia | SF3B1 | 2.53 | 1.60 | 4.17 | 4.41E-05 | 0.000888 | 116 | 636 | 22 | 305 |
| Thrombocytopenia | BCORL1 | 0.22 | 0.09 | 0.47 | 7.27E-05 | 0.001323 | 8 | 635 | 24 | 412 |
| Neutropenia | SF3B1 | 0.47 | 0.32 | 0.68 | 7.91E-05 | 0.001412 | 42 | 454 | 96 | 487 |
| Myeloid dysplasia | STAG2 | 2.81 | 1.66 | 4.98 | 9.41E-05 | 0.00159 | 56 | 528 | 18 | 477 |
| Neutropenia | IDH1 | 4.37 | 1.97 | 11.03 | 0.000223 | 0.003351 | 25 | 471 | 7 | 576 |
| High risk (vs low risk) | TET2 | 0.55 | 0.40 | 0.75 | 0.000252 | 0.003722 | 66 | 393 | 145 | 475 |
| High risk (vs low risk) | TP53 | 2.22 | 1.45 | 3.42 | 0.000317 | 0.004611 | 58 | 401 | 38 | 582 |
| Myeloid dysplasia | SRSF2 | 2.05 | 1.37 | 3.11 | 0.000445 | 0.006184 | 83 | 501 | 37 | 458 |
| Neutropenia | SRSF2 | 0.49 | 0.32 | 0.72 | 0.000438 | 0.006184 | 37 | 459 | 83 | 500 |
| Thrombocytopenia | TP53 | 2.30 | 1.44 | 3.81 | 0.000467 | 0.006396 | 73 | 570 | 23 | 413 |
| Erythroid dysplasia | EZH2 | 0.42 | 0.26 | 0.70 | 0.000865 | 0.010233 | 34 | 718 | 33 | 294 |
| MDS (vs MDS/MPN) | ASXL1 | 0.55 | 0.39 | 0.78 | 0.000955 | 0.011153 | 120 | 709 | 59 | 191 |
| Myelofibrosis | JAK2 | 2.97 | 1.55 | 5.54 | 0.001163 | 0.013097 | 17 | 187 | 26 | 849 |
| Myeloid dysplasia | SF3B1 | 0.55 | 0.38 | 0.79 | 0.00134 | 0.014735 | 57 | 527 | 81 | 414 |
| Neutropenia | CBL | 0.34 | 0.17 | 0.66 | 0.001486 | 0.016163 | 11 | 485 | 36 | 547 |
| Elevated MgK | STAG2 | 0.37 | 0.18 | 0.69 | 0.001623 | 0.017445 | 11 | 320 | 63 | 685 |
| MgK dysplasia | ASXL1 | 1.86 | 1.26 | 2.81 | 0.001983 | 0.02084 | 145 | 627 | 34 | 273 |
| Myelofibrosis | BCORL1 | 0.06 | Infinity | 0.49 | 0.002015 | 0.020943 | 0 | 204 | 32 | 843 |
| Elevated MgK | NRAS | 0.30 | 0.11 | 0.65 | 0.002546 | 0.024574 | 6 | 325 | 44 | 704 |
| MDS (vs MDS/MPN) | JAK2 | 0.36 | 0.20 | 0.68 | 0.002488 | 0.024574 | 24 | 805 | 19 | 231 |
| Myeloid dysplasia | TET2 | 1.62 | 1.19 | 2.21 | 0.002619 | 0.025022 | 134 | 450 | 77 | 418 |
| High risk (vs low risk) | KRAS | 0.19 | 0.04 | 0.55 | 0.002666 | 0.025219 | 3 | 456 | 21 | 599 |
| Elevated MgK | SF3B1 | 1.77 | 1.23 | 2.55 | 0.002897 | 0.02687 | 58 | 273 | 80 | 668 |
| Myelofibrosis | BCOR | 0.26 | 0.08 | 0.63 | 0.003344 | 0.029642 | 4 | 200 | 63 | 812 |
| MDS (vs MDS/MPN) | TP53 | 2.48 | 1.36 | 5.00 | 0.003384 | 0.029642 | 85 | 744 | 11 | 239 |
| Neutropenia | TET2 | 0.63 | 0.46 | 0.86 | 0.003448 | 0.029922 | 78 | 418 | 133 | 450 |
| Neutropenia | NRAS | 0.40 | 0.20 | 0.74 | 0.003555 | 0.030576 | 13 | 483 | 37 | 546 |
| Thrombocytopenia | JAK2 | 0.39 | 0.20 | 0.72 | 0.003784 | 0.031771 | 16 | 627 | 27 | 409 |
| Neutropenia | ASXL1 | 0.62 | 0.44 | 0.86 | 0.00516 | 0.042569 | 65 | 431 | 114 | 469 |
| MDS (vs MDS/MPN) | EZH2 | 0.48 | 0.29 | 0.81 | 0.006814 | 0.05327 | 42 | 787 | 25 | 225 |
| Anemia | ETV6 | 2.94 | 1.37 | 7.30 | 0.007257 | 0.055363 | 33 | 640 | 7 | 399 |
| Anemia | TET2 | 0.67 | 0.49 | 0.90 | 0.009031 | 0.067803 | 115 | 558 | 96 | 310 |
| Myeloid dysplasia | TP53 | 1.78 | 1.15 | 2.80 | 0.010013 | 0.073428 | 64 | 520 | 32 | 463 |
| Erythroid dysplasia | SRSF2 | 0.59 | 0.40 | 0.88 | 0.011132 | 0.079778 | 71 | 681 | 49 | 278 |
| Erythroid dysplasia | KRAS | 0.36 | 0.16 | 0.81 | 0.01355 | 0.094253 | 11 | 741 | 13 | 314 |
CI, confidence interval; MgK, megakaryocyte; OR, odds ratio.
Morphologic profiling
Univariate hypothesis testing identified significant pairwise associations among several morphologic and mutation features, warranting further interrogation of integrative subtypes. The morphologic characteristics evaluated tend to contribute redundant information describing the intersections among dysplastic features, cytopenias and monocytosis. Using more than 20 morphologic variables, unsupervised analysis based on the consensus clustering method demonstrated that these features describe only 5 distinct morphologic profiles (Figure 2A). Almost all the patients with HR subtypes clustered into profile 1 (P1; n = 283, 34%), whereas the other 4 profiles, mostly LR subtypes, each demonstrated unique morphologic (Figure 2B). Patients in P2 (n = 138; 17%) had trilineage dysplasia and pancytopenia; patients in P3 (n = 218; 17%) had trilineage dysplasia, 2-lineage cytopenia, and monocytosis; patients in P4 (n = 130; 16%) had 2-lineage dysplasia, 1-lineage cytopenia (anemia), and elevated megakaryocytes; and patients in P5 (n = 66; 8%) had erythroid dysplasia occasionally arising with anemia. Patients with P5 had better overall survival than those with P2, P3, and P4 (Figure 2C).

Morphologic profiles. (A) Consensus clustering applied to the discovery cohort reveals 5 morphologic profiles. (B) Distributions of each morphologic feature among the 5 morphologic profiles (P). Color is used to describe the prevalence of individual traits within each of the 5 profiles, with red depicting high and blue depicting low prevalence, respectively. (C) Kaplan-Meier curves for overall survival among patients identified by the 5 morphologic profiles. Survival differs significantly among the 5 profiles when evaluated using the log-rank test (*P < .05), demonstrating that the morphologic profiles confer prognostic utility. A, anemia; E, erythoroid dysplasia; Emg, elevated megakaryocytes; F, fibrosis; M, myeloid dysplasia; Mef, myelofibrosis; MgK, megakaryocytic dysplasia; Mnc, monocytosis; N, neutropenia; P, morphologic profiles; Th, thrombocytopenia.
Morphologic profiles. (A) Consensus clustering applied to the discovery cohort reveals 5 morphologic profiles. (B) Distributions of each morphologic feature among the 5 morphologic profiles (P). Color is used to describe the prevalence of individual traits within each of the 5 profiles, with red depicting high and blue depicting low prevalence, respectively. (C) Kaplan-Meier curves for overall survival among patients identified by the 5 morphologic profiles. Survival differs significantly among the 5 profiles when evaluated using the log-rank test (*P < .05), demonstrating that the morphologic profiles confer prognostic utility. A, anemia; E, erythoroid dysplasia; Emg, elevated megakaryocytes; F, fibrosis; M, myeloid dysplasia; Mef, myelofibrosis; MgK, megakaryocytic dysplasia; Mnc, monocytosis; N, neutropenia; P, morphologic profiles; Th, thrombocytopenia.
Genetic signatures
Patterns of cooccurrence among mutations and morphologic subtypes, were interrogated and then subsequently evaluated for association with patient outcomes. High-risk MDS patients mainly exhibited a common morphologic profile (P1). Thus, machine learning was used to interrogate prognostic signatures for survival among mutations observed in the HR cohort. Analyses revealed that patients classified clinically as high risk exhibited 1 of 6 genetic subtypes. By way of contrast, the morphologic characteristics of patients classified clinically as low risk varied by 4 distinct profiles P2-P5, which comprised N = 552 patients (85% were LR).
To elucidate patterns of cooccurrence among the morphologies and genetics of LR patients, Bayesian machine-learning techniques21-23 were applied (supplemental Figure 9). The models identified 8 genetic signatures: LR signature A (LR-SA) through signature H (LR-SH) (Figure 3A). For instance, LR-SA was enriched for TET2 mutations, LR-SB for TET2 and SRSF2 mutations, and LR-SG for SF3B1 mutations (Figure 3B). Focusing on patients with TET2 mutations, they were separated into different groups based on other accompanying mutations (LR-SB; SRSF2-mutated, LR-SD; JAK2-mutated, LR-SA; neither SRSF2- nor JAK2-mutated as well as corresponding morphologic profiles (P3, P4, and P2, respectively). In contrast, LR-SC was characterized by more heterogeneous mutational profiles compared with LR-SB and LR-SG (Figure 3C). These genetic signatures were also associated with differences in prognosis (eg, patient with LR-SA had better overall survival than those with LR-SC; P = .0011, Figure 3D). We then examined the linkage between LR genetic signatures (LR SA-SH) linked to morphologic profiles (P2-P5).
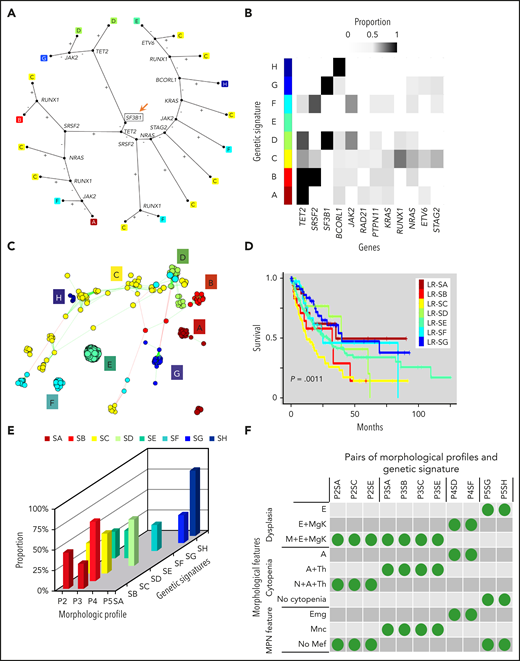
Genetic signatures in LR MDS demonstrate morphologic orientation. (A) Decision tree defining a genetic signature with 8 subtypes for LR MDS patients (LR-SA through LR-SH). The decision process initiates with SF3B1 (depicted with a square and orange arrow). Incidences of individual mutations are evaluated in a sequence to assign patients to the 8 subtypes. Plus and minus signs depict mutated and wild type, respectively. (B) Distributions of mutations within each genetic signature. Genes used in the decision tree are shown. (C) Network representation of the signature. Nodes define patients, color is used to describe their subtypes, and edges are drawn between neighboring patients with commensurate mutational patterns as defined by the statistical model. (D) Kaplan-Meier curves compare overall survival among patients assigned to the 8 genetic signatures; the P value is from the log-rank test. (E) Distribution of morphologic profiles among the genetic signatures. Frequent profiles (>30%) in each genetic signature are depicted. (F) The presence of specific morphologic features among morphologic profiles and genetic signatures. Morphologic parameters are ordered as rows; frequent pairs of morphologic profiles and genetic signature are displayed as columns.
Genetic signatures in LR MDS demonstrate morphologic orientation. (A) Decision tree defining a genetic signature with 8 subtypes for LR MDS patients (LR-SA through LR-SH). The decision process initiates with SF3B1 (depicted with a square and orange arrow). Incidences of individual mutations are evaluated in a sequence to assign patients to the 8 subtypes. Plus and minus signs depict mutated and wild type, respectively. (B) Distributions of mutations within each genetic signature. Genes used in the decision tree are shown. (C) Network representation of the signature. Nodes define patients, color is used to describe their subtypes, and edges are drawn between neighboring patients with commensurate mutational patterns as defined by the statistical model. (D) Kaplan-Meier curves compare overall survival among patients assigned to the 8 genetic signatures; the P value is from the log-rank test. (E) Distribution of morphologic profiles among the genetic signatures. Frequent profiles (>30%) in each genetic signature are depicted. (F) The presence of specific morphologic features among morphologic profiles and genetic signatures. Morphologic parameters are ordered as rows; frequent pairs of morphologic profiles and genetic signature are displayed as columns.
In total, we identified 11 frequent signature/profile (SP) combinations (Figure 3E; supplemental Figure 10). For example, SA was enriched for P2 (profiles characterized pancytopenia), SB, and SC for P3 features, most prominently monocytosis, SD for P4, elevated megakaryocytes, and SG and SH for P5, erythroid dysplasia (Figure 3F). P2-SA patients had better overall survival than P3-SB patients (supplemental Figure 11).
Genetic subtypes of high-risk MDS were interrogated through association with survival, which further defined the prognostic heterogeneity of the HR population. As explained in supplemental Figure 9, different methodologies were used to obtain the N × N proximity measures which was entirely based on the object of supervision. Survival random forest was applied to interrogate genetic mutations for association with survival among the high-risk cohort, which exhibited relative homogeneity with respect morphology. HR genetic signatures HR-SA through HR-SF (Figure 4A-B) had distinct mutational compositions (supplemental Figure 12): HR-SB was enriched for DNMT3A mutations, HR-SC for TP53 mutations, and HR-SF for U2AF1 mutations. Patients with HR-SA, HR-SB and HR-SD had better survival than those with HR-SF (Figure 4C). Eleven frequent SP combinations were also identified in HR (Figure 4D): For instance, the P1 profile, uniformly containing HR patients, showed 6 HR signatures (HR-SA through HR-SF), but HR-SA, HR-SC, HR-SD and HR-SF were also present in the less numerous patients with P3, whereas HR-SB was also found among those with the P4 profile (Figure 4E).

Genetic signatures in HR MDS. (A) Consensus clustering applied to genetic mutations identify a signature with 6 subtypes (HR-SA through HR-SF) among HR patients in the discovery cohort. (B) Distribution of mutations within each genetic signature. Frequently mutate genes (in ≥5% of patients) are shown. (C) Kaplan-Meier curves compare overall survival among patients identified by 6 genetic subtypes; the P value is from the log-rank test. (D) Distribution of morphologic profiles in each genetic signature. Frequent profiles (in >15% of patients) in each genetic signature are depicted. (E) The presence of specific morphologic features across morphologic profiles and genetic signatures. Representative morphologic features are shown in rows; frequent pairs of morphologic profiles and genetic signature are displayed as columns.
Genetic signatures in HR MDS. (A) Consensus clustering applied to genetic mutations identify a signature with 6 subtypes (HR-SA through HR-SF) among HR patients in the discovery cohort. (B) Distribution of mutations within each genetic signature. Frequently mutate genes (in ≥5% of patients) are shown. (C) Kaplan-Meier curves compare overall survival among patients identified by 6 genetic subtypes; the P value is from the log-rank test. (D) Distribution of morphologic profiles in each genetic signature. Frequent profiles (in >15% of patients) in each genetic signature are depicted. (E) The presence of specific morphologic features across morphologic profiles and genetic signatures. Representative morphologic features are shown in rows; frequent pairs of morphologic profiles and genetic signature are displayed as columns.
Validation analysis
Validation analyses considered the robustness of more novel associations identified among genetic mutations and morphologic profiles. The k-nearest neighbor algorithm was used to assign validation patients to subtypes based on their genetic mutations. The dissimilarity metrics considered binary distance functions mapping the set of mutation presence/absence into a distance. For each validation patient, the k = 5 nearest patients from the training cohort (least distant) were selected. A subtype was assigned based on majority rule (supplemental Table 2), which recapitulated the 5 morphologic profiles. Six of the 11 morphologic profiles/genetic signature combinations identified by the discovery cohort demonstrated commensurate statistical associations with the validation cohort (Figure 5A; supplemental Figure 13): SA and SE associated with P2; SA, SB, and SC associated with P3; and SD associated with P4 (Figure 5B). Representative variables of signature profile (SP) pairs included TET2mut/SRSF2wt (SA) with trilineage dysplasia and pancytopenia (P2) and SF3B1mut/JAK2mut (SD) with erythroid and megakaryocytic dysplasia (P4) (Figure 5C).
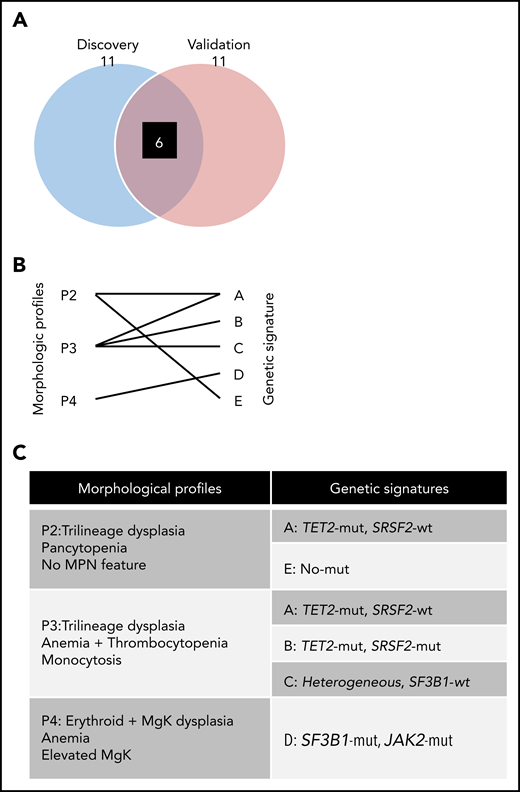
Validated morphologic profiles and genetic signatures. (A) Venn diagram depicts associations identified by LR patients for discovery and validation groups, respectively. After evaluating the significant pairs of morphologic profiles and genetic signatures in the discovery cohort for LR compared with those in the validation cohort for LR, 6 pairs (black box) were recapitulated in both. (B) Diagram depicting 6 validated morphologic and genetic associations. (C) Resultant validated associations between morphologic profiles and genetic signatures.
Validated morphologic profiles and genetic signatures. (A) Venn diagram depicts associations identified by LR patients for discovery and validation groups, respectively. After evaluating the significant pairs of morphologic profiles and genetic signatures in the discovery cohort for LR compared with those in the validation cohort for LR, 6 pairs (black box) were recapitulated in both. (B) Diagram depicting 6 validated morphologic and genetic associations. (C) Resultant validated associations between morphologic profiles and genetic signatures.
Discussion
Distinct morphologic features constitute the gold standard in the diagnosis of MDS. Although invariant pathognomonic morphology/genotype associations are not common, the few classical examples indicate that systematic and comprehensive analyses of morphologic and genomic features may reveal diagnostically and prognostically important relationships. Our study represents the first comprehensive analytic attempt to correlate individual morphologic features with the mutational profiles in MDSs. Our approach included a univariate analysis of binomial, mostly objective, features. Currently, the ubiquitously applied WHO classification is likely to be replaced by artificial intelligence– and machine learning–based analytics according to the image-recognition technologies, which have been already introduced in automated differential blood smear evaluation. We have then applied unsupervised clustering strategies to identify novel links between mutational signatures and morphologic profiles, that is, SP.
In addition to confirming previously known genotype/morphology/prognosis associations (eg, TP53mut with thrombocytopenia and higher blast counts30 and JAK2mut with myelofibrosis,31 SRSF2mut with granulopoietic hyperplasia, monocytosis and predictors for worsened overall survival,32,33 and U2AF1 mut with higher blast counts and higher hazard ratio14 ), new SP included STAG2mut and SRSF2mut with myeloid dysplasia and ASXL1mut with megakaryocytic dysplasia. Furthermore, anemia or thrombocytopenia were associated with ETV6 or TP53 mutations, respectively, concordant with patients with germline ETV6 mutations showing pathological abnormalities and cytopenia.34
We hypothesized that the combinatorial complexity of overlapping MDS morphologic and blood count features was not random, that is, that genetic defect combinations shape them. Patients’ similarities in morphologic variables were thus used to classified patients into groups with distinct profiles: P1 and P2 differed only in P1 being HR (of becoming leukemia) and P2 being LR, otherwise P1 and P2 had similar morphologic features of trilineage dysplasia and trilineage pancytopenia; P3 was enriched in patients with monocytosis; P4 had elevated megakaryocyte counts, and P5 had single-lineage erythroid dysplasia and some anemia. These groups had significant survival differences. They thus have biological relevance. LR patients were prevalent in P2-P5 and patients with P5 had a better prognosis than those in P2-P4. These results suggest that morphologic profiles with dichotomized elements can be useful for classifying MDS patients.
We then clustered patients based on mutational signatures (S) with elements dichotomized as wild type or mutant in a targeted gene. HR patients (defined by higher blast content) differed greatly from LR patients and were thus analyzed separately. Within LR patients, 8 genetic signatures (LR-SA to LR-SH) were identified using Bayesian graphical models with 14 defining genes in the decision tree. In HR MDS patients, 6 genetic signatures were revealed, including both previously known TP53 mutations14,35,36 in HR-SC, and novel associations, for example, in HR-SB DNMT3Amut and in HR-SF U2AF1mut. The signatures yielded different survival times within both the LR and HR groups, the former being particularly important, as such separations have previously been hard to define.
In a final step, we combined genetic signatures (S) and morphologic profiles (P) into SP links. In total, 11 frequent SP pairs were identified. These included LR-SB enriched in P3, LR-SD in P4 and LR-SH in P5. Elaborating upon only the first of these: P3 was characterized by monocytosis and LR-SB by both TET2- and SRSF2- mutations reflective of MDS/MPN classification.33 Some of the links were clinically prognostic: patients with LR-SA and P2 had better prognoses than those with LR-SB and P3.
Previous reports tended to use mutations as single variables. Our mutation signature-based classification strategy is appealing in that it is more reflective of the multihit molecular pathogenesis of MDS. It thus has greater potential as a tool for furthering MDS understanding. Using other cooccurring mutations enabled LR MDS patients with the specific genetic mutations to be divided into different groups. For example, patients with SRSF2 were divided 2 genetic signatures (LR-SB; TET2-mutated, LR-SF; JAK2-mutated) and they had unique corresponding morphologic profiles (P3 and P4, respectively), those with TET2 mutations were separated into different 3 groups as well. These results suggested this statistical approach, on its own, reflected the biology of cooperating and mutually exclusive mutations.
To focus on the most robust associations, subsequent analyses were applied to an independent validation cohort. Of 11 SP links identified in our discovery group, 6 were validated in a smaller group of patients suggesting overall reproducibility. The SP of LR-SD (SF3B1mut and JAK2 mutants) and P4 (erythroid and megakaryocytic dysplasia, anemia, and elevated megakaryocyte counts) was previously suggested for RARS-T.8 New here are 2 SP links of TET2-mutant and SRSF2-wild type (LR-SA), or wild type for recurrent mutations (LR-SE), to P2 (containing 2 different genetic signatures, ie, LR MDS patients with trilineage dysplasia pancytopenia without MPN features). Two other validated SP links are TET2mut and SRSF2mut (LR-SB) or RUNX1mut (LR-SC) with P3 (monocytosis).
Mutations in <10% of tumor cells (variant allele frequency <5% in copy number neutral regions) were removed from our analysis to raise the stringency of our genetic signatures to levels in morphology profiles where at least 2 of 20 marrow cells must be dysplastic to classify a lineage as dysplastic. This eliminated only 46 of 1975 mutations, that is, 1929 mutations (98%) were used. A limitation potentially more concerning is that our targeted panel lacks DDX41, SETBP1, CALR, and PPM1D. CALR mutations are enriched in MDS/MPN features,37 SETBP1 mutations associate with HR MDS and increasing blast counts,11 and germline mutations in DDX41 associate with hematopoietic phenotypes.38 Their inclusion could thus have revealed additional associations un-identified in our study. Orderings of successive hits could also be accounted for in future studies.
In sum, our study demonstrates that despite of the tremendous morphologic diversity of MDSs, nonrandom or even pathognomonic relationships between the MDS phenotype and genotype can be identified. Such relationships include mutual exclusivity certain invariant features and molecular lesions or a strong association specific mutational patterns and profiles of morphologic features. Although this analysis was conducted using classical morphologic classification criteria, we also envision future studies using unbiased image recognition tools for morphologic classification. In the future, operator-independent, automated, and fully objective methods assessed by image recognition by computerized image recognition technologies will replace subjectively biased, labor-intensive, and not precisely reproducible human assessment of dysplasia or blast, megakaryocyte, and other quantitative parameters. Ultimately, patients with uniquely distinctive morphologic profiles could supplant molecular testing and that will produce classifications that better reflect underlying true biological subgroupings of these MDS disease entities.
Genome data that support the findings of this study have been deposited in the National Center for Biotechnology Information (NCBI) Genotypes and Phenotypes (dbGaP) database (accession number phs001898.v1.p1). All other remaining data are available within the article and supplemental files or are available from the authors upon request.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by US National Institutes of Health National Heart, Lung, and Blood Institute grants R35 HL135795, R01HL123904, R01 HL118281, R01 HL128425, and R01 HL132071 (J.P.M.); the Edward P. Evans Foundation (J.P.M.); and a Japan Society for the Promotion of Science (JSPS) Overseas Research Fellow grant and JSPS KAKENHI grant JP 20K17412 (Y.N.).
Authorship
Contribution: Y.N., R.Z., H.A., and T.R. performed experiments of molecular study and data analysis; Y.N., R.Z., C.M.K., I.M., H.M., J.G.S., and B.P.H. were committed to bioinformatics analysis of sequencing data; S.K., A.N., and M.A.S. collected specimens and were involved in planning the project; Y.N., R.Z., B.P.H., and J.P.M. generated figures and tables, and wrote the manuscript; Y.N., B.P.H., and J.P.M. led the entire project; and all authors participated in discussions and interpretation of the data and results.
Conflict-of-interest disclosure: B.P.H. reports research funds from Amgen and is a scientific advisor for Presagia. The remaining authors declare no competing financial interests.
Correspondence: Jaroslaw P. Maciejewski, Taussig Cancer Institute, NE6-250, Cleveland Clinic, 2111 E. 96th St, Cleveland, OH 44195; e-mail: maciejj@ccf.org; Brian P. Hobbs, Taussig Cancer Institute and Lerner Research Institute, Cleveland Clinic, CA-60, 9500 Euclid Ave, Cleveland, OH 44195; e-mail: bphobbs@gmail.com; or Yasunobu Nagata, Department of Hematology, Nippon Medical School, 1-1-5 Sendagi, Bunkyo-Ku, Tokyo, 113-8603, Japan; e-mail: ysnagata-tky@umin.ac.jp.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal