

Key Points
FXII inhibition prevented the activation of kallikrein-kininogen and coagulation systems induced by heat-inactivated S aureus.
Blocking FXII decreased complement activation and inflammatory cytokines, preserved organ function, and saved S aureus–challenged baboons.

Abstract
Activation of coagulation factor (F) XI promotes multiorgan failure in rodent models of sepsis and in a baboon model of lethal systemic inflammation induced by infusion of heat-inactivated Staphylococcus aureus. Here we used the anticoagulant FXII-neutralizing antibody 5C12 to verify the mechanistic role of FXII in this baboon model. Compared with untreated control animals, repeated 5C12 administration before and at 8 and 24 hours after bacterial challenge prevented the dramatic increase in circulating complexes of contact system enzymes FXIIa, FXIa, and kallikrein with antithrombin or C1 inhibitor, and prevented cleavage and consumption of high-molecular-weight kininogen. Activation of several coagulation factors and fibrinolytic enzymes was also prevented. D-dimer levels exhibited a profound increase in the untreated animals but not in the treated animals. The antibody also blocked the increase in plasma biomarkers of inflammation and cell damage, including tumor necrosis factor, interleukin (IL)-1β, IL-6, IL-8, IL-10, granulocyte-macrophage colony-stimulating factor, nucleosomes, and myeloperoxidase. Based on clinical presentation and circulating biomarkers, inhibition of FXII prevented fever, terminal hypotension, respiratory distress, and multiorgan failure. All animals receiving 5C12 had milder and transient clinical symptoms and were asymptomatic at day 7, whereas untreated control animals suffered irreversible multiorgan failure and had to be euthanized within 2 days after the bacterial challenge. This study confirms and extends our previous finding that at least 2 enzymes of the contact activation complex, FXIa and FXIIa, play critical roles in the development of an acute and terminal inflammatory response in baboons challenged with heat-inactivated S aureus.
Introduction
Certain infections can provoke pathologic host reactions characterized by systemic inflammation, activation of coagulation, organ failure, and ultimately death. An estimated 48.9 million cases of sepsis occurred worldwide in 2017, including 11 million deaths (19.7% of all global deaths).1 Therapeutic interventions are still limited to: supportive care; antibacterial, antifungal, or antiviral therapies; and corticosteroids. Mortality remains high in patients with advanced forms of sepsis despite the use of potent antibiotics that rapidly kill bacteria and terminate systemic infections.2 Effective therapies directly targeting the underlying pathophysiology of sustained and progressive sepsis have remained elusive, as evidenced by past attempts to develop numerous drugs, including activated protein C, which failed to show an overall outcome benefit in clinical trials.3
Coagulation factor (F) XII is a serine protease positioned at the nexus of the kallikrein-kinin, complement, coagulation, and fibrinolysis systems. However, the true physiological role of FXII remains unknown, as individuals born without FXII have no apparent pathology such as bleeding diathesis or immunocompromise and are generally identified through incidental detection of a prolonged activated partial thromboplastin time (aPTT). However, hyperactivity of FXII seems to be pathologic, as mutations in exon 9 provoke a rare form of hereditary angioedema.4 Investigation into the role of FXII in the pathogenesis of bacterial sepsis was initiated by the pioneering work of Pixley and colleagues,5-7 who found that preadministration of the anti–β-FXIIa antibody C6B7 before lethal Escherichia coli challenge altered the clinical course of septic shock in baboons, even preventing death in 1 of 5 animals. The extent to which FXII was neutralized by C6B7 in these experiments is unknown, and the mechanistic role by which FXII contributes to sepsis remains to be elucidated; this experiment served as compelling groundwork, however, for the continued investigation of the contact system in models of inflammation and infection.
Later studies suggested that the contact activation system composed of FXII, FXI, high molecular weight kininogen (HK), and prekallikrein may contribute to systemic inflammation caused by certain infections. Our studies in mouse models of listeriosis and polymicrobial sepsis and in a baboon model of lethal heat-inactivated Staphylococcus aureus (HI-SA) provided experimental evidence that a contact activation system, in particular reciprocal interactions of FXI and FXII, plays a pathogenic role in the development of some forms of severe sepsis or bacterial component–induced acute shock.8-12 Additional data also indicate that the contact activation could have a pathogenic role in select forms of sepsis.13,14 These findings build on the earlier sepsis models by Pixley and colleagues,5-7 lending weight to the idea that targeting contact activation may prevent or reduce the progression of some forms of infections into terminal septic shock and death, even in cases in which antimicrobial therapies have stopped the infection.
The current article expands on our previous studies regarding the role of contact activation in inflammatory responses. Specifically, we investigated the mechanistic role of FXII in our baboon model of lethal challenge with HI-SA. This model is designed to study the effects of the pathogen-associated molecular patterns (PAMPs) released from dead S aureus rather than sepsis induced by live bacteria. We have shown previously that gram-positive PAMPs such as peptidoglycan are strong inducers of coagulation and complement cascades and can be major contributors to sepsis coagulopathy.15 To test the effects of selective pharmacologic inhibition of FXII activity, the potent neutralizing monoclonal antibody 5C12 was used to target the catalytic domain of human FXII.
Materials and methods
Preparation of anti-FXII antibody
The 5C12 function-blocking anti-FXII antibody was produced as previously described.13,16 Briefly, FXII knockout mice were immunized with a mixture of human and murine recombinant FXII, spleens were removed after 50 days, fused with myeloma cells, and subcloned to form a stable expressing hybridoma cell line.17 To purify 5C12, hybridoma cells were cultured and expanded in a CL1000 bioreactor (Corning Inc, Corning, NY), and supernatants were purified by 2-step chromatography using a cation exchange (mercaptoethyl-pyridine) column followed by affinity purification (protein A/G). The inhibitory effect of 5C12 was evaluated in aPTT assays and compared with the anti–β-FXIIa monoclonal antibody C6B7 (GeneTex, Irvine, CA). The antibodies (0-2 µM) were incubated first with human platelet-poor plasma (33% final) for 10 minutes, then with aPTT reagent for 3 minutes before adding CaCl2 (8.3 mM final) and measuring clotting time with a KC4 analyzer (Trinity Biotech, Bray, Ireland). The data in supplemental Figure 1 (available on the Blood Web site) show that 5C12 inhibits contact activation of coagulation at lower concentrations than C6B7, which behaves as a relatively poor anticoagulant in the aPTT assay.
Lethal challenge of baboons with HI-SA infusion
The experiments were approved by both the Interfaculty Animal Ethics Committee of the University of the Free State (Bloemfontein, South Africa) and the Institutional Animal Care and Use Committee of MD Anderson Cancer Center (Houston, TX). All experiments were conducted in compliance with the Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the National Institutes of Health Office of Laboratory Animal Welfare. We used healthy Papio species baboons (5 males and 3 females) ranging in body weight from 6 to 21 kg, with white blood cell counts <13 000/µL and hemoglobin >10 g/dL.
Although we have not historically observed sex differences in our model, we included both male and female animals in our study. However, because female animals are used for breeding, they are less available for experimental groups. Animals were distributed randomly between the control, untreated group (n = 4; 3 male and one female animal), and the treated group (n = 4; 2 male and 2 female animals). Both groups were challenged with a lethal dose (∼3 × 1010) of heat-inactivated S aureus (ATCC 49496) as previously described.12 All animals received bacteria via a 2-hour intravenous infusion. Baboons in the treatment group received 3 intravenous boluses of 5C12 antibody: (1) 10 mg/kg at 30 minutes (T-0.5) before challenge; (2) 10 mg/kg at 8 hours (T+8); and (3) 5 mg/kg at 24 hours (T+24) after challenge. Control animals did not receive 5C12.
The animals were returned to the cage 8 hours after bacteria infusion, continuously monitored, and humanely euthanized if they exhibited signs of unrecoverable organ failure, as described.12 Surviving baboons were euthanized at 7 days’ postchallenge. Blood samples were collected and physiological parameters measured as detailed. Coagulation parameters (fibrinogen, prothrombin time [PT], and aPTT) and hematologic parameters (red blood cell [RBC] counts, hematocrit, hemoglobin, platelet counts, and differential white blood cell counts) were monitored. At time of euthanasia, blood and tissue samples were collected for biochemical analyses, and select tissue samples were collected postmortem for histopathology.
Biochemical tests
Serum blood urea nitrogen, creatinine, alanine aminotransferase, amylase, and glucose levels were measured by using standard clinical diagnostic tests. Blood lactate levels were measured by using a Lactate Scout Analyzer (EKF Diagnostics GmbH, Barleben, Germany). Plasma myeloperoxidase (MPO) was measured by using the Fluoro MPO Detection Kit (Cell Technology, Fremont, CA). Tissue factor (TF) mRNA was determined by quantitative reverse transcription polymerase chain reaction as described elsewhere.15
ELISA assays
Factor XIIa-antithrombin (FXIIa-AT), FXIIa-C1 inhibitor (FXIIa-C1INH), kallikrein (Kal)-AT, Kal-C1INH, FXIa-AT, FIXa-AT, FVIIa-AT, FXa-AT complexes, and thrombin-AT (TAT) were quantified as previously described.12 The standards were prepared by incubating lepirudin-anticoagulated baboon plasma with dextran sulfate, except for FVIIa-AT, in which dextran sulfate was substituted with TF–phospholipid vesicles (PT reagent).
DuoSet ELISA Kits (R&D Systems, Minneapolis, MN) were used for assessing plasminogen activator inhibitor-1 (PAI-1) and tissue-type plasminogen activator (t-PA). Total and cleaved HK levels were measured by using the 3E8 monoclonal anti-kininogen capture antibody and 2B7 biotinylated monoclonal anti-kininogen detection antibody.18 D-dimer and plasmin–antiplasmin complexes were quantified as previously described.12 For nucleosome detection, a Cell Death Detection ELISA PLUS Kit (Roche Diagnostics GmbH, Mannheim, Germany) was used. C3b and C5b-9 levels were measured as previously described.19 Plasma cytokines were measured by using the MILLIPLEX MAP Non-Human Primate Cytokine Magnetic Bead Panel (EMD Millipore, Billerica, MA).
Microscopy
Harvested tissues were formalin fixed and embedded in paraffin or optimum cutting temperature medium. Histopathologic analysis and scoring of paraffin sections stained with hematoxylin-eosin or phosphotungstic acid–hematoxylin were performed by a veterinary pathologist blinded to the experimental conditions. For immunofluorescence confocal microscopy on optimum cutting temperature sections, the following antibodies were used: mouse monoclonal anti-human GPIIIa (clone SZ21; Beckman Coulter, Brea, CA), rabbit anti-human fibrinogen and mouse monoclonal anti-human CD68 (clone KP1), both from Agilent Technologies (Santa Clara, CA), rabbit antineutrophil elastase (MilliporeSigma, Burlington, MA), and mouse monoclonal anti-human C5b-9 neopeptide (clone aE11, Enzo Life Sciences Inc, Farmingdale, NY).
Statistical analysis
Data are depicted as mean ± standard error of the mean. Visualization was performed by using Prism version 9.0 (GraphPad Software, La Jolla, CA). The significance threshold was set at P < .05 (*P < .05; **P < .01; ***P < .001). For parameters measured at multiple time points, confidence intervals and P values were determined by using the generalized least squares (gls) function implemented in the nlme R package. The function implements a linear mixed effect model, tailored to the specific repeated measure design by proper specification of the random effect structure.20 An AR(1) correlation structure describes the variation of measured values on the same animal along a time path. An overall P value for the difference between 2 curves along the time path was retrieved from the analysis of variance table on the linear model. Statistical testing was limited to 0- to 8-hour intervals. Inference was preceded by a Box-Cox transformation21 when visual inspection of the diagnosis plots showed the need for better matching the model assumptions. For easier interpretation, the results are shown in the original linear scale. Temporal changes of the vital signs (temperature, respiration, heart rate, and mean systemic arterial pressure) were modeled in the framework of Generalized Additive Models, as implemented in the Generalized Additive Models function of the R package, mgcv.22 The function uses smoothing splines for fitting the temporal curves and can handle random effect specifications. The log-rank (Mantel-Cox) test was used to evaluate the differences in survival curves.
Results
Effect of 5C12 on baboon survival after lethal challenge with HI-SA
Intravenous infusion of 1010 HI-SA bacteria/kg body weight induces systemic inflammation and robust activation of coagulation, leading to disseminated intravascular coagulation (DIC).12 All control animals receiving bacteria were humanely euthanized within 10 to 34 hours’ postchallenge due to irreversible organ failure. All treated baboons that received 3 consecutive doses of the 5C12 antibody had only mild and transient clinical symptoms, recovered quickly, and reached the 7-day end point asymptomatic (Mantel-Cox test, P = .0067) (Figure 1).
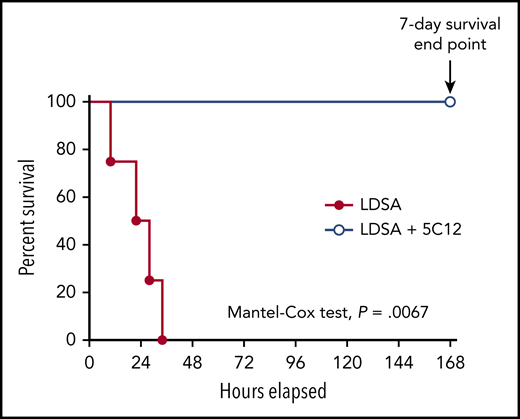
5C12 anti-FXII antibody treatment prevents the death of baboons injected with a lethal dose of HI-SA (LDSA). Survival plot of baboons treated with the anti-FXII antibody 5C12 and injected with LDSA (LDSA + 5C12, n = 4) compared with animals challenged with LDSA but lacking the 5C12 treatment (LDSA, n = 4). The survival rate of these 2 groups was determined by using the Mantel-Cox (log-rank) test. Results are significant at P < .01.
5C12 anti-FXII antibody treatment prevents the death of baboons injected with a lethal dose of HI-SA (LDSA). Survival plot of baboons treated with the anti-FXII antibody 5C12 and injected with LDSA (LDSA + 5C12, n = 4) compared with animals challenged with LDSA but lacking the 5C12 treatment (LDSA, n = 4). The survival rate of these 2 groups was determined by using the Mantel-Cox (log-rank) test. Results are significant at P < .01.
Effect of 5C12 on HI-SA–induced coagulopathy
HI-SA infusion led to pronounced prolongation of the aPTT and PT (Figure 2A-B); activation of FXII, FXI, and prekallikrein (Figure 2C-F, 2I-M); consumptive coagulopathy characterized by a decrease in fibrinogen level (Figure 3A) and platelet count (Figure 3B); microvascular thrombosis (Figure 3C-D); and activation of the fibrinolytic pathway (Figure 3E-H). aPTT was already elevated at 2 hours, and plasma levels of protease-serpin complexes (FXIIa-AT, FXIIa-C1INH, Kal-AT, Kal-C1INH, FXIa-AT, and FIXa-AT) increased early within 2 to 6 hours after bacterial challenge (Figure 2I-J). HI-SA also induced activation of the kallikrein-kinin system, as indicated by HK consumption and increased amount of cleaved HK during the first 4 hours’ postchallenge (Figure 2G-H).
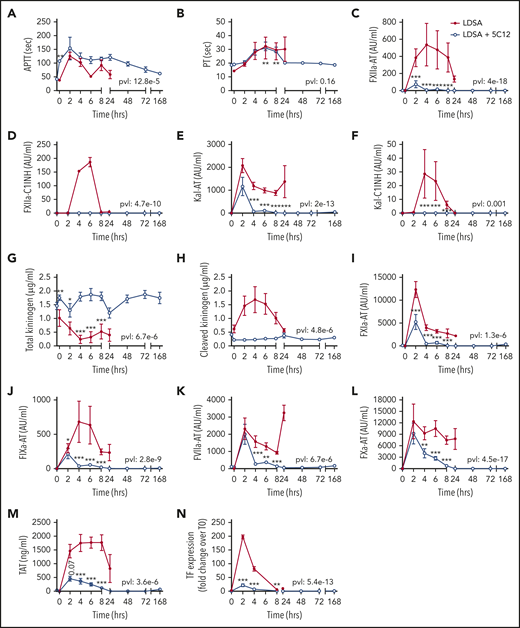
Effect of 5C12 anti-FXII antibody treatment on the activation of coagulation and coagulopathy in baboons challenged with a lethal dose of HI-SA (LDSA). Time course dynamics of aPTT (A), PT (B), complexes of FXIIa- AT (C), FXIIa-C1 inhibitor (D), kallikrein-AT (E), kallikrein-C1 inhibitor (F), total kininogen (G), cleaved kininogen (H), FXIa-AT (I), FIXa-AT (J), FVIIa-AT (K), FXa-AT (L), TAT (M), and TF mRNA in leukocytes (N) in HI-SA–challenged animals, with or without 5C12 antibody treatment. Same time points are compared between the treated and untreated baboons by using the generalized least squares (gls) function as described in the Statistical analysis section. *P < .05; **P < .01; ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant.
Effect of 5C12 anti-FXII antibody treatment on the activation of coagulation and coagulopathy in baboons challenged with a lethal dose of HI-SA (LDSA). Time course dynamics of aPTT (A), PT (B), complexes of FXIIa- AT (C), FXIIa-C1 inhibitor (D), kallikrein-AT (E), kallikrein-C1 inhibitor (F), total kininogen (G), cleaved kininogen (H), FXIa-AT (I), FIXa-AT (J), FVIIa-AT (K), FXa-AT (L), TAT (M), and TF mRNA in leukocytes (N) in HI-SA–challenged animals, with or without 5C12 antibody treatment. Same time points are compared between the treated and untreated baboons by using the generalized least squares (gls) function as described in the Statistical analysis section. *P < .05; **P < .01; ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant.
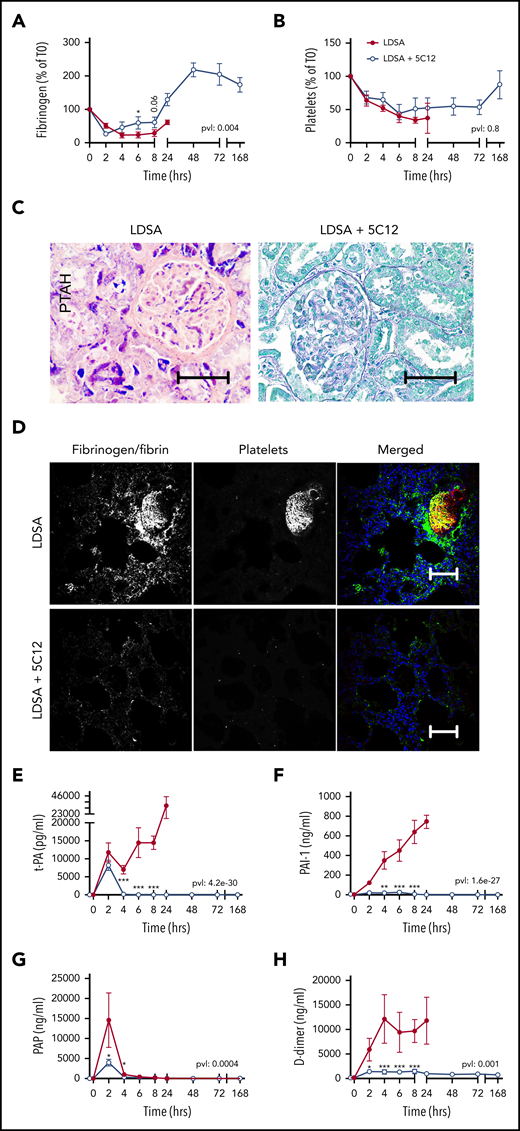
Effect of 5C12 anti-FXII antibody on fibrinogen and platelet consumption, fibrin deposition in the tissues, and activation of fibrinolysis after challenge with a lethal dose of HI-SA (LDSA). (A-B) Time course dynamics of plasma fibrinogen (A) and platelet numbers (B). (C) Phosphotungstic acid–hematoxylin (PTAH) staining of kidney sections of animals challenged with LDSA, not treated (left) or treated with the 5C12 antibody (right). (D) Immunofluorescence staining for fibrin/fibrinogen (left) and platelet marker CD41 (middle) in the lung of baboons challenged with LDSA that were either not treated (top) or treated with the 5C12 antibody (bottom); merged color images are shown in the right panels. Nuclei were stained with To-PRO-3 (blue). Scale bars, 100 µm. Confocal images were acquired by using a Nikon Eclipse TE2000-U microscope equipped with a Nikon C1 scanning head; the Nikon EZ-C1 software (version 3.80) was used for image acquisition. (E-H) Time course changes of fibrinolysis markers in the plasma: t-PA (E), PAI-1 (F), plasmin-antiplasmin (PAP) complexes (G), and D-dimer (H). Same time points are compared between the treated and untreated baboons by using the generalized least squares (gls) function. *P < .05; ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant.
Effect of 5C12 anti-FXII antibody on fibrinogen and platelet consumption, fibrin deposition in the tissues, and activation of fibrinolysis after challenge with a lethal dose of HI-SA (LDSA). (A-B) Time course dynamics of plasma fibrinogen (A) and platelet numbers (B). (C) Phosphotungstic acid–hematoxylin (PTAH) staining of kidney sections of animals challenged with LDSA, not treated (left) or treated with the 5C12 antibody (right). (D) Immunofluorescence staining for fibrin/fibrinogen (left) and platelet marker CD41 (middle) in the lung of baboons challenged with LDSA that were either not treated (top) or treated with the 5C12 antibody (bottom); merged color images are shown in the right panels. Nuclei were stained with To-PRO-3 (blue). Scale bars, 100 µm. Confocal images were acquired by using a Nikon Eclipse TE2000-U microscope equipped with a Nikon C1 scanning head; the Nikon EZ-C1 software (version 3.80) was used for image acquisition. (E-H) Time course changes of fibrinolysis markers in the plasma: t-PA (E), PAI-1 (F), plasmin-antiplasmin (PAP) complexes (G), and D-dimer (H). Same time points are compared between the treated and untreated baboons by using the generalized least squares (gls) function. *P < .05; ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant.
Treatment with 5C12 markedly decreased HI-SA–induced FXII activation, as indicated by the reduction in FXIIa-AT and FXIIa-C1INH complex formation (Figure 2C-D), and decreased activation of the kallikrein-kinin system (Figure 2E-H) compared with the control animals. FXII inhibition also reduced the formation of FVIIa-AT complexes (Figure 2K) and TF expression on circulating leukocytes (Figure 2N). 5C12 treatment led to decreased FXa-AT (Figure 2L) and TAT (Figure 2M), suggesting a general inhibition of coagulation.
Fibrinogen consumption (Figure 3A), decreased platelet counts (Figure 3B), and the aforementioned increase in clotting times all indicate the development of consumptive coagulopathy after HI-SA infusion, typical of some forms of bacterial or viral sepsis.23 Microscopic analysis of select vital organs, such as kidney (Figure 3C) and lung (Figure 3D) harvested from untreated control animals revealed increased fibrin deposition and the presence of fibrin- and platelet-rich microthrombi. As is common in sepsis DIC, HI-SA challenge induced t-PA release (Figure 3E) paralleled by strong induction of PAI-1 (Figure 3F). Peak plasmin generation (Figure 3G) was restricted to the first 2 hours, suggesting the suppression of fibrinolysis by PAI-1 during the 4 to 24 hours’ postchallenge period. D-dimer (Figure 3H) remained elevated during the entire time course of clinical deterioration in the HI-SA–challenged control animals. All 5C12-treated animals displayed decreased levels of the fibrinolytic markers.
Effect of 5C12 on the intravascular microenvironment and blood cells
HI-SA challenge led to an early increase in RBC and hematocrit (Figure 4A-B), suggesting hemoconcentration. This may reflect increased capillary permeability induced by bradykinin generated after HK cleavage (Figure 2H). Bacterial challenge also induced a rapid drop in circulating leukocytes, especially neutrophils and lymphocytes, which normalized within 24 hours’ postchallenge (Figure 4C-F). Immunofluorescence staining for neutrophil elastase in the lung indicated a striking accumulation of neutrophils (Figure 4G). Neutrophil activation was reflected in the marked increase of MPO released into the plasma (Figure 4H).
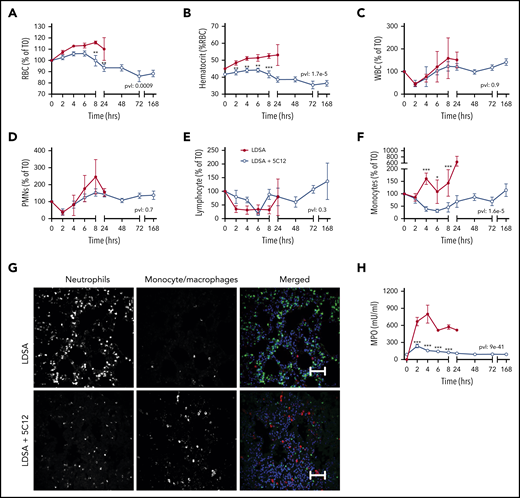
Effects of 5C12 anti-FXII antibody treatment on blood cell counts, leukocyte accumulation in tissues, and neutrophil activation markers in baboons following lethal challenge with HI-SA (LDSA). (A-F) Time course dynamics of RBCs (A), hematocrit (B), white blood cells (WBCs) (C), polymorphonuclear neutrophils (PMNs) (D), lymphocytes (E), and monocytes (F) in animals infused with LDSA only or infused with LDSA and treated with 5C12 (LDSA + 5C12). (G) Immunofluorescence staining for neutrophil elastase (left), and CD68 (middle) as markers for neutrophils and monocytes/macrophages, respectively, in lungs of baboons challenged with LDSA, without (top) or with 5C12 treatment (LDSA + 5C12; bottom). Merged color images are shown in the right panels. Nuclei were stained with To-PRO-3 (blue). Scale bars, 100 µm. Confocal images were captured by using a Nikon Eclipse TE2000-U microscope equipped with a Nikon C1 scanning head; the Nikon EZ-C1 software (version 3.80) was used for image acquisition. (H) Time course changes of MPO activity in plasma. Same time points are compared between the treated and untreated baboons by using the generalized least squares (gls) function. *P < .05; **P < .01; ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant.
Effects of 5C12 anti-FXII antibody treatment on blood cell counts, leukocyte accumulation in tissues, and neutrophil activation markers in baboons following lethal challenge with HI-SA (LDSA). (A-F) Time course dynamics of RBCs (A), hematocrit (B), white blood cells (WBCs) (C), polymorphonuclear neutrophils (PMNs) (D), lymphocytes (E), and monocytes (F) in animals infused with LDSA only or infused with LDSA and treated with 5C12 (LDSA + 5C12). (G) Immunofluorescence staining for neutrophil elastase (left), and CD68 (middle) as markers for neutrophils and monocytes/macrophages, respectively, in lungs of baboons challenged with LDSA, without (top) or with 5C12 treatment (LDSA + 5C12; bottom). Merged color images are shown in the right panels. Nuclei were stained with To-PRO-3 (blue). Scale bars, 100 µm. Confocal images were captured by using a Nikon Eclipse TE2000-U microscope equipped with a Nikon C1 scanning head; the Nikon EZ-C1 software (version 3.80) was used for image acquisition. (H) Time course changes of MPO activity in plasma. Same time points are compared between the treated and untreated baboons by using the generalized least squares (gls) function. *P < .05; **P < .01; ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant.
Treatment with 5C12 prevented the increase in RBC and hematocrit, and slightly yet significantly reduced the consumption of neutrophils. 5C12 treatment blunted MPO activity in plasma, which suggests decreased levels of neutrophil activation and degranulation. Immunostaining of tissues collected from treated animals revealed no significant pathology in the lung after 7 days’ postbacterial infusion and full clinical recovery (Figure 4G).
Effect of 5C12 on organ function and inflammation after HI-SA infusion in baboons
HI-SA challenge induced progressive multiorgan dysfunction leading to organ failure in all untreated animals. Baboons exhibited signs of early cardiorespiratory distress, with a drop in mean systemic arterial pressure, tachycardia, tachypnea, and a transient increase in body temperature (supplemental Figure 2A-D). 5C12 treatment blunted the drop in blood pressure and prevented the rise in body temperature.
Untreated animals gradually developed liver, pancreas, and kidney dysfunction or damage, as indicated by increased serum blood urea nitrogen, creatinine, alanine aminotransferase, and pancreatic amylase levels (Figure 5A-D). After an initial increase in blood glucose at 2 hours, animals became hypoglycemic within 4 to 8 hours’ postchallenge, suggesting metabolic disturbance (Figure 5E). Baboons treated with 5C12 were protected from acute organ damage, and hypoglycemia was less severe.
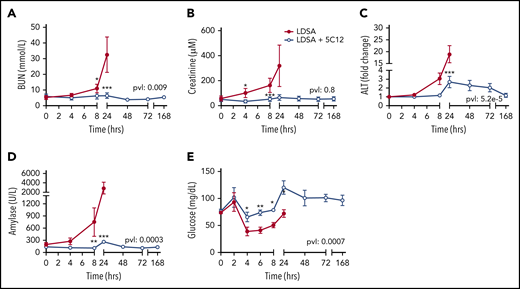
Effects of 5C12 anti-FXII antibody treatment on renal, hepatic, and pancreatic damage and function parameters in baboons infused with a lethal dose of HI-SA (LDSA). Time course changes of plasma levels of kidney function biomarkers: blood urea nitrogen (BUN) (A), and creatinine (B), liver injury biomarker alanine aminotransferase (ALT) (C), pancreas injury biomarker amylase (D), and glucose regulation (E). Same time points are compared between the control baboons (LDSA) and the 5C12-treated animals (LDSA + 5C12) using the generalized least squares (gls) function. *P < .05; **P < .01; ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant.
Effects of 5C12 anti-FXII antibody treatment on renal, hepatic, and pancreatic damage and function parameters in baboons infused with a lethal dose of HI-SA (LDSA). Time course changes of plasma levels of kidney function biomarkers: blood urea nitrogen (BUN) (A), and creatinine (B), liver injury biomarker alanine aminotransferase (ALT) (C), pancreas injury biomarker amylase (D), and glucose regulation (E). Same time points are compared between the control baboons (LDSA) and the 5C12-treated animals (LDSA + 5C12) using the generalized least squares (gls) function. *P < .05; **P < .01; ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant.
Consistent with the profound organ damage, untreated control animals had highly elevated levels of circulating nucleosomes (Figure 6A), a marker of cell death. FXII inhibition significantly reduced plasma nucleosomes, reflecting organ preservation.
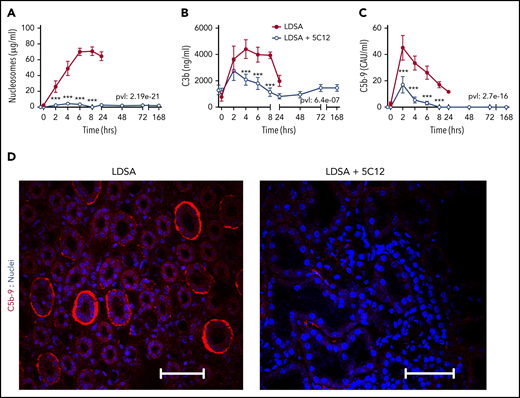
5C12 anti-FXII antibody treatment decreases complement activation after challenge with a lethal dose of HI-SA (LDSA). (A-C) Time course dynamics of plasma levels of nucleosomes, a cell death marker (A), and of complement activation markers C3b (B) and soluble C5b-9 (C). Same time points are compared between the control baboons (LDSA) and the 5C12-treated animals (LDSA + 5C12) using the generalized least squares (gls) function. *P < .05; **P < .01; ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant. (D) Immunofluorescence staining of C5b-9 terminal complement complex (red) in kidneys of baboons challenged with LDSA, without (left) or with 5C12 treatment (LDSA + 5C12; right). Nuclei were stained with To-PRO-3 (blue). Scale bars, 100 µm. Confocal images were captured by using a Nikon Eclipse TE2000-U microscope equipped with a Nikon C1 scanning head; the Nikon EZ-C1 software (version 3.80) was used for image acquisition.
5C12 anti-FXII antibody treatment decreases complement activation after challenge with a lethal dose of HI-SA (LDSA). (A-C) Time course dynamics of plasma levels of nucleosomes, a cell death marker (A), and of complement activation markers C3b (B) and soluble C5b-9 (C). Same time points are compared between the control baboons (LDSA) and the 5C12-treated animals (LDSA + 5C12) using the generalized least squares (gls) function. *P < .05; **P < .01; ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant. (D) Immunofluorescence staining of C5b-9 terminal complement complex (red) in kidneys of baboons challenged with LDSA, without (left) or with 5C12 treatment (LDSA + 5C12; right). Nuclei were stained with To-PRO-3 (blue). Scale bars, 100 µm. Confocal images were captured by using a Nikon Eclipse TE2000-U microscope equipped with a Nikon C1 scanning head; the Nikon EZ-C1 software (version 3.80) was used for image acquisition.
HI-SA induced robust complement activation, as shown by increased circulating C3b (Figure 6B) and soluble terminal complement complex C5b-9 (Figure 6C). Treatment with 5C12 markedly reduced both C3b and sC5b-9. Immunofluorescence staining for C5b-9 in the kidney showed increased staining, especially in peritubular locations in untreated animals (Figure 6D).
HI-SA infusion provoked systemic inflammation in untreated animals characterized by high levels of plasma cytokines, including tumor necrosis factor (TNF), interleukin (IL)-1β, IL-6, IL-8, IL-10, granulocyte-macrophage colony-stimulating factor; 5C12 treatment reduced the cytokine storm (Figure 7).
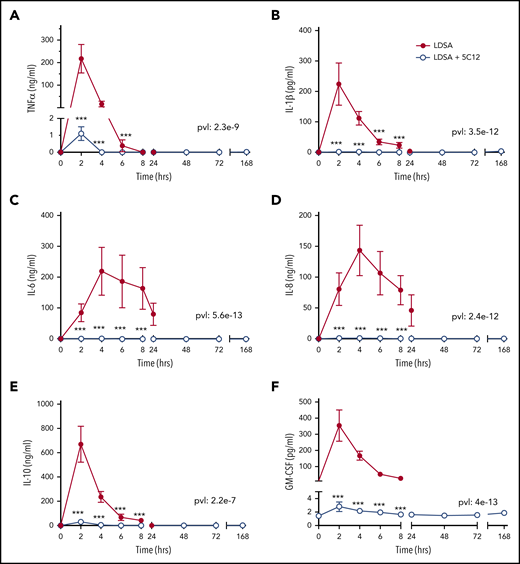
5C12 antibody treatment prevents or attenuates the increase of pro-inflammatory cytokines in baboons challenged with a lethal dose of HI-SA (LDSA). Time course changes of plasma levels of TNF-α (A), IL-1β (B), IL-6 (C), IL-8 (D), IL-10 (E), and granulocyte-macrophage colony-stimulating factor (GM-CSF) (F) in HI-SA–challenged animals, with or without 5C12 treatment. Data are presented as mean ± standard error of the mean. Same time points are compared between the 2 experimental groups by using the generalized least squares (gls) function. ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant.
5C12 antibody treatment prevents or attenuates the increase of pro-inflammatory cytokines in baboons challenged with a lethal dose of HI-SA (LDSA). Time course changes of plasma levels of TNF-α (A), IL-1β (B), IL-6 (C), IL-8 (D), IL-10 (E), and granulocyte-macrophage colony-stimulating factor (GM-CSF) (F) in HI-SA–challenged animals, with or without 5C12 treatment. Data are presented as mean ± standard error of the mean. Same time points are compared between the 2 experimental groups by using the generalized least squares (gls) function. ***P < .001. The overall P value (pvl) describing the difference between the 2 curves is shown on each graph. P < .05 is considered significant.
HI-SA–induced organ damage was confirmed by histopathology in select organs collected at necropsy. Although postmortem histopathologic evaluation cannot be temporally compared between animals because of varying times of death, essentially normal histology of survivors suggested either healing without residual damage or lack of severe damage to start with, consistent with the organ function and damage data collected during the time course of the experiments.
Kidneys of nontreated baboons euthanized at 10 to 34 hours’ postchallenge showed glomerular and peritubular fibrin deposition and microthrombosis, and widespread tubular necrosis characterized by nuclear pyknosis and karyolysis (supplemental Figure 3A). Livers exhibited slightly swollen hepatocytes and marked leukocyte infiltration in peri-sinusoidal spaces (supplemental Figure 3B). Lungs also showed leukocyte infiltration into the interalveolar wall, leukocyte aggregation in small to medium vessels, and marked congestion (supplemental Figure 3C). Spleens were congested and showed prominent leukocyte infiltration of the red pulp, as well as lymphoid follicular apoptosis in the central white pulp (supplemental Figure 3D). Organs from all 5C12-treated animals displayed no residual damage, indicating full recovery at day 7 postchallenge.
Discussion
This study showed that FXII plays a substantial role in promoting pathologic inflammatory host responses in baboons challenged with a lethal dose of HI-SA. This work builds on previous animal models of infection and/or inflammation by our group and others, including the study of the effect of an anti-FXI antibody in the same baboon model.12 Our results further solidify the roles of FXI and FXII in inflammation and coagulation activation during the host response to a severe challenge to homeostasis as produced by infusing heat-inactivated gram-positive bacteria.
Despite potent antimicrobial agents and improved supportive care, sepsis remains among the leading causes of death worldwide.1 After >150 clinical trials of experimental treatments, no new strategies proved safe and effective to interrupt the complex and often deadly host response to severe infection. Early recognition of the interplay between coagulation and inflammation drove the investigation of potential targets, including anticoagulants (eg, heparins, activated protein C, AT, TF pathway inhibitor, recombinant thrombomodulin), inhibitors of bacterial products, and pro-inflammatory cytokines (eg, TNF, IL-6). Clinical trial data showing little to no benefit or concerning safety signals deterred further consideration of these agents.24,25 Barriers to the identification of safe and effective therapeutic agents specific to sepsis include uncertainties about appropriate timing of novel drug administration and variability in host response to distinct pathogens. Equally plausible is that a therapeutic target that dampens coagulation and inflammation to a safe but effective degree is hard to identify. Based on our study, we posit that FXII of the contact activation system could achieve this ideal balance in certain cases. Key to this hypothesis is the clinical finding that humans deficient in FXII experience neither abnormal bleeding nor any noticeable immune deficiency, despite the position of FXII at the intersection of several critical biochemical pathways.26 Based on published animal and observational human data, the potential safety of pharmacologic FXII inhibition in humans looks encouraging.17,27
FXII is a 80-kDa precursor of the serine protease FXIIa, which can be activated by kallikrein, FXIa, or through autoactivation in the presence of negatively charged surfaces and HK.13 During infections, such charged surfaces may include: pathogen surfaces, components, and bacterial wall products, bacterial or platelet polyphosphate, subendothelial collagen and laminin, misfolded or denatured proteins, neutrophils, glycosaminoglycans, and nucleic acids released from dying cells, viruses, or from neutrophil extracellular traps.28 However, these mechanisms have not yet been well characterized in vivo.
Upon activation, FXII is cleaved, resulting in a heavy chain connected to a light chain via a disulfide bond.10,29 The resulting FXIIa protease is positioned at the nexus of the kinin-kallikrein, complement, coagulation, and fibrinolytic pathways. With respect to the kallikrein-kinin and complement systems, FXIIa can activate pre-kallikrein to kallikrein, which can then convert FXII to FXIIa in a feedback mechanism. Moreover, FXIIa can activate the complement cascade via activation of the C1qrs complex,30 whereas kallikrein also activates factors B, C3, and C5.31 FXIIa also plays a role in coagulation by activating FXI to FXIa, which accelerates production of thrombin that drives platelet activation and fibrin formation, and exerts effects on a variety of cells.29,32 When FXIIa is cleaved after Arg334, the resulting β-FXIIa can still generate kallikrein, which further cleaves HK to release bradykinin, a systemic vasoregulatory and inflammatory mediator.33,34 Nevertheless, β-FXIIa is unable to activate FXI and thus does not substantially participate in the coagulation system.29
Our data show that inhibiting FXIIa using 5C12 abrogates the increase in plasma markers of inflammation or cell damage and inhibits the activation of coagulation after lethal challenge with HI-SA. Specifically, no substantial increases in circulating TNF, IL-1β, IL-6, IL-8, IL-10, granulocyte-macrophage colony-stimulating factor, nucleosomes, or MPO levels were seen after treatment with 5C12 in our primate model of systemic inflammatory response. Notably, 5C12-mediated FXIIa inhibition strongly decreased TNF levels; conversely, this was not observed in our previous study using the anti-FXI monoclonal antibody 3G3, which inhibits the activation of FXI by FXIIa and the reciprocal FXIa-mediated FXII activation.12 Indeed, 5C12 more potently decreased the serum and physiological markers of cytokine storm compared with 3G3.12 However, similarly to 3G3,12 5C12 treatment reduced activity of all 3 arms of the traditional coagulation pathways: the intrinsic pathway shown by substantial reduction in FXIIa-AT and FXIIa-C1INH complexes; the extrinsic pathway via lower plasma FVIIa-AT complexes and reduced TF expression by circulating leukocytes; and the common pathway shown by reduced FXa-AT and TAT complexes. The inhibitory effects on levels of TF and the fibrinolysis inhibitor PAI-1 were more pronounced in animals treated with 5C12 than with 3G3,12 likely reflecting the robust decrease of TNF, IL-6, and IL-1β observed with 5C12, as these cytokines are known inducers of both TF35 and PAI-1.36
Targeting FXII as a sepsis therapy in baboon models was attempted >2 decades ago.5-7 Although direct comparison between our data and the historic studies using the anti–β-FXIIa C6B7 antibody cannot be made due to experimental and molecular target differences, there are several similarities and divergences between these studies. Both C6B7 treatment in E coli sepsis and 5C12 treatment in HI-SA challenge decreased activation of complement and fibrinolysis, reduced production of several proinflammatory cytokines, and blunted neutrophil activation.7 Because neutrophil accumulation is a landmark event seen in both clinical and experimental sepsis, including baboon models of localized infection,37 and may be partially explained by the intravenous delivery of bacterial PAMPs, it is of note that a role for FXII in upregulating neutrophil function and accumulation was recently highlighted in mice.38 Although these and other similarities were observed between the studies, several stark differences were noted. Only the anticoagulant anti–FXII/FXIIa 5C12 prevented the increase in TNF, TF, and PAI-1 levels. Moreover, C6B7 did not protect against sepsis DIC,5 possibly because the coagulopathy in gram-negative sepsis is driven mainly by lipopolysaccharide-induced TF expression. Inhibition of FXII or FXI12 activation in our model of gram-positive challenge blunted the activation of coagulation, likely resulting from the fact that the gram-positive PAMP peptidoglycan is an inducer of both intrinsic and extrinsic coagulation.15 Moreover, when comparing 5C12 and C6B7 in an aPTT assay, we confirmed that the anticoagulant 5C12 inhibited plasma coagulation at significantly lower concentrations than the C6B7 antibody. Consequently, C6B7 may not have offered sufficient inhibition of procoagulant FXIIa generation or activity in the early sepsis studies. Finally, both treatments improved physiological parameters, including blood pressure, but only 5C12 saved all challenged baboons, whereas C6B7 extended the life of animals but only saved 1 of 5 treated baboons.
We acknowledge limitations to our study, including the small number of test animals, use of heat-inactivated rather than live bacteria, and administration of 5C12 antibody before bacterial challenge. Our experiments indicate that prophylactic inhibition of FXII attenuated the acute effects of S aureus–derived PAMPs. Exposure to S aureus material would predictably occur in bacteremic patients when antibiotic treatment causes rapid bacteriolysis with release of cell debris into the circulation. Therefore, FXII inhibition before or at the time of initiation of antibiotic treatment could have a beneficial clinical effect. Use of PAMPs, but not live S aureus challenge, sacrificed some relevance in our disease model; however, it allowed us to standardize the dose of the lethal challenge and to avoid the effects of S aureus exotoxins that interfere with coagulation and fibrinolysis pathways.39 Chiefly important, we managed to achieve reproducible data and improve our ability to detect efficacy or safety signals using a small number of animals. In the future, using live bacteria and administration of an FXII inhibitor and antibiotics after the setup of a severe infection or sepsis could provide information on whether treatment of sepsis using FXIIa inhibitors would also generate outcome benefit.
In summary, we showed that FXII plays a significant role in promoting coagulation and inflammation in a baboon model of lethal HI-SA challenge, and that inhibiting FXII dampens these processes enough to reduce mortality and morbidity. FXII remains an attractive target for novel sepsis prevention or mitigation efforts in severe infections in which FXII activity has a pathogenic role.
Presented in abstract form at the International Society on Thrombosis and Hemostasis 2019 Congress, Melbourne, Australia, 6-10 July 2019.
Requests for original data may be submitted to the corresponding author (Florea Lupu; e-mail: florea-lupu@omrf.org).
The online version of the article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
There is a Blood Commentary on this article in this issue.
Acknowledgments
The technical support of Monalisa Choudhury is kindly acknowledged.
This work was supported by grants from the National Institutes of Health, National Institute of General Medical Sciences (GM116184 [O.J.T.M. and F.L.], GM121601, GM122775, and P30GM114731 [F.L.]), National Heart, Lung, and Blood Institute (HL101972 [O.J.T.M.], HL128016, and HL106919 [A.G., C.U.L., and E.I.T.], HL151367 [J.J.S.]), and the National Institute of Allergy and Infectious Diseases (AI157037 [O.J.T.M. and F.L.], U19AI062629 [F.L.] and AI088937 [A.G., C.U.L., and E.I.T.]). The Specific Pathogen Free Baboon Research Resource was supported by the National Institutes of Health, Office of the Director (P40OD024628, J.H.S.).
Authorship
Contribution: F.L., J.H.S., R.S., G.R., and R.S.K. performed animal experiments; C.U.L., S.S., C.L., G.R., R.S.K., and R.S. performed assays; A.G., C.U.L., D.G., E.I.T., and M.W. generated and characterized FXII antibodies; C.G. performed statistical analysis of the data; S.S. provided valuable reagents; A.G., F.L., and O.J.T.M. conceived and designed the study; O.J.T.M. and F.L. supervised the study; A.G., F.L., C.L., D.G., C.P., T.C.L.K., J.J.S., S.R.O., C.U.L., E.I.T., O.J.T.M., and R.S. wrote and edited the manuscript; and all authors analyzed the data and reviewed, read, and approved the final manuscript.
Conflict-of-interest disclosure: A.G., M.W., C.U.L., E.I.T., Aronora, and Oregon Health & Science University may have a financial interest in the results of this study. J.J.S. serves as a medical consultant for Aronora, Inc. The remaining authors declare no competing financial interests.
Correspondence: Florea Lupu, Cardiovascular Biology Research Program, Oklahoma Medical Research Foundation, 825 NE 13th St, Oklahoma City, OK 73104; e-mail: florea-lupu@omrf.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal