

Key Points
The homeostatic bone marrow localization of femoral and sternal HSCs reflects the abundance of microenvironmental cell types.
Juvenile HSCs show preferential association with Cxcl12 stroma cells.

Abstract
The exact localization of hematopoietic stem cells (HSCs) in their native bone marrow (BM) microenvironment remains controversial, because multiple cell types have been reported to physically associate with HSCs. In this study, we comprehensively quantified HSC localization with up to 4 simultaneous (9 total) BM components in 152 full-bone sections from different bone types and 3 HSC reporter lines. We found adult femoral α-catulin-GFP+ or Mds1GFP/+Flt3Cre HSCs proximal to sinusoids, Cxcl12 stroma, megakaryocytes, and different combinations of those populations, but not proximal to bone, adipocyte, periarteriolar, or Schwann cells. Despite microanatomical differences in femurs and sterna, their adult α-catulin-GFP+ HSCs had similar distributions. Importantly, their microenvironmental localizations were not different from those of random dots, reflecting the relative abundance of imaged BM populations rather than active enrichment. Despite their functional heterogeneity, dormant label-retaining (LR) and non-LR hematopoietic stem and progenitor cells both had indistinguishable localization from α-catulin-GFP+ HSCs. In contrast, cycling juvenile BM HSCs preferentially located close to Cxcl12 stroma and farther from sinusoids/megakaryocytes. We expect our study to help resolve existing confusion regarding the exact localization of different HSC types, their physical association with described BM populations, and their tissue-wide combinations.
Introduction
The BM microenvironment is important in controlling hematopoietic stem cell (HSC) fates throughout life.1-3 Bone marrow (BM) HSCs reside in specialized anatomical microenvironments.4 To date, HSCs have been reported to physically associate with a plethora of BM components.3,5,6 Early studies identified the association of HSCs with bone-lining osteoblasts after transplantation,7-9 whereas homeostatic HSCs have been reported close to nonmyelinated Schwann cells,10 nestin11 and Cxcl12-expressing stroma,12,13 megakaryocytes (MKs),14-16 BM vasculature,17 and specific vessels and perivascular cells (arterioles/periarteriolar18 or sinusoids/perisinusoidal13 ). These data mainly come from studies measuring HSC distances from only a limited number of single BM populations and often in different bone types, resulting in contradictory findings. Thus, the cellular composition of the BM microenvironment surrounding HSCs remains disputed.
The existence of distinct niches supporting different HSC subtypes also continues to be debated. Arteriolar and NG2+ periarteriolar cells were described as preferential niches for quiescent Ki67-HSCs,18 whereas another study reported no difference in localization of quiescent and cycling HSCs relative to arterioles, sinusoids, or transition zone vessels.13 Although most HSCs are quiescent at individual time points, only a few show long-term quiescence (dormancy).19 Those rare HSCs contain the highest regeneration potential after transplantation, and can be identified by long-term retention of fluorescent biomarkers (label-retaining; LR), whereas their dividing counterparts gradually dilute and lose fluorescence and repopulation potential (non-LR).19,20 A limited number of mouse models enables endogenous labeling of hematopoietic stem and progenitor cells (HSPCs).19,21 Visualizing genetically labeled LR and non-LR HSPCs in situ has been challenging, mainly because of their low frequency and need for quantitative BM imaging for their accurate classification.
Cell-cycle properties of HSCs are age dependent. BM HSCs retain fetal liver characteristics (active cycling) for the first 3 weeks after birth.22 A few days later, they switch to an adult phenotype showing the divisional properties and repopulation kinetics of adult HSCs. Whether pre- and postswitch HSCs occupy distinct niches is currently unclear.
To resolve existing uncertainties regarding HSC localization and better quantify the complexity of their 3-dimensional (3D) microenvironment, we used novel quantitative multicolor imaging of thick full-bone sections,23 in different bone types and developmental stages. To maximize the number of simultaneously imaged BM populations, we used 3 different reporter mouse systems that allowed for efficient 2-marker detection of HSCs/HSPCs (c-Kit and α-catulin-GFP+,13 c-Kit and Mds1GFP/+Flt3Cre,24 and c-Kit and doxycycline [DOX]-treated SCL/tTA;H2B-GFP19 mice). We comprehensively quantified HSC localization in relation to up to 4 simultaneous (9 total) BM components and their combinations at the tissue-wide level with single-cell resolution.
Methods
Mice
Mice were bred under specific pathogen-free conditions, and all procedures were approved by the cantonal veterinary office of Basel-Stadt, Switzerland; German authorities; and the Institutional Animal Care and Use Committee of Massachusetts General Hospital. For HSC identification, we used α-catulinGFP/+ and Mds1GFP/+Flt3Cre knockin lines13,24 or double-transgenic SCL-tTA;H2B-GFP animals treated with DOX for 150 days, as previously described.19 To reliably identify dormant label-retaining HSPCs, we compared BM sections from age-matched controls and treated mice immunostained in parallel and imaged sequentially with the same microscope and same settings. Cxcl12tm2.1Sjm/J mice were used for detection of Cxcl12-producing cells.25
Bone sectioning and immunostaining
Bones were processed as previously described.23 Supplemental Table 1, available on the Blood Web site, lists all antibodies used in the study. Optically cleared (2,2-thiodiethanol; Sigma-Aldrich) sections were mounted using 100% 2,2-thiodiethanol with 0.1 M N-propyl gallate (pH 8.5; refractive index = 1.518; Sigma-Aldrich) in silicon spacers (Grace Biolabs) and 0.13- to 0.16-mm-thick coverslips (No. 1; Ted Pella).
Confocal microscopy
Confocal imaging was performed with a Leica TCS SP8 microscope equipped with 3 photomultiplier tubes, 2 HyD detectors, and 5 laser lines: 405-nm blue diode, argon (458, 476, 488, 496, and 514 nm), and 3 helium neon (543, 594, and 633 nm). All imaging was performed with type F immersion liquid (Leica) matching the refractive index of our optically cleared sections to avoid mismatches, and a 20× multi-immersion objective (numerical aperture, 0.75; free working distance, 0.680 mm) at 400-Hz, bidirectional mode with 2.49 z-spacing. Eight-bit images were acquired with 0.85 zoom at 1024 × 1024 resolution. Optimal settings (laser power, detector bandwidth, gain, and offset) were based on fluorescence minus one (FMO) controls.
Analysis
Image analysis was performed with Imaris ver. 8.3.1, 9.1.2, or 9.5.1 (Bitplane) using the MeasurementPro and ImarisXT extensions. Distance transformations were performed in 16-bit transiently converted data to avoid truncation. To identify individual HSCs (cKit+α-catulin-GFP+), we first segmented cKit+ cells with the Imaris surface function (split touching objects), to distinguish cKit+ hematopoietic cells from BM endothelium (α-catulin-GFP+cKit−). cKit+ cells were separated based on GFP expression and filtered for cKit+ α-catulin-GFP+ objects (mean fluorescence intensity [MFI] of GFP above background based on FMO controls). Finally, we visually inspected all gated objects to confirm their immunophenotype (α-catulin-GFP+cKit+ double positive), size, morphology (small, round cells), and correct signal localization (homogenous GFP signal throughout the cell and membrane cKit staining). Only cKit+GFP+ objects/cells fulfilling all 4 criteria were classified as HSCα-cats, HSCMFGs, or LR/non-LR HSPCs (examples of excluded objects in supplemental Figure 2B). BM populations were segmented using the Imaris surface function, and individual structures were curated based on morphological features. To confine the NG2 signal to pericytes associated with arteries and exclude it from osteoblasts,26 we computationally masked NG2 signal inside the bone isosurface (masked with collagen 1 [col.1]/osteopontin [opn]) and away from Sca1+ arteries/arterioles to appear black. Random dots (RDs) were generated as described.23 RDs located inside vessels or bone structures were removed by Imaris. Final BM volume was defined by the less deep fluorescent channel. A self-written script was developed to enable calculation of available combinations of BM locations in full-bone sections (detailed description in supplemental Material).
Statistical analysis
Results were analyzed with GraphPad Prism, using the Kolmogorov-Smirnov test for distributions and the unpaired, nonparametric 2-tailed Mann-Whitney U test to compare individual bins unless otherwise stated in the figure legends (including the number of replicates and P values).
Code availability
The open-source software used for random dot generation is available at https://www.bsse.ethz.ch/csd/software/XiT.html.
Results
Multicolor imaging allows for simultaneous visualization of HSCs and multiple BM niche components
Our multicolor deep-tissue–imaging pipeline allows for simultaneous visualization of HSCs and up to 4 BM components in full-bone sections of different bones (Figure 1A-B; supplemental Figure 1; Video 1). To efficiently identify HSCs in situ, we used α-catulinGFP/+ mice expressing GFP from the Ctnnal1 promoter active in HSCs and BM endothelial cells.13 Detecting homogenous GFP signal surrounded by cKit cell-surface expression in small cells with round morphology enabled 2-color HSC identification (HSCα-cat), with purity comparable to the more complex marker combinations13 (supplemental Figure 2A-C). Furthermore, we imaged the recently established HSC reporter mouse Mds1GFP/+Flt3Cre (MFG) in which GFP expression is restricted in primitive HSCs (HSCMFGs).24 Combining all those criteria (immunophenotype, signal localization, cell size, and morphology), we examined >3.5 × 106 cKit+ cells with detectable GFP signal and visually confirmed 4067 HSCα-cats and 1336 HSCMFGs in 115 full-bone sections (see “Methods” for more details).

Multicolor simultaneous full-bone quantitative imaging of HSCs and multiple niche populations. (A) Experimental flow and duration of large-volume, multicolor, quantitative confocal imaging. See description in “Analysis” in “Methods.” (B) Six-color immunostaining of 220- to 250-μm–thick, full-bone, longitudinal sections of mouse femur (top) and sternum (bottom) from an α-catulinGFP/+ reporter mouse. Sections stained for collagen.1/osteopontin-CF405S (trabecular and cortical bone), GFAP fab-Alexa-594 (nonmyelinated Schwann cells), laminin-CF633 (vasculature), and GP1bβ-CF680 (MKs). HSCα-cats were marked by GFP (amplified by biotin-streptavidin 488) and cKit-CF555 expression. CF, cyanine-based fluorescent dyes, SA, streptavidin. (C) Overview of niche components, respective marker combinations, and staining schemes. The number of sections imaged for each niche population, marker, bone type, and HSC reporter system are shown. (D-G) High-magnification images of multicolor staining schemes for niche components. (D) Femoral area (enlargement of red dashed square appearing in panel B) showing nonmyelinated Schwann cells (Sc, yellow, GFAP); cortical bone (white, Col.1/Opn); MKs (magenta, GP1bβ); and different vessel types marked by laminin: central sinus, sinusoids (S), arteries (A), arterioles (a) and transition zone vessels (TZ). (E) Trabecular and cortical bone (white, Col.1/Opn), adipocytes (yellow; boron dipyrromethene, BODIPY TRx), and BM vasculature (cyan, laminin). (F) Cortical bone (white, Col.1/Opn), sinusoids (cyan, CD105), and Cxcl12+ stroma (yellow). (G) Trabecular bone (white, Col.1/Opn), Sca1+ arteries (cyan), and NG2+ periarteriolar cells (yellow) after computationally masking all NG2+ objects outside Sca1+ arteriolar signal to appear black. Scale bars: 1000 μm (B), 100 μm (D), 50 μm (E, G), and 30 μm (F).
Multicolor simultaneous full-bone quantitative imaging of HSCs and multiple niche populations. (A) Experimental flow and duration of large-volume, multicolor, quantitative confocal imaging. See description in “Analysis” in “Methods.” (B) Six-color immunostaining of 220- to 250-μm–thick, full-bone, longitudinal sections of mouse femur (top) and sternum (bottom) from an α-catulinGFP/+ reporter mouse. Sections stained for collagen.1/osteopontin-CF405S (trabecular and cortical bone), GFAP fab-Alexa-594 (nonmyelinated Schwann cells), laminin-CF633 (vasculature), and GP1bβ-CF680 (MKs). HSCα-cats were marked by GFP (amplified by biotin-streptavidin 488) and cKit-CF555 expression. CF, cyanine-based fluorescent dyes, SA, streptavidin. (C) Overview of niche components, respective marker combinations, and staining schemes. The number of sections imaged for each niche population, marker, bone type, and HSC reporter system are shown. (D-G) High-magnification images of multicolor staining schemes for niche components. (D) Femoral area (enlargement of red dashed square appearing in panel B) showing nonmyelinated Schwann cells (Sc, yellow, GFAP); cortical bone (white, Col.1/Opn); MKs (magenta, GP1bβ); and different vessel types marked by laminin: central sinus, sinusoids (S), arteries (A), arterioles (a) and transition zone vessels (TZ). (E) Trabecular and cortical bone (white, Col.1/Opn), adipocytes (yellow; boron dipyrromethene, BODIPY TRx), and BM vasculature (cyan, laminin). (F) Cortical bone (white, Col.1/Opn), sinusoids (cyan, CD105), and Cxcl12+ stroma (yellow). (G) Trabecular bone (white, Col.1/Opn), Sca1+ arteries (cyan), and NG2+ periarteriolar cells (yellow) after computationally masking all NG2+ objects outside Sca1+ arteriolar signal to appear black. Scale bars: 1000 μm (B), 100 μm (D), 50 μm (E, G), and 30 μm (F).
We established multicolor stainings for 9 distinct BM components (Figure 1C; supplemental Table 1). We used antibody stainings and morphological features to identify cortical and trabecular osteoblasts and bone matrix (Col.1/Opn), MKs (glycoprotein Ibβ [GP1bβ]), the entire vasculature (laminin) and distinct vessel types (CD105 for sinusoidal and Sca1 for arteriolar cells), periarteriolar cells (neuronal/glial antigen 2 [NG2]), adipocytes (BODIPY), stromal cells (Cxcl12-DsRed mouse),25 and nonmyelinated Schwann cells (glial fibrillary acidic protein [GFAP]; Figure 1D-G).
Localization of adult HSCs in relation to individual BM populations is conserved in different HSC reporters and bone types
To identify BM populations associated with HSCα-cats, we first quantified the distance of every HSCα-cat from individual BM components in full-bone sections (Figure 2). In the femurs, HSCα-cats were scattered throughout the BM as single cells, mostly locating in the diaphysis around the central sinus, which occupies a large percentage of the BM diaphyseal space in long bones (supplemental Figure 2D-H). We did not observe clusters, as most HSCα-cats were >40 μm away from their closest HSC neighbor (supplemental Figure 2D). To assess whether this HSC localization reflects the abundance of different BM populations, RDs of HSC size were computationally placed throughout the BM volume, excluding the insides of the bone matrix and blood vessels. Because the depth of detection varies between different fluorescent channels, depending on fluorochrome, excitation laser, and the epitope’s density, only those RDs located within the 3D volume defined by the shallowest marker fluorochrome were analyzed.23 This method was crucial for unbiased statistical analysis, because placing RDs in the entire imaging volume leads to noncomparable spatial distributions of RDs vs cells and wrong conclusions (supplemental Figure 2E).
Most HSCs were not close to trabecular and cortical bone, adipocytes, or nonmyelinated Schwann cells; rather, they were >90 μm away (Figure 2B-D; supplemental Figure 3). Instead, a proportion of HSCα-cats were close to MKs, and most were in proximity to the Cxcl12 stroma and vasculature. Importantly, none of these relationships were significantly different from RD localization (Figure 2E-G). HSCs were distant from Sca1+ arteriolar and NG2+ periarteriolar cells, but close to CD105+ sinusoids (Figure 2H-J; supplemental Figure 3G-I). Only a small proportion of femoral HSCα-cats were adjacent to Sca1+ arteriolar and NG2+ periarteriolar cells (6% ± 3% and 1% ± 2%, respectively), whereas 80% located >20 μm away. The percentage of HSCα-cats found close to BM populations and their distance distribution were similar to that of the RDs in all cases, indicating that femoral HSCα-cats were close to MKs, Cxcl12 stroma, and sinusoids because of the high frequency of those cell types rather than as the result of active enrichment. Similar results were obtained in MFG mice, where the localization of cKit+GFP+ HSCMFGs, including the frequency of those within 5 μm of individual BM populations, was also indistinguishable from RDs (Figure 2). Notably, individual HSCs can be found close to any cell type, if assessed in small-volume BM snapshots. However, quantifying many HSCs throughout thick full-bone sections elucidates that some relationships are only infrequent. Therefore deep, large-volume, full-bone quantitative imaging is indispensable for accurate assessment of HSC localization.

Despite anatomical differences, femoral and sternal HSCα-cats and femoral HSCMFGs mainly associate with sinusoids and Cxcl12 stroma cells, reflecting frequencies of imaged BM. (A) Overview of HSC reporter mouse lines and corresponding bones used for full-bone, multicolor, quantitative imaging and analysis. (B-J) HSC-niche distance quantification in femurs and sterna of α-catulinGFP/+ and Mds1GFP/+Flt3Cre reporter mice. (B-D) BODIPY+ adipocytes (B; n = 4 each); Col.1+/Opn+ bone surfaces (C; α-catulinGFP/+ femur, n = 13; Mds1GFP/+Flt3Cre femur, n = 12; α-catulinGFP/+ sternum, n = 14); and GFAP+ Schwann cells (D; n = 4 each) were not associated with HSCα-cats. (E-G) Both femoral and sternal HSCα-cats randomly associated with GP1bβ MKs (E; α-catulinGFP/+ femur, n = 12; Mds1GFP/+Flt3Cre femur, n = 9; α-catulinGFP/+ sternum, n = 8); Cxcl12 stroma (F; α-catulinGFP/+ femur, n = 5; Mds1GFP/+Flt3Cre femur and α-catulinGFP/+ sternum, n = 4); and laminin+ entire BM vasculature (F-G; α-catulinGFP/+ femur, n = 9; Mds1GFP/+Flt3Cre femur and α-catulinGFP/+ sternum, n = 8). (H-J) Only a minority of femoral and sternal HSCα-cats localized near Sca1+ arteriolar (H; n = 4 each) and NG2+ periarteriolar niches (I; n = 4 each), whereas most were randomly associated with CD105+ sinusoids (J; α-catulinGFP/+ femur, n = 12; Mds1GFP/+Flt3Cre femur, n = 4; α-catulinGFP/+ sternum, n = 8). Data in panels B-J represent mean ± standard deviation. Statistical significance for the 0- to 5-μm bin was assessed by 2-tailed nonparametric Mann-Whitney U test. *P < .05; **P < .01; ***P < .001.
Despite anatomical differences, femoral and sternal HSCα-cats and femoral HSCMFGs mainly associate with sinusoids and Cxcl12 stroma cells, reflecting frequencies of imaged BM. (A) Overview of HSC reporter mouse lines and corresponding bones used for full-bone, multicolor, quantitative imaging and analysis. (B-J) HSC-niche distance quantification in femurs and sterna of α-catulinGFP/+ and Mds1GFP/+Flt3Cre reporter mice. (B-D) BODIPY+ adipocytes (B; n = 4 each); Col.1+/Opn+ bone surfaces (C; α-catulinGFP/+ femur, n = 13; Mds1GFP/+Flt3Cre femur, n = 12; α-catulinGFP/+ sternum, n = 14); and GFAP+ Schwann cells (D; n = 4 each) were not associated with HSCα-cats. (E-G) Both femoral and sternal HSCα-cats randomly associated with GP1bβ MKs (E; α-catulinGFP/+ femur, n = 12; Mds1GFP/+Flt3Cre femur, n = 9; α-catulinGFP/+ sternum, n = 8); Cxcl12 stroma (F; α-catulinGFP/+ femur, n = 5; Mds1GFP/+Flt3Cre femur and α-catulinGFP/+ sternum, n = 4); and laminin+ entire BM vasculature (F-G; α-catulinGFP/+ femur, n = 9; Mds1GFP/+Flt3Cre femur and α-catulinGFP/+ sternum, n = 8). (H-J) Only a minority of femoral and sternal HSCα-cats localized near Sca1+ arteriolar (H; n = 4 each) and NG2+ periarteriolar niches (I; n = 4 each), whereas most were randomly associated with CD105+ sinusoids (J; α-catulinGFP/+ femur, n = 12; Mds1GFP/+Flt3Cre femur, n = 4; α-catulinGFP/+ sternum, n = 8). Data in panels B-J represent mean ± standard deviation. Statistical significance for the 0- to 5-μm bin was assessed by 2-tailed nonparametric Mann-Whitney U test. *P < .05; **P < .01; ***P < .001.
To investigate whether reported controversies in HSC localization13,18 stem from different types of bones imaged previously, we extended our study to the mouse sternum. Anatomically, the sternum consists of individual bone segments that lack central diaphyseal areas of long bones, but are rich in trabecular bone areas, similar to the femoral epi-/metaphysis (Figure 1B). Despite the microanatomical differences, HSCα-cats physically associated with the same BM populations in femurs and sterna (Figure 2; supplemental Figure 4). A marginally higher proportion of sternal HSCα-cats located close to MKs, but so did the RDs (Figure 2E).
HSCs reside in complex combinatorial microenvironments reflecting cellular abundance of BM populations
To better dissect the HSC microenvironment complexity, we quantified HSCα-cat localization in relation to combinations of up to 4 BM components simultaneously. Most of the HSCα-cats located within 10 μm (∼1 HSC diameter) from vasculature. Simultaneous quantification of distance from MKs revealed that most of those HSCα-cats were >10 μm away from the MKs, whereas some were close to both (Figure 3A-B; Q2 vs Q1). Conversely, of the 1 in 3 HSCα-cats locating within 10 μm of the MKs, almost all were also close to the vasculature. Only a small proportion of HSCα-cats located within 10 μm of the bone. Consistent with bone surface vascularization13 almost all of those are also proximal to the vasculature (Figure 3C; Q1 vs Q4). Interestingly, 66% of HSCα-cats located within 10 μm of both sinusoids and Cxcl12 stroma, whereas 29% were close only to Cxcl12 stroma (Figure 3D; Q1 and Q4), showing that HSCα-cats did not homogenously associate with 1 or a combination of BM populations, but occupied multiple combinatorial BM environments composed of vasculature and/or MKs or sinusoids and/or Cxcl12 stroma (Figure 3E; supplemental Figure 5).
![HSCα-cats occupy different BM locations composed of discrete cell types. (A-D) Quantification of HSCα-cat localization in relation to combinatorial BM niches. Scatterplot showing 2-dimensional [2D]) distance quantification of single HSCs in relation to the entire vasculature and MKs (A) and the corresponding high-magnification images (B) or entire vasculature and bone (C). Most HSCα-cats located proximal to the vasculature (A; Q2 and Q1) and did not simultaneously associate with MKs (A; Q2 vs Q1); almost all HSCα-cats found close to MKs (A; Q1 and Q4) were also close to the vasculature (A; Q1 vs Q4). Dots represent 261 individual HSCs (orange; n = 6) and 584 RDs (gray; n = 6). Data points highlighted with blue circles are shown in panel B. To highlight the cells of interest in panel B (cKit+GFP+ cell), cKit and GFP signal outside the cells were computationally masked to appear black (mcKit/mGFP). (D) Scatterplot showing 2D distance quantification of single HSCs in relation to sinusoids and Cxcl12 stroma (D; 251 HSCs and 440 RDs, n = 5). (E) Graphic depiction of HSC localization in relation to dual and triple niches and quantification of their frequency in full-bone sections (n = 3). Statistical significance for panels A and C-D was assessed by 2-tailed nonparametric Mann-Whitney U test. *P < .05. Scale bars,10 μm (B). Cxcl12, Cxcl12 stroma.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/20/10.1182_blood.2020006574/1/m_bloodbld2020006574f3.png?Expires=1767697867&Signature=Ok7d0mbe79hpluqr0Ba85~eof4nZGNwTd4QJl06xcwX-Bxw70cq5gUGeQVXQBvUjBiZY4TVZ5qZYrESahtk2OR5NJdLuzssPNHSRKEmsSUItK07EEriLYySafkUFgqTomm36jPGePs7qlvSn2yWL-TKA7KFLN~lwkJNV~PO7nDtVNTk8jwysD6HHZNjCnky3hzE8LwzqEiLkE8L9RXqd0pa62tctMNaU5Pv-1UPT1PWMymlvr3LB955rPtmbIM71bBLGMPLVTBR5yeDV706pEahvEcFP3f6X-JK8WGVFFLquLtvQSzBw~DlKrgAjAujt9G3amv~tkZecmK3nqynvEg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HSCα-cats occupy different BM locations composed of discrete cell types. (A-D) Quantification of HSCα-cat localization in relation to combinatorial BM niches. Scatterplot showing 2-dimensional [2D]) distance quantification of single HSCs in relation to the entire vasculature and MKs (A) and the corresponding high-magnification images (B) or entire vasculature and bone (C). Most HSCα-cats located proximal to the vasculature (A; Q2 and Q1) and did not simultaneously associate with MKs (A; Q2 vs Q1); almost all HSCα-cats found close to MKs (A; Q1 and Q4) were also close to the vasculature (A; Q1 vs Q4). Dots represent 261 individual HSCs (orange; n = 6) and 584 RDs (gray; n = 6). Data points highlighted with blue circles are shown in panel B. To highlight the cells of interest in panel B (cKit+GFP+ cell), cKit and GFP signal outside the cells were computationally masked to appear black (mcKit/mGFP). (D) Scatterplot showing 2D distance quantification of single HSCs in relation to sinusoids and Cxcl12 stroma (D; 251 HSCs and 440 RDs, n = 5). (E) Graphic depiction of HSC localization in relation to dual and triple niches and quantification of their frequency in full-bone sections (n = 3). Statistical significance for panels A and C-D was assessed by 2-tailed nonparametric Mann-Whitney U test. *P < .05. Scale bars,10 μm (B). Cxcl12, Cxcl12 stroma.
HSCα-cats occupy different BM locations composed of discrete cell types. (A-D) Quantification of HSCα-cat localization in relation to combinatorial BM niches. Scatterplot showing 2-dimensional [2D]) distance quantification of single HSCs in relation to the entire vasculature and MKs (A) and the corresponding high-magnification images (B) or entire vasculature and bone (C). Most HSCα-cats located proximal to the vasculature (A; Q2 and Q1) and did not simultaneously associate with MKs (A; Q2 vs Q1); almost all HSCα-cats found close to MKs (A; Q1 and Q4) were also close to the vasculature (A; Q1 vs Q4). Dots represent 261 individual HSCs (orange; n = 6) and 584 RDs (gray; n = 6). Data points highlighted with blue circles are shown in panel B. To highlight the cells of interest in panel B (cKit+GFP+ cell), cKit and GFP signal outside the cells were computationally masked to appear black (mcKit/mGFP). (D) Scatterplot showing 2D distance quantification of single HSCs in relation to sinusoids and Cxcl12 stroma (D; 251 HSCs and 440 RDs, n = 5). (E) Graphic depiction of HSC localization in relation to dual and triple niches and quantification of their frequency in full-bone sections (n = 3). Statistical significance for panels A and C-D was assessed by 2-tailed nonparametric Mann-Whitney U test. *P < .05. Scale bars,10 μm (B). Cxcl12, Cxcl12 stroma.
Quantification of the absolute number of available BM locations and their HSC occupancy further confirmed these conclusions. We computationally calculated the number of all available 3-dimensional BM locations composed of different combinations of BM components within which a single HSC could physically fit (3D areas of a 10-μm diameter; see supplemental Methods for details). Per full-femur section, we estimated an average of 114 521 (79%) locations close to both sinusoids and Cxcl12+ stroma, 27 641 (19%) close to Cxcl12 stroma only, 1 213 (<1%) close to sinusoids only, and 435 (<1%) close to neither. The frequencies of these 4 BM locations (79% vs 19% vs <1% vs <1%, respectively) resembled the frequencies of HSCs found close to them (Figure 3E). The same holds true when quantifying BM locations consisting of 3 populations: entire vasculature, bone, and/or MKs (Figure 3E). This result confirmed that the proximity of HSCs to specific BM populations was governed by the abundance of these cell types and did not reflect active enrichment of the populations imaged here. In addition, available absolute numbers of those BM locations were not limiting, given that only 0.03% were occupied by HSCs. Our data provide quantitative evidence that HSC localization within the BM microenvironments reflects the abundance of those microenvironment populations.
Quantitative imaging enables reliable identification of endogenously labeled dormant LR and non-LR adult HSPCs in situ
To investigate whether those complex BM locations host different HSC subtypes, we quantified the location of dormant and nondormant adult HSPCs. We used double-transgenic SCL-tTA;H2B-GFP mice treated with DOX for 150 days and aged-matched SCL-tTA and H2B-GFP single transgenic animals as controls (Figure 4A-C). This system enabled DOX-dependent labeling of homeostatic HSPCs with histone H2B-GFP.19 When DOX was administered (chase period), H2B-GFP transcription stopped, resulting in progressive GFP dilution in HSPCs with a history of proliferation (non-LR), whereas dormant HSPCs retained their fluorescence (LR; Figure 4C-D). Dormant LR HSPCs contain most of the transplanted stem cell activity, whereas non-LR cells mainly have limited self-renewing capacity.19 Because hematopoietic progenitors divide more frequently than HSCs, the 150-day-long chase period ensured HSC-specific labeling (Figure 4E).27
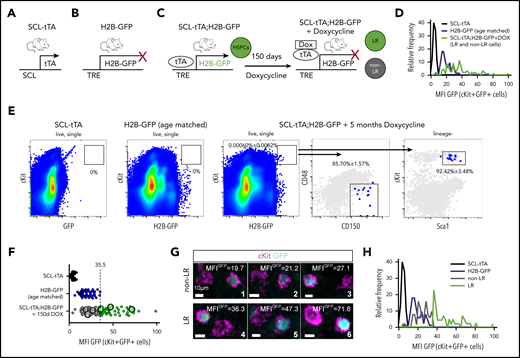
Identification of dormant label-retaining and nondormant HSPCs in situ by quantitative BM imaging. (A-C) Representation of the experimental approach for endogenous labeling of dormant LR HSPCs. Transgenic mice expressing tetracycline transactivator in HSPCs from the scl gene locus (A; SCL-tTA) and the histone 2B-GFP fusion protein (H2B-GFP) under the control of the tetracycline-responsive regulatory element (B; TRE; H2B-GFP) were crossed to confine GFP expression to the HSPCs (C, left; SCL-tTA; H2B-GFP). In the presence of DOX, H2B-GFP transcription was inhibited, resulting in dilution of GFP signal by proliferating cells. (D-G) Identification of BM LR and non-LR HSPCs in situ by quantitative imaging. (D) Frequency distribution of GFP levels (MFI) of cKit+GFP+ (see “Methods” for identification criteria) cells from imaging of SCL-tTA (110 cells), H2B-GFP control (67 cells), and DOX-treated SCL-tTA;H2B-GFP mice (96 cells). (E) Flow cytometry of SCL-tTA, H2B-GFP, and double transgenic SCL-tTA;H2B-GFP mice treated for 150 days with DOX revealed the high purity of HSC marker expression in cKit+GFPhigh LR cells (n = 3). (F) Dot plot showing the absolute GFP MFI (MFIGFP) of cKit+GFP+ single cells from control and treated mice shown in panel D (SCL-tTA: MGIGFP = 4.8; H2B-GFP: MGIGFP = 19.2; DOX-treated SCL-tTA;H2B-GFP: MGIGFP = 35.99). Note the leakiness of GFP expression in the cKit+GFP+ BM cells of the H2B-GFP mouse. LR HSPC identification therefore required precise GFP quantification. LR HSPCs were identified as cKit+ cells with higher MFIGFP than background levels in the single H2B-GFP mouse line (H2B-GFP: maximum MGIGFP = 35.3). (G) High-resolution images of BM LR and non-LR HSPCs, illustrating the need for computational quantification. (H) Frequency distribution of LR and non-LR HSPCs after thresholding based on H2B-GFP background fluorescence from aged-matched animals.
Identification of dormant label-retaining and nondormant HSPCs in situ by quantitative BM imaging. (A-C) Representation of the experimental approach for endogenous labeling of dormant LR HSPCs. Transgenic mice expressing tetracycline transactivator in HSPCs from the scl gene locus (A; SCL-tTA) and the histone 2B-GFP fusion protein (H2B-GFP) under the control of the tetracycline-responsive regulatory element (B; TRE; H2B-GFP) were crossed to confine GFP expression to the HSPCs (C, left; SCL-tTA; H2B-GFP). In the presence of DOX, H2B-GFP transcription was inhibited, resulting in dilution of GFP signal by proliferating cells. (D-G) Identification of BM LR and non-LR HSPCs in situ by quantitative imaging. (D) Frequency distribution of GFP levels (MFI) of cKit+GFP+ (see “Methods” for identification criteria) cells from imaging of SCL-tTA (110 cells), H2B-GFP control (67 cells), and DOX-treated SCL-tTA;H2B-GFP mice (96 cells). (E) Flow cytometry of SCL-tTA, H2B-GFP, and double transgenic SCL-tTA;H2B-GFP mice treated for 150 days with DOX revealed the high purity of HSC marker expression in cKit+GFPhigh LR cells (n = 3). (F) Dot plot showing the absolute GFP MFI (MFIGFP) of cKit+GFP+ single cells from control and treated mice shown in panel D (SCL-tTA: MGIGFP = 4.8; H2B-GFP: MGIGFP = 19.2; DOX-treated SCL-tTA;H2B-GFP: MGIGFP = 35.99). Note the leakiness of GFP expression in the cKit+GFP+ BM cells of the H2B-GFP mouse. LR HSPC identification therefore required precise GFP quantification. LR HSPCs were identified as cKit+ cells with higher MFIGFP than background levels in the single H2B-GFP mouse line (H2B-GFP: maximum MGIGFP = 35.3). (G) High-resolution images of BM LR and non-LR HSPCs, illustrating the need for computational quantification. (H) Frequency distribution of LR and non-LR HSPCs after thresholding based on H2B-GFP background fluorescence from aged-matched animals.
As expected, the GFP MFI of cKit+ cells in SCL-tTA control mice was at background levels. H2B-GFP mice (without tTa expression) showed low but detectable leaky fluorescence (Figure 4D). DOX-treated SCL-tTA;H2B-GFP mice displayed a range of GFP intensities, including those higher than H2B-GFP controls. We used the maximum GFP intensity of cKit+GFP+ cells from age-matched H2B-GFP controls as the threshold,28 above which cells were classified as LR (Figure 4F). Notably, the number of LR HSPCs per full-bone section was twofold lower than in the HSCα-cats. The remaining cKit+GFP+ cells were classified as non-LR and were considered nondormant HSPCs with a history of proliferation.19-21 Digital GFP quantification was essential for accurate distinction between LR and non-LR HSPCs, because visual decision could lead to misclassification (Figure 4G). Our approach enabled in situ identification of endogenously labeled dormant and nondormant HSPCs in the same section (Figure 4H).
Dormant and nondormant adult HSPCs and HSCα-cats have indistinguishable BM localization
To address whether dormant LR and nondormant, non-LR HSPCs are associated with distinct BM microenvironments, we quantified their BM localization. Consistent with our HSCα-cat data, LR and non-LR HSPCs did not locate near adipocytes, bone surfaces, or nonmyelinated Schwann cells, with <5% being within 5 μm of these structures and most being >90 μm away (Figure 5A-C). Although few LR and non-LR cells located close to the MKs, most were close to the BM vasculature, similar to RDs and HSCα-cats (Figure 5D-E). Distance quantification from distinct vessels and pericytes revealed that only a small fraction of LR and non-LR HSPCs were close to Sca1+ arteries and NG2 periarteriolar cells, similar to RDs (Figure 5F-G). Instead, almost 60% of LR and non-LR cells located >50 μm from Sca1+ arteries (supplemental Figure 6F) and most were >90 μm away from NG2 periarteriolar cells (Figure 5G). In contrast, >60% of LR and non-LR HSPCs located within 5 μm of the CD105+ sinusoids (RD, 59% ± 6%; Figure 5H). Importantly, the percentage of femoral LR and non-LR HSPCs found close to individual BM populations and their distance distribution was statistically indistinguishable (Figure 5A-I; supplemental Figure 6). This result was further confirmed by quantifying their proximity to dual BM microenvironments (Figure 5I-J). Similar to HSCα-cats, the localization of LR and non-LR HSPCs was conserved between femurs and sterna (supplemental Figure 7). Thus, dormant LR and nondormant non-LR HSPCs were associated with the same imaged BM populations and had statistically indistinguishable localizations as HSCα-cats in both femurs and sterna.

HSCα-cats, dormant LR HSCs, and nondormant non-LR HSPCs all have similar BM niche signatures. (A-H) Comparative niche distance quantification of LR and non-LR HSPCs in femurs of DOX-treated SCL-tTA;H2B-GFP mice or of HSCα-cats in α-catulinGFP/+ reporter mice. (A-C) BODIPY+ adipocytes (A; n = 4 each); Col.1+/Opn+ bone surfaces (B; n = 13 each); and GFAP+ Schwann cells (C; n = 4 each) were not physically associated with HSPCs. (D-G) Some LR, non-LR HSPCs and HSCα-cats located randomly near MKs (D; LR/non-LR, n = 4; HSCα-cat, n = 12). Most were randomly associated with laminin+ vasculature (E; LR/non-LR, n = 7; HSCα-cat, n = 9), whereas a few associated with Sca1+ arteries and arterioles (F; n = 4 each) or periarteriolar niches (G; n = 4 each). (H) LR and non-LR HSPCs, as well as HSCα-cats randomly associated with CD105+ sinusoids (H; LR/non-LRC, n = 4; HSCα-cat, n = 12). (I, J) Scatterplots showing 2-dimentional distance quantification of single LR HSPCs (green), non-LR HSPCs (gray dots), and RDs (purple dots) in relation to the entire vasculature and MKs (I; 90 LR, 270 non-LR and 383 RDs) or bone (J; 90 LR, 270 non-LR and 383 RDs). Data in panels A-H represent mean ± standard deviation. Statistical significance in the 0- to 5-μm bin was assessed by 2-tailed nonparametric Mann-Whitney U test. ns, not significant.
HSCα-cats, dormant LR HSCs, and nondormant non-LR HSPCs all have similar BM niche signatures. (A-H) Comparative niche distance quantification of LR and non-LR HSPCs in femurs of DOX-treated SCL-tTA;H2B-GFP mice or of HSCα-cats in α-catulinGFP/+ reporter mice. (A-C) BODIPY+ adipocytes (A; n = 4 each); Col.1+/Opn+ bone surfaces (B; n = 13 each); and GFAP+ Schwann cells (C; n = 4 each) were not physically associated with HSPCs. (D-G) Some LR, non-LR HSPCs and HSCα-cats located randomly near MKs (D; LR/non-LR, n = 4; HSCα-cat, n = 12). Most were randomly associated with laminin+ vasculature (E; LR/non-LR, n = 7; HSCα-cat, n = 9), whereas a few associated with Sca1+ arteries and arterioles (F; n = 4 each) or periarteriolar niches (G; n = 4 each). (H) LR and non-LR HSPCs, as well as HSCα-cats randomly associated with CD105+ sinusoids (H; LR/non-LRC, n = 4; HSCα-cat, n = 12). (I, J) Scatterplots showing 2-dimentional distance quantification of single LR HSPCs (green), non-LR HSPCs (gray dots), and RDs (purple dots) in relation to the entire vasculature and MKs (I; 90 LR, 270 non-LR and 383 RDs) or bone (J; 90 LR, 270 non-LR and 383 RDs). Data in panels A-H represent mean ± standard deviation. Statistical significance in the 0- to 5-μm bin was assessed by 2-tailed nonparametric Mann-Whitney U test. ns, not significant.
Distinct BM localization signature between juvenile and postswitch HSCs
Because dormant and nondormant HSPCs share the same localization in adult animals, we next investigated the BM localization of juvenile 3-week-old HSCs retaining fetal liver characteristics (high proliferation rates) and compared them with postswitch (5-week-old) and adult (12-week-old) HSCs in α-catulinGFP/+ mice. The BM localization of postswitch 5-week-old and adult 12-week-old HSCα-cats was largely identical, except that 5-week-old HSCα-cats preferentially located close to Cxcl12 stroma (Figure 6A-E; supplemental Figure 8A-E). Comparing pre- and postswitch HSCα-cats, we found no difference in their localization relative to adipocytes, Schwann cells, BM vasculature, and Sca1 arteriolar and NG2 periarteriolar cells (supplemental Figures 8A-E, 9, and 10). However, significantly fewer preswitch HSCα-cats were located close to MKs, a twofold decrease compared with postswitch HSCα-cats (Figure 6B). Similarly, significantly fewer preswitch HSCα-cats located proximal to CD105 sinusoids compared with RDs and postswitch HSCα-cats (Figure 6C). Instead, preswitch HSCα-cats preferentially located close to Cxcl12 stroma irrespective of the distance bin applied and exhibited a marginally higher preference toward bone surfaces, shared equally between trabecular and cortical bone (Figure 6D-F; supplemental Figure 8F-G). We next evaluated whether observed differences between pre- and postswitch HSCα-cat localization reflected age-related alterations in BM composition and cell abundance. We quantified the BM volume occupied by MKs and Cxcl12 stroma and indeed found a gradual increase with age (Figure 6G-H) that was most likely linked with bone growth (supplemental Figure 8H). Combinatorial localization analysis revealed that significantly fewer preswitch HSCα-cats were distant from Cxcl12 stroma and close to sinusoids compared with RDs (Q2; Figure 6I). In addition, preswitch HSCα-cats residing proximal to bone surfaces were also proximal to endosteal vessels (Figure 6J-K), showing that bone surfaces hosting HSCs are vascularized. Thus, most juvenile and adult HSCs associated with sinusoids and Cxcl12 stroma. However, in contrast to adult HSCs, preswitch HSCs were significantly nonrandomly enriched within 5 μm of Cxcl12+ stoma or bone surfaces, and 6 to 15 μm away from CD105 sinusoids. A detailed representation of the distinct BM populations occupied by 3-week-old HSCα-cats is shown in Figure 6L-M. Taking all evidence together, we report, for the first time to our knowledge, that pre- and postswitch HSCs not only have distinct intrinsic programs, but also have distinct localization in relation to imaged BM populations.
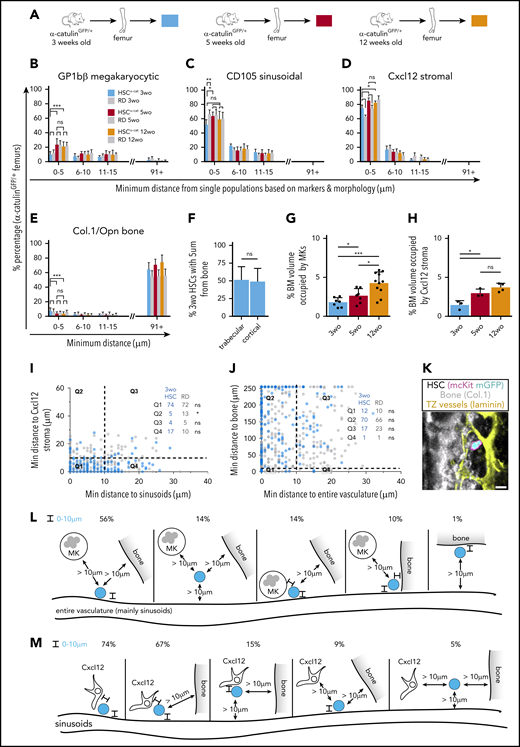
Distinct niche signatures between 3-week-old preswitch and 5- and 12-week-old postswitch HSCα-cats. (A) Representation of the age groups used for isolation of α-catulinGFP/+ femurs. (B-E) Three-week-old (wo) HSCα-cats were less associated with MKs (B; 3 wo, n = 8; 5 wo, n = 7; 12 wo, n = 12) and CD105+ sinusoids (C; 3 wo, n = 8; 5 wo, n = 6; 12 wo, n = 12), compared with postswitch HSCα-cats, but were preferentially associated with Cxcl12 stroma (D; 3 wo, n = 4; 5 wo, n = 4; 12 wo, n = 5) and showed a marginally higher preference toward Col.1/Opn+ osteoblasts and bone matrix (E; 3 wo, n = 17; 5 wo, n = 10; 12 wo, n = 13). (F) Percentage of 3-wo HSCα-cats found within 5 μm of the trabecular or cortical bone surfaces (n = 12). (G-H) Comparison of BM volume occupied by MKs (G; 3 wo, n = 7; 5 wo, n = 7; 12 wo, n = 11) and Cxcl12 stroma (H; 3 wo, n = 3; 5 wo, n = 3; 12 wo, n = 4) in juvenile and adult α-catulinGFP/+ femurs. (I-J) Scatterplots showing 2-dimensional (2D) distance quantification of single 3-wo HSCα-cat in relation to CD105+ sinusoids and Cxlc12 stroma (I; 192 HSCs and 414 RDs, n = 4), as well as entire vasculature and bone surfaces (J; 197 HSCs and 427 RDs, n = 4). (K) Representative image of a single 3-wo HSCα-cat that was adjacent to bone surface and transition zone (TZ) vessels simultaneously. (L-M) Quantification of HSC frequency occupying niches with distinct cellular composition in 3-wo α-catulinGFP/+ femurs. Graphic depiction of 3-wo HSCα-cat localization in relation to triple niches, such as entire vasculature, bone, and MKs (L; n = 4) and sinusoids, bone, and Cxcl12 stroma (M; n = 4). Data in panels B-H represent mean ± standard deviation. The 2-tailed nonparametric Mann-Whitney U test was used to assess statistical significance in panels A-D (for the 0-5-μm bin) and H-I. Statistical significance for panels E-G was assessed by 1-tailed Mann-Whitney U test. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Distinct niche signatures between 3-week-old preswitch and 5- and 12-week-old postswitch HSCα-cats. (A) Representation of the age groups used for isolation of α-catulinGFP/+ femurs. (B-E) Three-week-old (wo) HSCα-cats were less associated with MKs (B; 3 wo, n = 8; 5 wo, n = 7; 12 wo, n = 12) and CD105+ sinusoids (C; 3 wo, n = 8; 5 wo, n = 6; 12 wo, n = 12), compared with postswitch HSCα-cats, but were preferentially associated with Cxcl12 stroma (D; 3 wo, n = 4; 5 wo, n = 4; 12 wo, n = 5) and showed a marginally higher preference toward Col.1/Opn+ osteoblasts and bone matrix (E; 3 wo, n = 17; 5 wo, n = 10; 12 wo, n = 13). (F) Percentage of 3-wo HSCα-cats found within 5 μm of the trabecular or cortical bone surfaces (n = 12). (G-H) Comparison of BM volume occupied by MKs (G; 3 wo, n = 7; 5 wo, n = 7; 12 wo, n = 11) and Cxcl12 stroma (H; 3 wo, n = 3; 5 wo, n = 3; 12 wo, n = 4) in juvenile and adult α-catulinGFP/+ femurs. (I-J) Scatterplots showing 2-dimensional (2D) distance quantification of single 3-wo HSCα-cat in relation to CD105+ sinusoids and Cxlc12 stroma (I; 192 HSCs and 414 RDs, n = 4), as well as entire vasculature and bone surfaces (J; 197 HSCs and 427 RDs, n = 4). (K) Representative image of a single 3-wo HSCα-cat that was adjacent to bone surface and transition zone (TZ) vessels simultaneously. (L-M) Quantification of HSC frequency occupying niches with distinct cellular composition in 3-wo α-catulinGFP/+ femurs. Graphic depiction of 3-wo HSCα-cat localization in relation to triple niches, such as entire vasculature, bone, and MKs (L; n = 4) and sinusoids, bone, and Cxcl12 stroma (M; n = 4). Data in panels B-H represent mean ± standard deviation. The 2-tailed nonparametric Mann-Whitney U test was used to assess statistical significance in panels A-D (for the 0-5-μm bin) and H-I. Statistical significance for panels E-G was assessed by 1-tailed Mann-Whitney U test. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Discussion
Our study contributes to resolving existing confusion regarding exact HSC localization in relation to 9 BM components and their combinations, previously described to function as HSC niches, using quantitative multicolor imaging in 152 full-bone sections from 3 different HSC reporter mouse lines. We have provided a comprehensive data set quantifying the frequencies of HSCs found in close association with those BM populations (supplemental Figure 11). Fourteen percent of our data agree with previous reports, 4% differ, and 82% present novel findings. We showed that HSCs locate within multiple, spatially distinct BM microenvironments mainly composed of sinusoids and Cxcl12 stroma (Figure 3E). The need for physical interaction between HSCs and those cells was supported molecularly, because endothelial and Cxcl12 or LepR perisinusoidal stromal cells are the main sources of stem cell factor, a key protein promoting HSC maintenance.29,30 To ensure access to membrane-bound stem cell factor, HSCs require direct contact with these cell types. We and others previously reported substantial overlap between Cxcl12 and LepR cells.26,29 We have now shown a proportion of HSCs locating within 5 μm of MKs, confirming previous results.14,15 Mechanistically, MKs inhibit HSC proliferation through secreted proteins, including Tgfb115 and Cxcl414 ; therefore, direct contact may not be required.
We identified BM populations with limited physical association with HSCs. Fewer than 6% of femoral and 10% of sternal HSCs locate near bone, compared with almost 20% reported earlier.18 Similarly, <6% of HSCs were adjacent to Sca1+ arteriolar cells, in agreement with a previous study,13 but almost twofold lower than that in another study.18 Consequently, HSCs do not preferentially locate close to NG2+ periarteriolar cells, suggesting that reported effects on HSC function18 are mediated by secreted factors, as previously described.31 We found almost no spatial association of HSCs with GFAP+ Schwann cells, mainly located around Sca1+ diaphyseal arteries.26 Despite lacking physical association, the importance of those BM populations as functional homeostatic niches stands, but is most likely mediated by secreted factors. Finally, most HSCs did not associate with adipocytes. Importantly, HSC localization was conserved between both femurs and sterna, despite microanatomical differences. Notably, the hematopoietic cell type imaged is pivotal, because populations mainly consisting of multipotent progenitors (cKit+Sca1+ lineage) locate closer to the bone matrix and GFAP+ Schwann cells, in a random and nonrandom fashion, respectively.23
Although we found HSCs close to the cell types described herein, our analyses revealed that the localization of HSCα-cats reflected the cellular abundance of those BM populations, suggesting that HSC localization is mostly determined by microanatomical BM properties, rather than being actively selected by HSCs. Femoral HSC localization from sinusoids and bone in a recently developed HSC reporter mouse further confirmed this observation.24 Furthermore, the frequency with which HSCs were found within specific niches correlated with the abundance of those niches. A large number of anatomical microenvironments were vacant during homeostasis and thus were not a limiting factor, a result that was further supported by recent findings that transplanting a high number of HSCs into nonconditioned recipients results in their successful long-term engraftment, without replacing host HSCs.32 Empty niches may lack additional cell types or function under inflammation or stress. Alternatively, occupied niches may consist of currently unexplored subsets of sinusoidal, perisinusoidal, or stromal cells reflecting the recently reported heterogeneity of those populations33,34 or being induced by HSCs themselves.35,36 HSCs may modulate the gene and protein expression profile of specific BM cell types, making them functional niche cells only after interacting with HSCs. Such an HSC-niche unit is established only in the presence of both cell types, explaining why the number of potential niches vastly exceeds that of HSCs. This observation is in line with those in a previous study reporting differential transcriptional profiles of osteolineage cells, depending on their proximity to HSCs in situ.37 Furthermore, it is well accepted that tumor cells can reprogram their microenvironment to favor disease progression,34,38,39 most likely by hijacking the cross-talk mechanism between normal HSCs and their niche.40
The existence of distinct niches supporting HSCs with different divisional histories has also been debated. Previous studies using Ki67 as a proliferation marker not only led to contradictory results,13,18 but were limited to a static view of the cell-cycle status providing no information on the divisional history of HSCs. We therefore used DOX-treated, double-transgenic SCL-tTA;H2B-GFP mice19 to identify dormant (long-term quiescent) HSPCs compared with age-matched H2B-GFP controls.28 The advantage of endogenous labeling over 5-ethynyl-2′-deoxyuridine assays is the reliable marking of most immunophenotypic HSCs,19 whereas a 150-day-long chase allows for efficient exclusion of labeled hematopoietic progenitors otherwise included in shorter chase periods.19,41 Herein, we report that femoral and sternal dormant and nondormant adult HSPCs shared identical BM localization signatures, suggesting that functional differences between those subtypes may be niche independent and reflect intrinsic predetermined programs. This observation is in agreement with previous studies reporting identical behavior of clonally related adult HSCs in lineage contribution and repopulation kinetics after serial transplantations.42-44 We do not exclude the possibility that other (ie, lineage-biased) adult HSC types have different niche signatures,45 or that the same niches affect other hematopoietic cell types differently, such as instructing lymphoid progenitors46 or inducing niche signature changes during aging and stress.41 In line with these possibilities, we report that juvenile preswitch BM HSCs had a distinct BM localization signature: they preferentially associated with Cxcl12 stroma, potentially reflecting recently reported transcriptional differences between juvenile and adult Cxcl12 stroma cells.47 They also located farther from sinusoids and MKs (33% of adult vs 15% of preswitch HSCs were close to both types simultaneously), in line with a possible inhibitory effect of MKs on HSC proliferation.14,15 Further studies are needed to validate the functional relevance of those localization differences in juvenile HSCs.
Our study is based on mouse reporters yielding HSCs of a purity obtained by the latest purification methods. Approximately half of these cells failed to be classified as long-term, self-renewing HSCs in transplantation experiments, the current standard for assessing HSC function, despite increasing concerns about its accuracy in enumerating stem cell potential and reflecting unperturbed hematopoiesis.48 It thus is possible that actual distribution of pure HSCs may deviate from those reported previously and in this study. However, our finding that dormant and nondormant cells have statistically indistinguishable distance distributions from all the studied niches does not support this possibility.
The quantification of 152 full-bone sections of different bone types and the 3 different HSC reporter systems used in this study help resolve existing confusion regarding the physical association of HSCs with putative BM-niche cells and the possible mechanisms of interaction (cell adhesion molecules or secreted factors) at tissue-wide level and single-cell resolution.
Data are available in .ims format upon e-mail request to either of the corresponding authors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Sean J. Morrison for quickly providing α-catulinGFP/+ mice; M. D. Hussherr and G. Camenisch for animal husbandry and care; T. Horn, A. Ponti, and E. Montani (ETH Zurich) and Handley Maris (Center for Regenerative Medicine, Massachusetts General Hospital) for technical imaging support; and members of the T.S. and D.T.S. laboratories for valuable input on the manuscript.
This work was supported by Swiss National Science Foundation grants 31003A-179490 and CRSII5-186271 (T.S.). K.D.K. is a recipient of a Human Frontier Science Program (HFSP) long-term fellowship and a Swiss National Science Foundation (SNSF) postdoctoral mobility fellowship.
Authorship
Contribution: K.D.K. conceived the project, planned and performed all the experiments, analyzed the data, and wrote the manuscript with T.S.; L.K. programmed the script for estimating the number of available niches, with input from K.D.K.; N.C.-W., S.R., and A.T. performed the label-retaining experiments and provided bones from the SCL-tTA, H2B-GFP, and SCL-tTA;H2BGFP double-transgenic mice; C.C. and F.C. developed and provided bones from the Mds1GFP/+Flt3Cre reporter mouse; D.T.S. funded and supervised the project; T.S. funded, planned, and supervised the project; and all authors read and commented on the manuscript.
Conflict-of-interest disclosure: D.T.S. is a director and equity holder of Agios Pharmaceuticals, Magenta Therapeutics, Editas Medicines, ClearCreekBio, and Life-VaultBio; a founder of Fate Therapeutics and Magenta Therapeutics; and a consultant to FOG Pharma and VCanBio. The remaining authors declare no competing financial interests.
Correspondence: Konstantinos D. Kokkaliaris, Department of Biosystems Science and Engineering, ETH Zurich, Mattenstrasse 26, 4058 Basel, Switzerland, e-mail: kkokkaliaris@mgh.harvard.edu; and Timm Schroeder, Department of Biosystems Science and Engineering, ETH Zurich, Mattenstrasse 26, 4058 Basel, Switzerland; e-mail: timm.schroeder@bsse.ethz.ch.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal