

Key Points
MDM2 inhibition sensitized AML cells to allogeneic T-cell–mediated cytotoxicity through the restoration of TRAIL-R1/2 and MHC-II production.
MDM2 inhibitor-mediated effects were p53-dependent because p53 knockdown abolished TRAIL-R1/2 and MHC-II upregulation.

Abstract
Patients with acute myeloid leukemia (AML) often achieve remission after allogeneic hematopoietic cell transplantation (allo-HCT) but subsequently die of relapse driven by leukemia cells resistant to elimination by allogeneic T cells based on decreased major histocompatibility complex II (MHC-II) expression and apoptosis resistance. Here we demonstrate that mouse-double-minute-2 (MDM2) inhibition can counteract immune evasion of AML. MDM2 inhibition induced MHC class I and II expression in murine and human AML cells. Using xenografts of human AML and syngeneic mouse models of leukemia, we show that MDM2 inhibition enhanced cytotoxicity against leukemia cells and improved survival. MDM2 inhibition also led to increases in tumor necrosis factor-related apoptosis-inducing ligand receptor-1 and -2 (TRAIL-R1/2) on leukemia cells and higher frequencies of CD8+CD27lowPD-1lowTIM-3low T cells, with features of cytotoxicity (perforin+CD107a+TRAIL+) and longevity (bcl-2+IL-7R+). CD8+ T cells isolated from leukemia-bearing MDM2 inhibitor-treated allo-HCT recipients exhibited higher glycolytic activity and enrichment for nucleotides and their precursors compared with vehicle control subjects. T cells isolated from MDM2 inhibitor-treated AML-bearing mice eradicated leukemia in secondary AML-bearing recipients. Mechanistically, the MDM2 inhibitor-mediated effects were p53-dependent because p53 knockdown abolished TRAIL-R1/2 and MHC-II upregulation, whereas p53 binding to TRAILR1/2 promotors increased upon MDM2 inhibition. The observations in the mouse models were complemented by data from human individuals. Patient-derived AML cells exhibited increased TRAIL-R1/2 and MHC-II expression on MDM2 inhibition. In summary, we identified a targetable vulnerability of AML cells to allogeneic T-cell–mediated cytotoxicity through the restoration of p53-dependent TRAIL-R1/2 and MHC-II production via MDM2 inhibition.
Introduction
Allogeneic hematopoietic cell transplantation (allo-HCT) is a curative therapy for acute myeloid leukemia (AML). However, a significant proportion of patients experience relapses, and leukemia relapse is responsible for 57% of the mortality of patients undergoing allo-HCT.1,2 Mechanisms that lead to escape of AML cells from immune-mediated elimination include, among others,3 the loss of mismatched HLA,4 immune checkpoint ligand upregulation,5 reduced interleukin-15 (IL-15) production,6,7 IDO1 (indoleamine 2,3-dioxygenase 1) expression,8 leukemia-derived lactic acid release,9 and loss of surface expression of human leukocyte antigen (HLA) -DR, -DQ, and -DP on leukemia cells because of downregulation of the HLA class II regulator CIITA5,10 and a PRC2-dependent reduction in chromatin accessibility.11 These data suggest that therapeutic approaches that increase MHC-II expression could enhance antileukemia immunity after allo-HCT. A cellular therapy concept for relapse treatment is donor lymphocyte infusions (DLIs) alone or combined with pharmacological approaches.12,13 A retrospective analysis on response to DLIs reported complete response rates of 17%,14 and another large retrospective, multicenter study reported a 2-year overall survival on DLI treatment of 20% vs 9% on chemotherapy-only treatment.13 These reports indicate that donor T cells can exert graft-versus-leukemia (GVL) effects in a fraction of patients, but also that strategies to improve these GVL effects are urgently needed. Current pharmacological approaches for the prevention and treatment of AML relapse include the use of FLT3 kinase inhibitors,15,16 immune checkpoint inhibitors,17,18 demethylating agents,12,19 BCL-2 inhibitors,20 and others.21 Mouse-double-minute-2(MDM2) inhibitors22,23 can induce p53-dependent apoptosis in AML; however, their role in the setting after allo-HCT has not been evaluated to date.
The MDM2 protein functions as an ubiquitin ligase that recognizes the N-terminal transactivation domain of p53. This inhibits p53 transcriptional activation and facilitates proteasomal p53 degradation. MDM2 overexpression, in cooperation with oncogenic Ras, promotes the transformation of primary rodent fibroblasts, whereas MDM2 inhibition increases p53 activity.23 Besides its antioncogenic effect, p53 can also increase the expression of certain immune-related genes, and TP53 abnormalities correlate with immune infiltration in the bone marrow (BM)24 and associate with response to immunotherapy in patients with AML.25 Previous studies have shown that p53 can activate the type-I interferon(IFN) pathway in the setting of antiviral immunity.26 In addition, p53 increases MHC-I expression on pancreatic cancer cells.27 Recent studies have shown that the ubiquitin ligase MDM2 sustains STAT5 stability in T cells that enhance T-cell–mediated antitumor immunity in solid tumors.28 Since MDM2 inhibition with RG-7112 causes upregulation of intracellular MDM2 as a positive feedback loop in response to the inhibition,29 the increase of MDM2 in T cells can directly enhance their antitumor activity.28 In addition, p53 can induce a viral mimicry pathway and tumor inflammation signature genes in patients with melanoma.30 These data suggest that MDM2 targeting may be an attractive strategy to improve immunotherapy in solid tumors.
To render AML cells more susceptible to donor T-cell–mediated cytotoxicity after allo-HCT, we aimed to restore p53 activity by blocking MDM2. We observed that MDM2 inhibition increased MHC-II expression and improved survival using xenografts of human AML and syngeneic mouse models of leukemia. MDM2 inhibition also led to increases in tumor-necrosis factor related apoptosis-inducing ligand receptor-1 and -2 (TRAIL-R1/2) on leukemia cells and higher frequencies of nonexhausted CD8+ T cells, with features of cytotoxicity that eradicated leukemia in secondary AML-bearing recipients. Mechanistically, the MDM2-mediated effects were p53-dependent because p53 knockdown abolished TRAIL-R1/2 and MHC-II upregulation, whereas p53 binding to TRAILR1/2 promotors increased upon MDM2 inhibition. These findings indicate that MDM2 inhibition is a novel strategy to enhance the sensitivity of AML cells to T cell–mediated cytotoxicity after allo-HCT, through the restoration of p53-dependent TRAIL-R1/2 and MHC-II production.
Materials and methods
Human sample collection
Human sample collection and analysis were approved by the Institutional Ethics Review Board of the Medical Center, University of Freiburg, Freiburg, Germany (protocol number 100/20). Written informed consent was obtained from each patient. All analyses of human data were carried out in compliance with relevant ethical regulations. The characteristics of patients are listed in the supplemental Tables in the data supplement (available on the Blood Web site).
Mice
Animal protocols were approved by the animal ethics committee Regierungspräsidium Freiburg, Freiburg, Germany (protocol numbers: G17-093 and G-20/96).
GVL mouse models
GVL experiments were performed as previously described.6 Briefly, recipients were IV-injected with leukemia cells and donor BM cells (when indicated) after (sub) lethal irradiation using a 137Cs source. CD3+ T cells were isolated from donor spleens or, when indicated, peripheral blood of healthy donors and enriched by negative selection using Pan T Cell Isolation Kit II (Miltenyi Biotech) as previously reported.31 Obtained T-cell purity was ≥90% as assessed by flow cytometry. Allogeneic CD3+ T cells were given on day 2 after BM transplantation. We assessed graft-versus-host disease(GVHD) severity by histology as previously reported.32
AMLMLL-PTD FLT3-ITD leukemia model
For the AMLMLL-PTD FLT3-ITD leukemia model, C57BL/6 recipients were transplanted with 5 × 103 AMLMLL-PTD FLT3-ITD cells and 5 × 106 BALB/c BM cells via IV after lethal irradiation with 12 Gy in 2 equally split doses performed 4 hours apart. A total of 3 × 105 BALB/c (allogeneic model) splenic CD3+ T cells were introduced via IV on day 2 after initial transplantation, as previously reported.31,32
WEHI-3B leukemia model
For the WEHI-3B leukemia model, BALB/c recipients were transplanted with 5 × 103 AML (WEHI-3B) cells and 5 × 106 C57/BL6 BM cells via IV after lethal irradiation with 10 Gy in 2 equally split doses performed 4 hours apart. A total of 2 × 105 C57/BL6 (allogeneic model) splenic CD3+ T cells were introduced via IV on day 2 after initial transplantation.
OCI-AML3 xenograft model
For the OCI-AML3 xenograft model,33Rag2−/−Il2rγ−/− recipients were transplanted with 2 × 105 OCI-AML3 (wild type [WT] or TRAIL-R2 knockout) or 1 x 106 OCI-AML3 (WT or p53-deficient) cells via IV as indicated after sublethal irradiation with 5 Gy. A total of 1 × 105 human CD3+ T cells isolated from peripheral blood of healthy donors were introduced via IV on day 2 after initial transplantation.
Primary human AML xenograft model
For the primary human AML xenograft model,33Rag2−/−Il2rγ−/− recipients were used. Primary human AML cells were isolated by FICOLL density centrifugation and depleted from CD3+ cells by magnetic separation. A total of 10 × 106 CD3+-depleted primary human AML cells were transplanted via IV after sublethal irradiation with 5 Gy. A total of 5 × 104 human CD3+ T cells isolated from peripheral blood of healthy donors were introduced via IV on day 2 after initial transplantation.
MOLM-13 AMLFLT3 ITD xenograft model
Rag2−/−Il2rγ−/− recipients were transplanted with 0.1 × 106 MOLM-13Luc cells via IV after sublethal irradiation with 3.5 Gy 1 day prior.19 A total of 1 × 105 human CD3+ T cells per mouse isolated from peripheral blood of healthy donors were introduced via IV on day 2 after initial transplantation.
Statistical analysis
For the sample size in the murine GVL survival experiments, a power analysis was performed. A sample size of at least n = 8 per group was determined by 80% power to reach a statistical significance of 0.05 to detect an effect size of ≥1.06. Differences in animal survival (Kaplan-Meier survival curves) were analyzed by the Mantel-Cox test. The experiments were performed in a nonblinded fashion. For statistical analysis, an unpaired t test (2-sided) was applied. Data are presented as mean and standard error of the mean (error bars). Differences were considered significant when P < .01.
Results
MDM2 inhibition leads to a p53-dependent increase of MHC class I and II expression, enhances cytotoxicity against leukemia cells, and improves survival in multiple GVL mouse models
To test the on-target activity of the MDM2 inhibitor RG-7112, we studied the concentrations of p53 as the MDM2 target gene in primary human leukemia cells. MDM2 inhibition increased p53 (supplemental Figure 1A-C; supplemental Table 1 in the data supplement) in primary human AML cells collected from patients at primary diagnosis before treatment was started, indicating on-target activity of RG-7112. We also studied the concentrations of MDM2 itself, which was shown to be upregulated in an autoregulatory loop upon MDM2 inhibition.29 In human MOLM-13, OCI-AML3, and murine WEHI-3B, as well as 32D cells, we observed 2 MDM2 isoforms at 75 kDa and 90 kDa (supplemental Figure 2A-C), which had been previously described in other cell types.34 MDM2 inhibition increased the 75 kDa MDM2 isoform in the leukemia cell lines (supplemental Figure 2A-C), indicating on-target activity of RG-7112.
Based on the central role of HLA downregulation for leukemia relapse after allo-HCT,5,10 we studied the impact of MDM2 inhibition on HLA expression. We observed that MDM2 inhibition caused upregulation of HLA class I and II on human leukemia cells (OCI-AML3 cell line) on the mRNA level (Figure 1A) and protein level (Figure 1B-E). Consistent with the finding in human AML cells, MDM2 inhibition increased p53 and MHC-II expression by mouse AML (WEHI-3B) cells using 2 different MDM2 inhibitors (RG-7112 and HDM201) (supplemental Figure 3A-F). RG-7112 and HDM201 both inhibit p53 degradation by preventing MDM2 binding.
![MDM2 inhibition leads to increased MHC expression and improves survival in xenograft mouse models. (A) Microarray-based analysis of the expression level of HLA class I and II in OCI-AML3 cells after treatment with DMSO, RG-7112 (1 µM), or HDM-201 (200 nM) for 24 hours is shown as tile display from Robust Multichip Average (RMA) signal values, (Z-score), n = 6 biologically independent samples per group. The adjusted P value was calculated using the Benjamini & Hochberg method. *indicates significant regulation (P < .05) compared with DMSO. (B-C) Representative flow cytometry histograms show the mean fluorescence intensity (MFI) for (B) HLA-DR or (C) HLA-A,B,C expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours. (D) The bar diagram shows the MFI for HLA-DR expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours. Bar graphs show the mean ± standard error of the mean (SEM) from n = 5 to 6 independent experiments. P values were calculated using the 2-sided Student unpaired t test. (E) The bar diagram shows the MFI for HLA-A,B,C expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours. Bar graphs show the mean ± SEM from n = 5 to 6 independent experiments. P values were calculated using the 2-sided Student unpaired t test. (F-G) Percentage survival of Rag2−/−Il2rγ−/−-recipient mice after the transfer of primary human AML cells from 2 different patients is shown. As indicated, mice were injected with additional human CD3+ T cells (isolated from the peripheral blood of an HLA nonmatched healthy donor) and treated with either vehicle or MDM2 inhibitor RG-7112. Shown are n = 10 or 8 independent animals, and P values were calculated using the 2-sided Mantel-Cox test. (H) Percentage of specific lysis of isolated, CD3/28, and IL-2 expanded human T cells in contact with OCI-AML3 cells is shown. OCI-AML3 cells were pretreated with either DMSO or the MDM2-inhibitor RG-7112, and the E:T (effector [T-cell] to target [OCI-AML3 cell]) ratio was titrated between 10:1 and 1:1 as indicated. One representative experiment of 3 independent experiments is shown. (I) Representative Western blots showing the activation of caspase-3 and loading control (β-actin) in OCI-AML3 cells. OCI-AML3 cells exposed to DMSO or RG-7112 (1 µM) were cocultured with activated T cells at an E:T ratio of 10:1 for 4 hours. (J) The bar diagram indicates the ratio of the cleaved caspase-3 to pro–caspase-3 normalized to β-actin. The values were normalized to the T-cell–only group (set as “1”). (K-L) The graph shows fold change of MFI for (K) HLA-DR and (I) HLA-A,B,C expression on WT OCI-AML3 (WT) or p53 knockdown (p53 KD) OCI-AML3 cells after treatment with RG-7112 (2 µM) for 72 hours as mean ± SEM from n = 4 independent experiments. MFI of control-treated cells was set as 1.0. P values were calculated using a 2-sided Student unpaired t test. (M) Percentage survival of Rag2−/−Il2rγ−/−-recipient mice after transfer of human WT or p53 knockdown (p53 KD) OCI-AML3 cells are shown. As indicated, mice were injected with additional human T cells (isolated from the peripheral blood of an HLA nonmatched healthy donor) and treated with either vehicle or MDM2-inhibitor RG-7112. Shown are n = 10, 11, or 21 independent animals per group from ≥2 independent experiments, and P values were calculated using the 2-sided Mantel-Cox test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/10/10.1182_blood.2022016082/3/m_bloodbld2022016082f1.png?Expires=1770746544&Signature=ztL9WQYZRfYMBYZwgDuphhFImK0~WqGRL1kHQ~T1kMUnnFL3pRI2Oq1vX-FW01POHPzhdJsjII6TdK7irTss3-gyUu2odB9l7xwrBUcUHsBpmGj6eXUw0E1KzfGoT98W3zkuOjsafxUdhAKA4iUBhA5FChvkkM4lZMmFfVBSpyoS1xZ34KNgIorKr1WOm4ezMv4zdu9AOc4a3m2kNXOwyv2v8G0VIWwgR~0e9lJHSpdcD2vE0Z36bPBtX0j~y9mj6DJ6FiKplE5-u4WVXAEK5hj1sbNTiArRGItCX6bJVzago4uYVti09qZzXKQbcyFLUnJA4E-C1S37epZrBwheSg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MDM2 inhibition leads to increased MHC expression and improves survival in xenograft mouse models. (A) Microarray-based analysis of the expression level of HLA class I and II in OCI-AML3 cells after treatment with DMSO, RG-7112 (1 µM), or HDM-201 (200 nM) for 24 hours is shown as tile display from Robust Multichip Average (RMA) signal values, (Z-score), n = 6 biologically independent samples per group. The adjusted P value was calculated using the Benjamini & Hochberg method. *indicates significant regulation (P < .05) compared with DMSO. (B-C) Representative flow cytometry histograms show the mean fluorescence intensity (MFI) for (B) HLA-DR or (C) HLA-A,B,C expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours. (D) The bar diagram shows the MFI for HLA-DR expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours. Bar graphs show the mean ± standard error of the mean (SEM) from n = 5 to 6 independent experiments. P values were calculated using the 2-sided Student unpaired t test. (E) The bar diagram shows the MFI for HLA-A,B,C expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours. Bar graphs show the mean ± SEM from n = 5 to 6 independent experiments. P values were calculated using the 2-sided Student unpaired t test. (F-G) Percentage survival of Rag2−/−Il2rγ−/−-recipient mice after the transfer of primary human AML cells from 2 different patients is shown. As indicated, mice were injected with additional human CD3+ T cells (isolated from the peripheral blood of an HLA nonmatched healthy donor) and treated with either vehicle or MDM2 inhibitor RG-7112. Shown are n = 10 or 8 independent animals, and P values were calculated using the 2-sided Mantel-Cox test. (H) Percentage of specific lysis of isolated, CD3/28, and IL-2 expanded human T cells in contact with OCI-AML3 cells is shown. OCI-AML3 cells were pretreated with either DMSO or the MDM2-inhibitor RG-7112, and the E:T (effector [T-cell] to target [OCI-AML3 cell]) ratio was titrated between 10:1 and 1:1 as indicated. One representative experiment of 3 independent experiments is shown. (I) Representative Western blots showing the activation of caspase-3 and loading control (β-actin) in OCI-AML3 cells. OCI-AML3 cells exposed to DMSO or RG-7112 (1 µM) were cocultured with activated T cells at an E:T ratio of 10:1 for 4 hours. (J) The bar diagram indicates the ratio of the cleaved caspase-3 to pro–caspase-3 normalized to β-actin. The values were normalized to the T-cell–only group (set as “1”). (K-L) The graph shows fold change of MFI for (K) HLA-DR and (I) HLA-A,B,C expression on WT OCI-AML3 (WT) or p53 knockdown (p53 KD) OCI-AML3 cells after treatment with RG-7112 (2 µM) for 72 hours as mean ± SEM from n = 4 independent experiments. MFI of control-treated cells was set as 1.0. P values were calculated using a 2-sided Student unpaired t test. (M) Percentage survival of Rag2−/−Il2rγ−/−-recipient mice after transfer of human WT or p53 knockdown (p53 KD) OCI-AML3 cells are shown. As indicated, mice were injected with additional human T cells (isolated from the peripheral blood of an HLA nonmatched healthy donor) and treated with either vehicle or MDM2-inhibitor RG-7112. Shown are n = 10, 11, or 21 independent animals per group from ≥2 independent experiments, and P values were calculated using the 2-sided Mantel-Cox test.
MDM2 inhibition leads to increased MHC expression and improves survival in xenograft mouse models. (A) Microarray-based analysis of the expression level of HLA class I and II in OCI-AML3 cells after treatment with DMSO, RG-7112 (1 µM), or HDM-201 (200 nM) for 24 hours is shown as tile display from Robust Multichip Average (RMA) signal values, (Z-score), n = 6 biologically independent samples per group. The adjusted P value was calculated using the Benjamini & Hochberg method. *indicates significant regulation (P < .05) compared with DMSO. (B-C) Representative flow cytometry histograms show the mean fluorescence intensity (MFI) for (B) HLA-DR or (C) HLA-A,B,C expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours. (D) The bar diagram shows the MFI for HLA-DR expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours. Bar graphs show the mean ± standard error of the mean (SEM) from n = 5 to 6 independent experiments. P values were calculated using the 2-sided Student unpaired t test. (E) The bar diagram shows the MFI for HLA-A,B,C expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours. Bar graphs show the mean ± SEM from n = 5 to 6 independent experiments. P values were calculated using the 2-sided Student unpaired t test. (F-G) Percentage survival of Rag2−/−Il2rγ−/−-recipient mice after the transfer of primary human AML cells from 2 different patients is shown. As indicated, mice were injected with additional human CD3+ T cells (isolated from the peripheral blood of an HLA nonmatched healthy donor) and treated with either vehicle or MDM2 inhibitor RG-7112. Shown are n = 10 or 8 independent animals, and P values were calculated using the 2-sided Mantel-Cox test. (H) Percentage of specific lysis of isolated, CD3/28, and IL-2 expanded human T cells in contact with OCI-AML3 cells is shown. OCI-AML3 cells were pretreated with either DMSO or the MDM2-inhibitor RG-7112, and the E:T (effector [T-cell] to target [OCI-AML3 cell]) ratio was titrated between 10:1 and 1:1 as indicated. One representative experiment of 3 independent experiments is shown. (I) Representative Western blots showing the activation of caspase-3 and loading control (β-actin) in OCI-AML3 cells. OCI-AML3 cells exposed to DMSO or RG-7112 (1 µM) were cocultured with activated T cells at an E:T ratio of 10:1 for 4 hours. (J) The bar diagram indicates the ratio of the cleaved caspase-3 to pro–caspase-3 normalized to β-actin. The values were normalized to the T-cell–only group (set as “1”). (K-L) The graph shows fold change of MFI for (K) HLA-DR and (I) HLA-A,B,C expression on WT OCI-AML3 (WT) or p53 knockdown (p53 KD) OCI-AML3 cells after treatment with RG-7112 (2 µM) for 72 hours as mean ± SEM from n = 4 independent experiments. MFI of control-treated cells was set as 1.0. P values were calculated using a 2-sided Student unpaired t test. (M) Percentage survival of Rag2−/−Il2rγ−/−-recipient mice after transfer of human WT or p53 knockdown (p53 KD) OCI-AML3 cells are shown. As indicated, mice were injected with additional human T cells (isolated from the peripheral blood of an HLA nonmatched healthy donor) and treated with either vehicle or MDM2-inhibitor RG-7112. Shown are n = 10, 11, or 21 independent animals per group from ≥2 independent experiments, and P values were calculated using the 2-sided Mantel-Cox test.
To validate if the findings made in humans in murine cell lines have a correlation in patients, we analyzed human primary AML cells. We observed that MDM2 inhibition increased HLA-DR expression on primary human AML cells (supplemental Figure 3G-H) of patients that were treatment-naïve (supplemental Table 2). To understand if MDM2 was connected to CIITA, the positive regulator of MHC-II expression in human AML cells, we analyzed the gene expression profiles of AML blasts in 2 independent patient cohorts that included cases from the Beat AML (Personalized Medicine for Acute Myeloid Leukemia Based on Functional Genomics) initiative (n = 451)35 and cases from TCGA (The Cancer Genome Atlas) initiative (n = 200).36 In agreement with a negative regulation of MDM2 on the CIITA/MHC-II axis, a negative correlation between the transcript levels of MDM2 mRNA and CIITA mRNA in human AML cells was detected (supplemental Figure 3I-J). Pearson correlation was −0.22 for the TCGA data and −0.16 for Beat AML data, indicating a correlation. The Beat AML data also include cases that were not profiled at diagnosis but during therapy.35 Therefore, we performed the same analysis on 338 samples from patients who did not receive a previous allo-HCT and found a comparable pattern (Pearson, −0.17) (supplemental Figure 3K). Based on the potential role of p53 for MHC-I and MHC-II induction by MDM2 inhibition, we determined the number of p53-mutant cases in the 2 patient cohorts. TP53-mutant cases accounted for 42 of 451 cases in the Beat AML dataset (9%) and 16 of 200 cases in the TCGA dataset (8%). Therefore, p53-mutant cases were too rare to separately test for the association between the MDM2 and CIITA transcript levels in these subgroups. We analyzed posttransplantation relapse samples from 3 different stem cell transplant centers located in Freiburg, Tübingen, and Milano to determine if the MDM2 inhibitor effect was reproducible in the context of AML relapse. We observed that HLA-A,B,C increased upon exposure to the MDM2 inhibitor (supplemental Figure 3L-M). The increase of HLA-DR upon MDM2 inhibition we had seen in freshly isolated AML samples was not observed when we used frozen AML relapse samples. We could expose the frozen cells for only 24 hours to the MDM2 inhibitor, as the cell viability decreased below 50% after 24 hours of cell culture. A longer exposure time to the MDM2 inhibitor might also induce HLA-DR expression in freshly isolated AML cells from relapse patients.
To test the functional role of MDM2 inhibition in xenograft models, we next isolated PBMC from 2 AML patients that were treatment-naïve (supplemental Table 3) and depleted the CD3+ T cells (supplemental Figures 4A and 5A). Immunodeficient mice bearing the human AML cells and allogeneic T cells experienced improved survival upon MDM2 inhibition (Figure 1F-G). In vitro, cytotoxicity of allogeneic human T cells was higher when OCI-AML3 cells were exposed to MDM2 inhibition (Figure 1H). Consistently, cleaved caspase-3 was highest in OCI-AML3 cells when T cells were combined with MDM2 inhibition (Figure 1I-J). The synergistic effect was dependent on intact p53 because neither HLA-A,B,C, nor HLA-DR increased with MDM2 inhibition using RG-7112 (Figure 1K-L) or HDM201 (supplemental Figure 6A-D) in human p53-knockdown OCI-AML3 cells. MDM2 inhibition and allogeneic T cells improved the survival of OCI-AML3 cell-bearing immunodeficient mice (Figure 1M). Conversely, p53-knockdown OCI-AML3 AML cells enriched for the transgenic cells (supplemental Figure 6E) were resistant to the MDM2 inhibitor/allo–T-cell combination in vivo (Figure 1M). To test if increasing p53 independent of MDM2 would lead to comparable effects, we used the MDMX inhibitor XI-006.37 We observed that MDMX inhibition also increased HLA-A,B,C and HLA-DR on OCI-AML3 cells (supplemental Figure 7A-B). The upregulation of HLA-A,B,C and HLA-DR was reproducible for MOLM-13 cells (supplemental Figure 7C-D). MDM2 inhibition induced HLA-A,B,C in MV4-11 cells (supplemental Figure 7E). HLA-DR upregulation was not observed in MV4-11 cells upon MDM2 inhibition (supplemental Figure 7F). We had previously shown that AML cells in relapsed patients release lactic acid (LA).9 To understand if LA interferes with the expression of HLA molecules upon MDM2 inhibition, we repeated the studies and included LA in the culture. We found that despite the presence of LA, MDM2 inhibition caused an increase of HLA-A,B,C and HLA-DR on OCI-AML3 cells (supplemental Figure 7G-H).
These findings indicate that MDM2 inhibition leads to a p53-dependent increase of MHC class I and II expression and improves survival in patient xenograft-based and human cell line-based GVL mouse models.
MDM2 inhibition enhances in vivo leukemia elimination in syngeneic AML-bearing mice and TRAIL-R1/2 upregulation on leukemia cells
To validate the observation that MDM2 inhibition can synergize with the allogeneic immune response in xenografts, we treated syngeneic AML-bearing mice with allo-HCT using BM alone or in combination with T cells. In AML-bearing mice, the addition of T cells to the allogeneic BM graft improved survival (Figure 2A). Treatment of leukemia-bearing mice with MDM2 inhibitor in the absence of donor T cells improved survival but did not lead to long-term protection (Figure 2A). Conversely, when T cells were combined with MDM2 inhibitor treatment, the majority of the mice (>80%) showed long-term protection (Figure 2A). A comparable survival pattern was seen in the AMLMLL-PTD/FLT3-ITD model (Figure 2B). The T cell/MDM2 inhibitor combination reduced the leukemia burden (Figure 2C-E) without increasing acute GVHD severity compared with T cells/vehicle (supplemental Figure 8A-C).

MDM2 inhibition enhances GVL effects in syngeneic AML models and TRAIL-R1/2 upregulation on leukemia cells. (A) Percentage survival of BALB/c recipient mice after transfer of AML WEHI-3B cells (BALB/c background) and allogeneic C57BL/6 BM is shown. Mice were injected with additional allogeneic T cells (Tc; C57BL/6) and/or treated with either vehicle or MDM2 inhibitor RG-7112 when indicated. A total of n = 9 to 10 independent animals per group are shown, and P values were calculated using the 2-sided Mantel-Cox test. (B) Percentage survival of C57BL/6-recipient mice after transfer of AMLMLL-PTD FLT3-ITD cells (C57BL/6 background) and allogeneic BALB/c BM is shown. Mice were injected with additional allogeneic T cells (BALB/c) and/or treated with either vehicle or MDM2-inhibitor RG-7112 when indicated. A total of n = 10 biologically independent animals from 2 experiments are shown, and P values were calculated using the 2-sided Mantel-Cox test. (C) Representative bioluminescence imaging (BLI) on day 14 after transplantation with LUC+ MOLM-13 leukemia cells showing the expansion of the leukemia cells in Rag2−/−Il2rγ−/− mice. When indicated, mice received human CD3+ T cells (isolated from the peripheral blood of an HLA nonmatched healthy donor) and were treated with either vehicle or MDM2 inhibitor RG-7112. Images are of a representative mouse from each of the groups. (D) The graph shows the quantification of the BLI signal at day 14 after allo-HCT for each treatment group. Data from 3 independent experiments were pooled. The mean ± standard error of the mean (SEM) for each treatment group is shown. The P values were calculated using the 2-sided Mann-Whitney U test. (E) The graph shows the percent of human CD45+CD3−CD8− cells (representing the MOLM-13 AML cells) of all viable cells in the BM of Rag2−/−Il2rγ−/− mice 23 days after transplantation with LUC+ MOLM-13 leukemia cells and human CD3+ T cells. Data are shown as mean ± SEM from n = 3 independent experiments. P values were calculated using a 2-sided Student unpaired t test. (F) Microarray-based analysis of the expression level of TRAIL-R1 and TRAIL-R2 (TNFRSF10A and TNFRSF10B) in OCI-AML3 cells after treatment with DMSO, RG-7112 (1 µM), or HDM-201 (200 nM) for 24 hours is shown as a tile display from Robust Multichip Average (RMA) signal values (Z-score), n = 6 biologically independent samples per group. The adjusted P value was calculated using the Benjamini & Hochberg method. For both genes, a significant upregulation (both P < .001) compared with DMSO was observed. (G) The graph shows the fold change of MFI for TRAIL-R1 expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2-inhibitor RG-7112 for 72 hours. Data are shown as mean ± SEM from n = 3 independent experiments. P values were calculated using a 2-sided Student unpaired t test. (H) The graph shows the fold change of MFI for TRAIL-R2 expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours as mean ± SEM from n = 5 independent experiments. P values were calculated using a 2-sided Student unpaired t test. (I-J) The graph shows fold change of MFI for (I) TRAIL-R1 or (J) TRAIL-R2 expression on WT OCI-AML3 or p53 KD OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours as mean ± SEM from n = 4 independent experiments. MFI of control-treated cells was set as 1.0. P values were calculated using the 2-sided Student unpaired t test.
MDM2 inhibition enhances GVL effects in syngeneic AML models and TRAIL-R1/2 upregulation on leukemia cells. (A) Percentage survival of BALB/c recipient mice after transfer of AML WEHI-3B cells (BALB/c background) and allogeneic C57BL/6 BM is shown. Mice were injected with additional allogeneic T cells (Tc; C57BL/6) and/or treated with either vehicle or MDM2 inhibitor RG-7112 when indicated. A total of n = 9 to 10 independent animals per group are shown, and P values were calculated using the 2-sided Mantel-Cox test. (B) Percentage survival of C57BL/6-recipient mice after transfer of AMLMLL-PTD FLT3-ITD cells (C57BL/6 background) and allogeneic BALB/c BM is shown. Mice were injected with additional allogeneic T cells (BALB/c) and/or treated with either vehicle or MDM2-inhibitor RG-7112 when indicated. A total of n = 10 biologically independent animals from 2 experiments are shown, and P values were calculated using the 2-sided Mantel-Cox test. (C) Representative bioluminescence imaging (BLI) on day 14 after transplantation with LUC+ MOLM-13 leukemia cells showing the expansion of the leukemia cells in Rag2−/−Il2rγ−/− mice. When indicated, mice received human CD3+ T cells (isolated from the peripheral blood of an HLA nonmatched healthy donor) and were treated with either vehicle or MDM2 inhibitor RG-7112. Images are of a representative mouse from each of the groups. (D) The graph shows the quantification of the BLI signal at day 14 after allo-HCT for each treatment group. Data from 3 independent experiments were pooled. The mean ± standard error of the mean (SEM) for each treatment group is shown. The P values were calculated using the 2-sided Mann-Whitney U test. (E) The graph shows the percent of human CD45+CD3−CD8− cells (representing the MOLM-13 AML cells) of all viable cells in the BM of Rag2−/−Il2rγ−/− mice 23 days after transplantation with LUC+ MOLM-13 leukemia cells and human CD3+ T cells. Data are shown as mean ± SEM from n = 3 independent experiments. P values were calculated using a 2-sided Student unpaired t test. (F) Microarray-based analysis of the expression level of TRAIL-R1 and TRAIL-R2 (TNFRSF10A and TNFRSF10B) in OCI-AML3 cells after treatment with DMSO, RG-7112 (1 µM), or HDM-201 (200 nM) for 24 hours is shown as a tile display from Robust Multichip Average (RMA) signal values (Z-score), n = 6 biologically independent samples per group. The adjusted P value was calculated using the Benjamini & Hochberg method. For both genes, a significant upregulation (both P < .001) compared with DMSO was observed. (G) The graph shows the fold change of MFI for TRAIL-R1 expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2-inhibitor RG-7112 for 72 hours. Data are shown as mean ± SEM from n = 3 independent experiments. P values were calculated using a 2-sided Student unpaired t test. (H) The graph shows the fold change of MFI for TRAIL-R2 expression on OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours as mean ± SEM from n = 5 independent experiments. P values were calculated using a 2-sided Student unpaired t test. (I-J) The graph shows fold change of MFI for (I) TRAIL-R1 or (J) TRAIL-R2 expression on WT OCI-AML3 or p53 KD OCI-AML3 cells after treatment with the indicated concentrations of MDM2 inhibitor RG-7112 for 72 hours as mean ± SEM from n = 4 independent experiments. MFI of control-treated cells was set as 1.0. P values were calculated using the 2-sided Student unpaired t test.
In addition to the effects on MHC, gene expression analysis revealed upregulation of TNF-related apoptosis-inducing ligand receptor-1 (TRAIL-R1) and TRAIL-R2 by leukemia cells upon MDM2 inhibition (Figure 2F). Downregulation of proapoptotic receptors, including TRAIL-R1 and TRAIL-R2, was shown to be connected to therapy resistance and relapse in AML.38 Consistent with the gene expression data, both TRAIL-R1/TRAIL-R2 proteins and TRAIL-R1/TRAIL-R2 RNAs increased dose-dependently upon MDM2 inhibition in human OCI-AML3 cells (Figure 2G-H; supplemental Figure 9A-J). This was also the case in mouse WEHI-3B cells using 2 different MDM2 inhibitors (RG-7112 and HDM201) (supplemental Figure 10A-H). To test if increasing p53 independent of MDM2 would lead to comparable effects, we used the MDMX inhibitor XI-006.37 We observed that MDMX inhibition also increased TRAIL-R1/TRAIL-R2 proteins on OCI-AML3 cells (supplemental Figure 11A-C). The upregulation of TRAIL-R1/2 was reproducible using additional human AML cell lines, including MOLM-13 and MV4-11 (supplemental Figure 11D-G). We found that despite the presence of LA, MDM2 inhibition caused an increase of TRAIL-R1/TRAIL-R2 proteins on OCI-AML3 cells (supplemental Figure 11H-I). To determine if LA-exposed T cells were resistant to the MDM2 inhibitor effect, we studied the survival of leukemia-bearing allo-HCT mice treated with LA-exposed T cells and vehicle or MDM2 inhibitor. We observed that despite the exposure of the donor T cells to LA, the group treated with the MDM2 inhibitor experienced an improved survival compared with the vehicle group (supplemental Figure 11J).
To understand if MDM2 inhibition has similar effects on primary human AML cells, we used patient-derived AML cells. MDM2 inhibition increased concentrations of TRAIL-R1/TRAIL-R2 proteins (supplemental Figure 12A-D; supplemental Table 4 [22 patients]). We also analyzed posttransplantation relapse samples to determine if the MDM2 inhibitor effect on TRAIL-R1/2 expression was reproducible in the context of AML relapse. We observed that TRAIL-R1, TRAIL-R2, and p53 expression increased upon exposure to MDM2 inhibitors (supplemental Figure 12E-G).
In addition, MDM2 inhibition increased concentrations of TRAIL-R1 and TRAIL-R2 RNAs (supplemental Figure 13A-D; supplemental Table 5 [12 patients]) in primary human AML cells. We used p53-knockdown OCI-AML3 cells (supplemental Figure 6A) to test whether increased TRAIL-R1/2 expression after MDM2 inhibition was p53-dependent. TRAIL-R1/2 expression increased upon MDM2 inhibition on WT-AML but not on p53-knockdown AML using RG-7112 (Figure 2I-J) or HDM201 (supplemental Figure 14A-B) as MDM2-inhibitors. These findings indicate that MDM2 inhibition enhances in vivo leukemia elimination and p53-dependent TRAIL-R1/2 upregulation on leukemia cells.
The p53/TRAIL-R1/2 axis is required for enhanced T-cell–mediated antileukemia cytotoxicity upon MDM2 inhibition
To understand if p53 was required for TRAIL-R1/2 dependent cytotoxicity, we studied the impact of soluble recombinant TRAIL plus MDM2 inhibitor on the viability of human OCI-AML3 leukemia cells. We observed that soluble recombinant TRAIL plus MDM2 inhibitors induced less apoptosis in p53-knockdown AML cells compared with WT AML cells (Figure 3A). This supports the concept that upon MDM2 inhibition, the resulting p53 increase induces TRAIL-R1/2 expression. To directly test this connection, we exposed human OCI-AML3 leukemia cells to a vehicle or MDM2 inhibitor. Chromatin immunoprecipitation revealed increased p53 binding to the TRAILR1/2 promoters upon MDM2 inhibition in human OCI-AML3 leukemia cells (Figure 3B-C). To functionally test the role of TRAIL-R1/2, we generated clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-mediated TRAILR2-knockout and TRAILR1/2-knockout AML cells (supplemental Figure 15A-B).
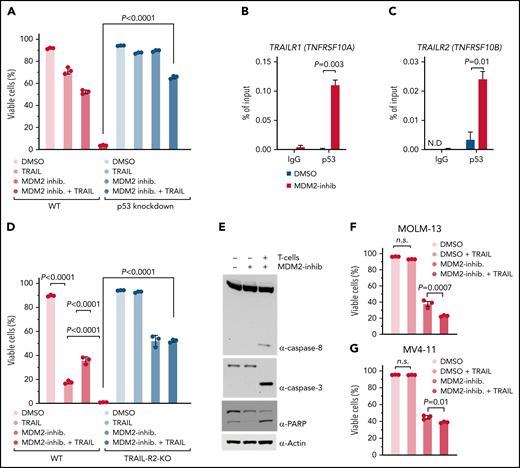
MDM2 inhibition increases p53 binding to TRAIL-R1/2 promoters and sensitizes AML cells to TRAIL-mediated apoptosis. (A) The graph shows the percentage of viable cells. Where indicated, WT OCI-AML3 or p53 KD OCI-AML3 were incubated with 1 µM MDM2 inhibitor RG-7112. After 48 hours, limiting concentrations of hTRAIL (TNFSF 10) were added for 24 hours, where indicated. The viability of cells was measured by flow cytometry. The mean of triplicates ± standard error of the mean (SEM) is displayed. P values were calculated using a 2-sided Student unpaired t test. (B-C) ChIP-qPCR analysis in OCI-AML3 cells treated with DMSO or 2 µM RG-7112 for 12 hours to detect the binding of p53 to the promoter of (B) TRAIL-R1 (TNFRSF10A) and (C) TRAIL-R2 (TNFRSF10B). Data are represented as percent input and are representative of 3 experiments; error bars, SEM from 3 technical replicates. N.D., not detected. (D) The bar diagram shows the viability of WT or TRAIL-R2−/− OCI-AML3 cells (TRAIL-R2−/−) that were incubated with 1 µM of the MDM2 inhibitor RG-7112, where indicated. After 48 hours, limiting concentrations of hTRAIL (TNFSF 10) were added for 24 hours, where indicated. The viability of the AML cells was measured by flow cytometry. The mean of triplicates ± SEM is displayed. P values were calculated using a 2-sided Student unpaired t test. (E) Representative Western blots showing caspase-8, caspase-3, PARP, and loading control (β-actin) in human OCI-AML3 cells. OCI-AML3 cells exposed to DMSO or RG-7112 (1 µM) were cocultured with activated T cells at an E:T ratio of 10:1 for 4 hours. (F) The bar diagram shows the viability of MOLM-13 cells pretreated with DMSO or 1 µM MDM2 inhibitor for 24 hours, as indicated. hTRAIL (TNFSF10) was added for an additional 24 hours before analysis by flow cytometry. P values were calculated using the one-way ANOVA test. (G) The graph shows the viability of MV4-11 cells pretreated with 1 µM MDM2 inhibitor for 24 hours. Limiting concentrations of hTRAIL (TNFSF10) were added for an additional 24 hours before analysis by flow cytometry. P values were calculated using the one-way ANOVA test. ns, not significant.
MDM2 inhibition increases p53 binding to TRAIL-R1/2 promoters and sensitizes AML cells to TRAIL-mediated apoptosis. (A) The graph shows the percentage of viable cells. Where indicated, WT OCI-AML3 or p53 KD OCI-AML3 were incubated with 1 µM MDM2 inhibitor RG-7112. After 48 hours, limiting concentrations of hTRAIL (TNFSF 10) were added for 24 hours, where indicated. The viability of cells was measured by flow cytometry. The mean of triplicates ± standard error of the mean (SEM) is displayed. P values were calculated using a 2-sided Student unpaired t test. (B-C) ChIP-qPCR analysis in OCI-AML3 cells treated with DMSO or 2 µM RG-7112 for 12 hours to detect the binding of p53 to the promoter of (B) TRAIL-R1 (TNFRSF10A) and (C) TRAIL-R2 (TNFRSF10B). Data are represented as percent input and are representative of 3 experiments; error bars, SEM from 3 technical replicates. N.D., not detected. (D) The bar diagram shows the viability of WT or TRAIL-R2−/− OCI-AML3 cells (TRAIL-R2−/−) that were incubated with 1 µM of the MDM2 inhibitor RG-7112, where indicated. After 48 hours, limiting concentrations of hTRAIL (TNFSF 10) were added for 24 hours, where indicated. The viability of the AML cells was measured by flow cytometry. The mean of triplicates ± SEM is displayed. P values were calculated using a 2-sided Student unpaired t test. (E) Representative Western blots showing caspase-8, caspase-3, PARP, and loading control (β-actin) in human OCI-AML3 cells. OCI-AML3 cells exposed to DMSO or RG-7112 (1 µM) were cocultured with activated T cells at an E:T ratio of 10:1 for 4 hours. (F) The bar diagram shows the viability of MOLM-13 cells pretreated with DMSO or 1 µM MDM2 inhibitor for 24 hours, as indicated. hTRAIL (TNFSF10) was added for an additional 24 hours before analysis by flow cytometry. P values were calculated using the one-way ANOVA test. (G) The graph shows the viability of MV4-11 cells pretreated with 1 µM MDM2 inhibitor for 24 hours. Limiting concentrations of hTRAIL (TNFSF10) were added for an additional 24 hours before analysis by flow cytometry. P values were calculated using the one-way ANOVA test. ns, not significant.
The combination of MDM2 inhibition and soluble recombinant TRAIL caused higher cytotoxicity compared with soluble recombinant TRAIL alone (Figure 3D), which is in agreement with the upregulation of TRAIL-R1/2 on leukemia cells upon MDM2 inhibition, which renders cells more susceptible to TRAIL-mediated apoptosis. MDM2 inhibitor/TRAIL-mediated killing was reduced in TRAIL-R2-knockout and TRAIL-R1/2-knockout AML cells compared with WT AML cells (Figure 3D; supplemental Figure 15C-D). Consistent with a central role of TRAIL-mediated apoptosis induction during allogeneic antileukemia immune responses, the combination of MDM2 inhibition and allogeneic T cells caused activation of the TRAIL-R1/2 downstream pathway (caspase-8, caspase-3, and poly (ADP-ribose) polymerase (PARP)) in human AML cells (Figure 3E). The observation that MDM2 inhibition increased TRAIL-mediated apoptosis was reproducible using the human AML cell lines MOLM-13 and MV4-11 (Figure 3F-G). These findings indicate that the p53/TRAIL axis is required for enhanced leukemia elimination upon MDM2 inhibition.
TRAIL-R1/2 contributes to enhanced GVL effects upon MDM2 inhibition in mice
To determine to what extent TRAIL-R1/2 expression in AML cells contributes to enhanced GVL effects upon MDM2 inhibition in vivo, we treated mice with an anti-TRAIL antibody. This intervention reduced the protective effect of the allo–T-cell/MDM2 inhibition against AML (Figure 4A). Consistent with the role of TRAIL expression on the alloreactive T cells, the transfer of TRAIL-deficient donor T cells (Tnfsf10tm1b(KOMP)Wtsi/MbpMmucd) reduced the protective effect of MDM2 inhibition (Figure 4B). Furthermore, in vitro anti-TRAIL antibody reduced the cytotoxicity of allogeneic T cells against MDM2 inhibitor-exposed leukemia cells (Figure 4C-E). CRISPR/Cas9-mediated TRAIL-R2-knockout AML cells (supplemental Figure 15A-B) were less susceptible to the allo–T-cell/MDM2 inhibition effect in vivo (Figure 4F). To understand which donor cell type was responsible for the immunomodulatory effect of MDM2 inhibition, we selectively depleted CD8+ T cells or natural killer (NK) cells. We observed that depletion of CD8+ T cells but not NK cells (supplemental Figure 16A-B) abrogated the protective MDM2 inhibition effect (Figure 4G), indicating that the antileukemia effect is mediated mainly by CD8+ T cells. To understand whether recall immunity developed under MDM2 inhibitor treatment, we isolated donor-type CD8+ T cells from leukemia-bearing mice treated with vehicle or MDM2 inhibitor (supplemental Figure 17A). T cells derived from MDM2 inhibitor-treated, leukemia-bearing mice caused improved control of leukemia in secondary leukemia-bearing mice (Figure 4H), indicating an antileukemia recall response. These findings indicate that TRAIL contributes to enhanced GVL effects upon MDM2 inhibition in mice and that leukemia-reactive donor T-cell clones evolve.
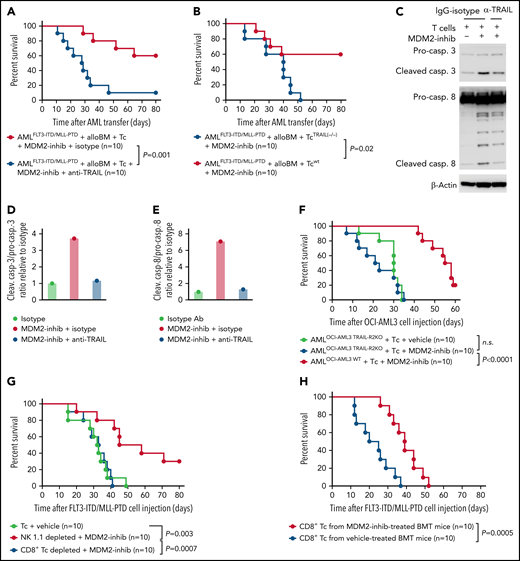
TRAIL contributes to enhanced GVL effects upon MDM2 inhibition. (A) Percentage survival of C57BL/6 recipient mice after transfer of AMLMLL-PTD FLT3-ITD cells (C57BL/6 background) and allogeneic BALB/c BM is shown. Mice were injected with additional allogeneic T cells (BALB/c) and treated with the MDM2-inhibitor RG-7112 and either anti-TRAIL antibody or IgG isotype, as indicated. A total of n = 10 independent animals from 2 experiments are shown, and P values were calculated using the 2-sided Mantel-Cox test. (B) Percentage survival of C57BL/6-recipient mice after transfer of AMLMLL-PTD FLT3-ITD cells (C57BL/6 background) and allogeneic BALB/c BM is shown. Mice were injected with additional allogeneic T cells (BALB/c), either WT T cells or TRAIL−/− T cells. A total of n = 10 independent animals from 2 experiments are shown, and P values were calculated using the 2-sided Mantel-Cox test. (C) Western blots showing the activation of caspase-3, caspase-8, and loading control (β-actin) in OCI-AML3 cells. Activated T cells were pretreated with 10 µg/mL anti-TRAIL neutralizing antibody or IgG control for 1 hour and were cocultured with OCI-AML3 cells exposed to DMSO or RG-7112 (1 µM) at an E:T ratio of 10:1 for 4 hours. (D) Quantification of the ratio of cleaved caspase-3/total caspase-3 normalized to isotype control. One representative experiment of 3 experiments with comparable results is shown. (E) Quantification of the ratio of cleaved caspase-8/total caspase-8 normalized to isotype control. One representative experiment of 3 experiments with comparable results is shown. A total of n = 3 independent experiments were performed. (F) Survival of Rag2−/−Il2rγ−/− mice receiving WT OCI-AML3 cells or CRISPR/Cas-mediated TRAIL-R2 knockout (TRAIL-R2−/−) OCI-AML3 cells. Mice were additionally injected with primary human T cells isolated from healthy donors and treated with vehicle or MDM2 inhibitor RG-7112. A total of n = 10 animals from 2 independent experiments are shown. P values were calculated using the 2-sided Mantel-Cox test. (G) Percentage survival of C57BL/6-recipient mice after transfer of AMLMLL-PTD FLT3-ITD cells (C57BL/6 background) and BMT using allogeneic BALB/c BM is shown. Mice were injected with additional allogeneic T cells (BALB/c) on day 2 after BMT. When indicated, CD8 T cells or NK cells were depleted from the allogeneic T-cell transplant. A total of n = 10 independent animals from 2 experiments are shown, and P values were calculated using the 2-sided Mantel-Cox test. (H) Percentage survival of C57BL/6-recipient mice after transfer of AMLMLL-PTD FLT3-ITD cells (C57BL/6 background) and allogeneic BALB/c BM is shown. Mice were injected with additional allogeneic T cells (BALB/c) derived from previously challenged and treated (MDM2 inhibitor or vehicle) mice. Shown are n = 10 independent animals from 2 experiments, and P values were calculated using the 2-sided Mantel-Cox test.
TRAIL contributes to enhanced GVL effects upon MDM2 inhibition. (A) Percentage survival of C57BL/6 recipient mice after transfer of AMLMLL-PTD FLT3-ITD cells (C57BL/6 background) and allogeneic BALB/c BM is shown. Mice were injected with additional allogeneic T cells (BALB/c) and treated with the MDM2-inhibitor RG-7112 and either anti-TRAIL antibody or IgG isotype, as indicated. A total of n = 10 independent animals from 2 experiments are shown, and P values were calculated using the 2-sided Mantel-Cox test. (B) Percentage survival of C57BL/6-recipient mice after transfer of AMLMLL-PTD FLT3-ITD cells (C57BL/6 background) and allogeneic BALB/c BM is shown. Mice were injected with additional allogeneic T cells (BALB/c), either WT T cells or TRAIL−/− T cells. A total of n = 10 independent animals from 2 experiments are shown, and P values were calculated using the 2-sided Mantel-Cox test. (C) Western blots showing the activation of caspase-3, caspase-8, and loading control (β-actin) in OCI-AML3 cells. Activated T cells were pretreated with 10 µg/mL anti-TRAIL neutralizing antibody or IgG control for 1 hour and were cocultured with OCI-AML3 cells exposed to DMSO or RG-7112 (1 µM) at an E:T ratio of 10:1 for 4 hours. (D) Quantification of the ratio of cleaved caspase-3/total caspase-3 normalized to isotype control. One representative experiment of 3 experiments with comparable results is shown. (E) Quantification of the ratio of cleaved caspase-8/total caspase-8 normalized to isotype control. One representative experiment of 3 experiments with comparable results is shown. A total of n = 3 independent experiments were performed. (F) Survival of Rag2−/−Il2rγ−/− mice receiving WT OCI-AML3 cells or CRISPR/Cas-mediated TRAIL-R2 knockout (TRAIL-R2−/−) OCI-AML3 cells. Mice were additionally injected with primary human T cells isolated from healthy donors and treated with vehicle or MDM2 inhibitor RG-7112. A total of n = 10 animals from 2 independent experiments are shown. P values were calculated using the 2-sided Mantel-Cox test. (G) Percentage survival of C57BL/6-recipient mice after transfer of AMLMLL-PTD FLT3-ITD cells (C57BL/6 background) and BMT using allogeneic BALB/c BM is shown. Mice were injected with additional allogeneic T cells (BALB/c) on day 2 after BMT. When indicated, CD8 T cells or NK cells were depleted from the allogeneic T-cell transplant. A total of n = 10 independent animals from 2 experiments are shown, and P values were calculated using the 2-sided Mantel-Cox test. (H) Percentage survival of C57BL/6-recipient mice after transfer of AMLMLL-PTD FLT3-ITD cells (C57BL/6 background) and allogeneic BALB/c BM is shown. Mice were injected with additional allogeneic T cells (BALB/c) derived from previously challenged and treated (MDM2 inhibitor or vehicle) mice. Shown are n = 10 independent animals from 2 experiments, and P values were calculated using the 2-sided Mantel-Cox test.
MDM2 inhibition promotes cytotoxicity and longevity of donor T cells
Donor (C57BL/6) CD8+ T cells isolated from leukemia-bearing MDM2 inhibitor-treated allo-HCT recipients (BALB/c) displayed higher expression of the antitumor cytotoxicity markers perforin, TRAIL, and CD107a, and of IFN-γ and TNF in leukemia-bearing MDM2 inhibitor-treated allo-HCT recipients compared with those receiving vehicle (Figure 5A-H; supplemental Figure 18A). In addition, CD69 and TRAIL expression were higher on donor (C57BL/6) CD8+ T cells isolated from leukemia-bearing MDM2 inhibitor-treated allo-HCT recipients (BALB/c) without there being an increase in total CD8+ T cells (supplemental Figure 19A-C).
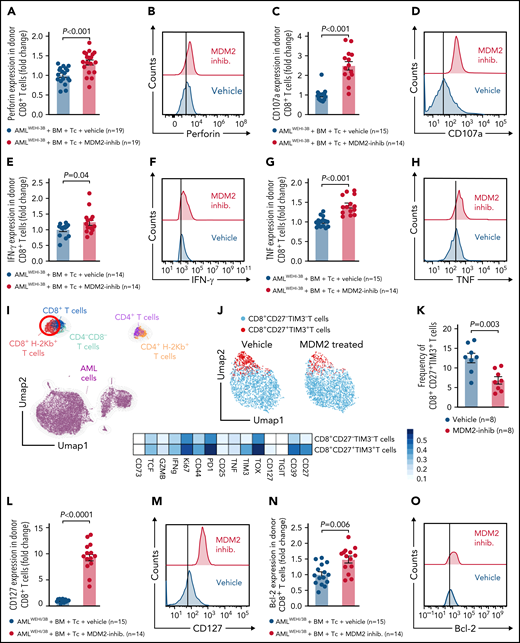
MDM2 inhibition promotes cytotoxicity and longevity of donor T cells. (A-H) Scatterplots and representative histograms show expression of (A-B) perforin, (C-D) CD107a, (E-F) IFN-γ, and (G-H) TNF of CD8+ T cells isolated from the spleen on day 12 following allo-HCT of WEHI-3B leukemia-bearing BALB/c mice transplanted with C57BL/6 BM plus allogeneic C57BL/6 T cells and treated with either vehicle or MDM2 inhibitor RG-7112. Mean value ± standard error of the mean (SEM) from n = 14 to 19 biologically independent animals per group from 2 experiments are shown, and P values were calculated using the 2-sided Mann-Whitney U test. (I) UMAP showing the FlowSOM-guided manual metaclustering of splenic live CD45+ cells from leukemia-bearing BALB/c mice after allo-HCT. (J) UMAP showing the FlowSOM-guided manual metaclustering (top) and heatmap showing median marker expression (bottom) of donor-derived (H-2kb+) TCRβ+ CD8+ T cells from leukemia-bearing BALB/c mice after allo-HCT treated with RG-7112 or vehicle, as indicated. (K) Quantification of donor-derived (H-2kb+) TCRβ+ CD8+ CD27+ TIM3+ T cells from leukemia-bearing BALB/c mice after allo-HCT treated with RG-7112 or vehicle, as indicated. (L-O) Scatterplots and representative histograms show expression of (L-M) CD127 and (N-O) Bcl-2 of CD8+ T cells isolated from spleen on day 12 following allo-HCT of WEHI-3B leukemia-bearing BALB/c mice transplanted with C57BL/6 BM plus allogeneic C57BL/6 T cells and treated with either vehicle or MDM2 inhibitor RG-7112. Mean value ± SEM from n = 14 to 19 biologically independent animals per group from 2 experiments are shown. P value was calculated using the 2-sided Mann-Whitney U test.
MDM2 inhibition promotes cytotoxicity and longevity of donor T cells. (A-H) Scatterplots and representative histograms show expression of (A-B) perforin, (C-D) CD107a, (E-F) IFN-γ, and (G-H) TNF of CD8+ T cells isolated from the spleen on day 12 following allo-HCT of WEHI-3B leukemia-bearing BALB/c mice transplanted with C57BL/6 BM plus allogeneic C57BL/6 T cells and treated with either vehicle or MDM2 inhibitor RG-7112. Mean value ± standard error of the mean (SEM) from n = 14 to 19 biologically independent animals per group from 2 experiments are shown, and P values were calculated using the 2-sided Mann-Whitney U test. (I) UMAP showing the FlowSOM-guided manual metaclustering of splenic live CD45+ cells from leukemia-bearing BALB/c mice after allo-HCT. (J) UMAP showing the FlowSOM-guided manual metaclustering (top) and heatmap showing median marker expression (bottom) of donor-derived (H-2kb+) TCRβ+ CD8+ T cells from leukemia-bearing BALB/c mice after allo-HCT treated with RG-7112 or vehicle, as indicated. (K) Quantification of donor-derived (H-2kb+) TCRβ+ CD8+ CD27+ TIM3+ T cells from leukemia-bearing BALB/c mice after allo-HCT treated with RG-7112 or vehicle, as indicated. (L-O) Scatterplots and representative histograms show expression of (L-M) CD127 and (N-O) Bcl-2 of CD8+ T cells isolated from spleen on day 12 following allo-HCT of WEHI-3B leukemia-bearing BALB/c mice transplanted with C57BL/6 BM plus allogeneic C57BL/6 T cells and treated with either vehicle or MDM2 inhibitor RG-7112. Mean value ± SEM from n = 14 to 19 biologically independent animals per group from 2 experiments are shown. P value was calculated using the 2-sided Mann-Whitney U test.
Patients undergoing allo-HCT receive calcineurin inhibitors (CNI), and it is unclear how this GVHD prophylaxis may impact the effect of the MDM2 inhibitor. Therefore, we pretreated mice undergoing allo-HCT with cyclosporine A (CSA) using a previously reported CNI dosage39 and then analyzed the donor T cells. We observed that the perforin and TNF expression increased in splenic CD8+ T cells derived from the group treated with the MDM2 inhibitor compared with the group treated with vehicle, despite CSA treatment in both groups (supplemental Figure 19D). MDM2 inhibition prolonged survival of AML-bearing mice despite CSA treatment compared with AML-bearing mice that received T cells and vehicle (supplemental Figure 19E). In addition, we isolated T cells from patients after allo-HCT that had received CSA and exposed them to a vehicle or MDM2 inhibitor in the presence of OCI-AML3 cells. We observed that the frequencies of CD8+CD27lowPD-1lowTIM-3low T cells increased (supplemental Figure 19F), whereas CD8+CD27+TIM3+ T cells and CD8+PD1+TIM3+ T cells declined upon treatment with the MDM2 inhibitor (supplemental Figure 19G-H). In addition, bcl-2, IFN-γ, and TNF production by human CD8+ T cells derived from patients treated with CSA increased upon treatment with MDM2 inhibitor compared with vehicle treatment (supplemental Figure 19I-K). These findings are consistent with the data from the mouse models.
In naïve mice, CD107a, TNF, and bcl-2 increased in CD8+ T cells upon MDM2 inhibition (supplemental Figure 20A-D). Effector T cells lacking CD27 display a high antigen-recall response,40 and we observed a lower frequency of CD8+CD27+TIM3+ donor T cells (TOX+TIM3+PD1+) in MDM2 inhibitor-treated recipients (Figure 5I-K; supplemental Figure 21A-B). T cells in MDM2 inhibitor-treated leukemia-bearing mice exhibited features of longevity,41 including high Bcl-2 and IL-7R (CD127) expression (Figure 5L-O).
These findings indicate that MDM2 inhibition enhances cytotoxicity and longevity of donor T cells in leukemia-bearing mice.
MDM2 inhibition enhances the glycolytic activity and enriches nucleotides and their precursors in donor T cells
Based on our previous observation that glycolysis is linked to GVL effects,6,9 we next analyzed the metabolic activity of the donor T cells of leukemia-bearing mice. We observed that donor (C57BL/6) CD8+ T cells isolated from leukemia-bearing MDM2 inhibitor-treated allo-HCT recipients (BALB/c) exhibited higher glycolytic activity compared with vehicle control subjects (Figure 6A-B). Increased glycolytic flux was confirmed by the elevated incorporation of U-13C-glucose into several glycolysis intermediates (supplemental Figure 22A). In addition, nucleotides and their precursors, particularly of the pyrimidine biosynthesis pathway, were enriched in T cells isolated from MDM2 inhibitor-treated mice (Figure 6C-E). Purine and pyrimidine de novo synthesis was shown to positively regulate cell cycle progression and maintain the survival of activated T cells.42 Increased glycolytic flux and nucleotide biosynthesis are indicative of strong T-cell activation, corresponding to higher GVL activity.9
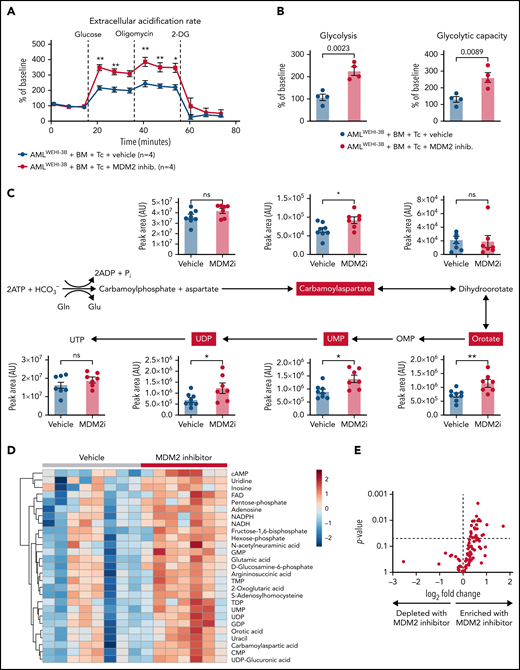
Metabolic analysis of H-2kb+CD8+ T cells derived from the vehicle or MDM2 inhibitor-treated leukemia-bearing mice. (A) Extracellular acidification rate (ECAR) of CD8+ T cells isolated from the spleen on day 12 following allo-HCT (C57BL/6 BM plus C57BL/6 T cells) of WEHI-3B leukemia-bearing BALB/c mice. Recipient mice were treated either with vehicle or MDM2 inhibitor RG-7112, as indicated. For each replicate, normalization to the ECAR baseline value was performed. Mean value ± standard error of the mean (SEM) from n = 4 biologically independent replicates; each replicate was generated by pooling the spleens from 2 mice. P values were calculated using a 2-sided unpaired Student t test. (B) Glycolysis (calculated as the difference between ECAR after glucose injection and basal ECAR) and glycolytic capacity (calculated as the difference between ECAR after oligomycin injection and basal ECAR) of CD8+ T cells isolated from BMT recipients as described in (A). Mean value ± SEM from n = 4 biologically independent replicates; each replicate was generated by pooling the spleens from 2 mice. P values were calculated using a 2-sided unpaired Student t test. (C) CD8+ T cells were enriched from the spleens of allo-HCT recipient mice and treated with an MDM2 inhibitor. Polar metabolites were extracted and measured by Liquid chromatography–mass spectrometry (LC-MS) as described in the supplemental Methods from n = 8 mice treated with vehicle and n = 7 mice treated with MDM2 inhibitor. Absolute abundance of metabolites from the pyrimidine biosynthesis pathway. Pathway scheme created with Biorender.com. *P < .05 and **P < .01. (D) CD8+ T cells were enriched from the spleens of allo-HCT recipient mice and treated with an MDM2 inhibitor. Polar metabolites were extracted and measured by LC-MS as described in the supplemental Methods from n = 8 mice treated with vehicle and n = 7 mice treated with an MDM2-inhibitor. Heatmap of the 27 significantly regulated metabolites between the MDM2 inhibitor and vehicle (P < .05). The color scale indicates the normalized concentration in each sample. (E) Volcano plot of 100 metabolites analyzed with a targeted approach isolated from CD8+ T cells that were enriched from the spleens of allo-HCT–recipient mice, treated with MDM2 inhibitor or vehicle. Polar metabolites were extracted and measured by LC-MS as described in the supplemental Methods from n = 8 mice treated with vehicle and n = 7 mice treated with an MDM2-inhibitor. P values were calculated using the unpaired 2-tailed Student t-test.
Metabolic analysis of H-2kb+CD8+ T cells derived from the vehicle or MDM2 inhibitor-treated leukemia-bearing mice. (A) Extracellular acidification rate (ECAR) of CD8+ T cells isolated from the spleen on day 12 following allo-HCT (C57BL/6 BM plus C57BL/6 T cells) of WEHI-3B leukemia-bearing BALB/c mice. Recipient mice were treated either with vehicle or MDM2 inhibitor RG-7112, as indicated. For each replicate, normalization to the ECAR baseline value was performed. Mean value ± standard error of the mean (SEM) from n = 4 biologically independent replicates; each replicate was generated by pooling the spleens from 2 mice. P values were calculated using a 2-sided unpaired Student t test. (B) Glycolysis (calculated as the difference between ECAR after glucose injection and basal ECAR) and glycolytic capacity (calculated as the difference between ECAR after oligomycin injection and basal ECAR) of CD8+ T cells isolated from BMT recipients as described in (A). Mean value ± SEM from n = 4 biologically independent replicates; each replicate was generated by pooling the spleens from 2 mice. P values were calculated using a 2-sided unpaired Student t test. (C) CD8+ T cells were enriched from the spleens of allo-HCT recipient mice and treated with an MDM2 inhibitor. Polar metabolites were extracted and measured by Liquid chromatography–mass spectrometry (LC-MS) as described in the supplemental Methods from n = 8 mice treated with vehicle and n = 7 mice treated with MDM2 inhibitor. Absolute abundance of metabolites from the pyrimidine biosynthesis pathway. Pathway scheme created with Biorender.com. *P < .05 and **P < .01. (D) CD8+ T cells were enriched from the spleens of allo-HCT recipient mice and treated with an MDM2 inhibitor. Polar metabolites were extracted and measured by LC-MS as described in the supplemental Methods from n = 8 mice treated with vehicle and n = 7 mice treated with an MDM2-inhibitor. Heatmap of the 27 significantly regulated metabolites between the MDM2 inhibitor and vehicle (P < .05). The color scale indicates the normalized concentration in each sample. (E) Volcano plot of 100 metabolites analyzed with a targeted approach isolated from CD8+ T cells that were enriched from the spleens of allo-HCT–recipient mice, treated with MDM2 inhibitor or vehicle. Polar metabolites were extracted and measured by LC-MS as described in the supplemental Methods from n = 8 mice treated with vehicle and n = 7 mice treated with an MDM2-inhibitor. P values were calculated using the unpaired 2-tailed Student t-test.
To understand if CSA impacts the effect of the MDM2 inhibitor on metabolic changes in T cells, we analyzed the glycolytic activity of human T cells that were treated with the MDM2 inhibitor in the presence vs absence of CSA. We observed that glycolytic activity was not different in MDM2 inhibitor-treated T cells exposed to the vehicle compared with MDM2 inhibitor-treated T cells exposed to CSA (supplemental Figure 22B-C). Enrichment for nucleotides and their precursors was not different in the MDM2 inhibitor/vehicle-treated T cells compared with MDM2 inhibitor/CSA-treated T cells (supplemental Figure 23A).
These findings indicate that besides the phenotypical changes of T cells upon MDM2 inhibition, glycolytic activity and nucleotide synthesis increased in donor T cells upon MDM2 inhibition.
Discussion
The curative potential of allo-HCT for AML is limited by relapse, causing 57% of the mortality of patients undergoing this intervention.1,2 The GVL effect depends on donor T cells that recognize foreign MHC molecules on the AML cells. Therefore, major mechanisms of immune evasion include the genetic loss of mismatched MHC4 and reduced expression of MHC molecules on leukemia cells.5,10 These data suggest that therapeutic approaches that increase MHC-II expression could enhance antileukemia immunity after allo-HCT. Here, we identified a pharmacological approach to enhance MHC-II expression on AML cells using MDM2 inhibition. The increased MHC-II expression was connected to increased expression of markers of cytotoxicity and activation, whereas exhaustion markers were reduced on T cells.
In addition, MDM2 inhibition induced TRAIL-R1/2 expression and sensitized AML cells to TRAIL-mediated apoptosis, which could be induced by soluble TRAIL or TRAIL-expressing T cells. Upon TRAIL ligation, TRAIL death receptors induce the assembly of the death-inducing signaling complex, composed of FAS (CD95/TNFRSF6)-associated protein with death domain and procaspase-8/10 at their intracellular death domain,43 thus TRAIL-R activation exerts antitumor activity.44 In addition, we found that MDM2 inhibition increased markers of cytotoxicity and activation on T cells in naïve mice in the absence of leukemia.
These findings indicate that targeting MDM2 enhances antileukemia immunity after allo-HCT in mice and humans via 3 converging mechanisms involving MHC-II upregulation on AML cells, increased TRAIL-R1/2 expression on leukemia cells, and direct T-cell activation (supplemental Figure 24).
Our finding that p53 plays a central role in response to immunotherapy is in agreement with studies showing that p53 increases MHC class-I expression on pancreatic cancer cells, rendering them more immunogenic.27 We show that MDM2 inhibition does not lead to upregulation of HLA-DR and HLA-A,B,C, as well as TRAIL-R1/2 in p53-mutant leukemia, which is relevant for a fraction of patients with TP53-mutated AML.45 The direct effect of MDM2 inhibition on T cells that we observed in mice without leukemia will still be intact despite the lack of functional p53 in the AML cells. Consistent with our results, others have described the direct effects of MDM2 inhibition on T cells used to treat solid tumors in mouse models.28 MDM2 inhibition leads to upregulation of MDM2, and high MDM2 concentrations in T cells sustain STAT5 stability, which enhances T-cell–mediated antitumor immunity against solid tumors.28 Consistent with this report, we also observed increased MDM2 concentrations in leukemia cells and increased T-cell activation in leukemia-free mice upon MDM2 inhibition. In addition to the effects of the MDM2/STAT5 axis in T cells, MDM2-induced p53 can activate a viral mimicry pathway and tumor inflammation signature genes in melanoma patients.30 These studies and our study support the concept of a central role for p53 in immune activation for cancer immunotherapy. However, p53-mutant AML is per se not resistant to immunotherapy, as previous work has shown that TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy, a bispecific dual-affinity retargeting (DART) antibody to CD3epsilon and CD123, in AML,24 indicating that the role of functional p53 in immunotherapy is dependent on the immunotherapeutic intervention and that the bispecific DART antibody flotetuzumab acts possibly independent of functional p53. Type 1 IFNs cause upregulating of MHC-II and were recently tested in a trial for the prevention of leukemia relapse after allo-HCT.46 Patients with treatment-resistant AML not in remission or those with poor-risk leukemia received pegylated IFNα.46 The authors reported that the incidence of relapse was 39% at 6 months, which was sustained through 1 year after allo-HCT.46
The finding that MDM2 inhibition installs MHC-II and TRAIL-R1/2 on the surface of AML cells is of particular interest because MDM2 inhibitors are well tolerated not only in our experimental AML models but also in patients with cancer.47 At advanced stages of leukemia, immune surveillance is disturbed by multiple mechanisms such as LA release and impaired T-cell function.9 Therefore, the immune-sensitizing effect of MDM2 inhibition on AML cells is likely most effective when applied in the context of a functionally intact immune system, in particular at minimal residual disease stage after allo-HCT in which MDM2 inhibition may allow for sensitizing the AML cells to TRAIL-mediated elimination by alloreactive T cells activated by MHC-I and II. This treatment concept will be clinically explored in a phase 1/2 proof-of-concept trial (siremadlin [HDM201] for post–allo-HCT for AML relapse prevention, Novartis-sponsored study CHDM201K12201). In summary, we identified a targetable vulnerability of AML cells to T-cell–mediated cytotoxicity through the restoration of p53-dependent TRAIL-R1/2 and MHC-II production by AML cells and donor T-cell activation via MDM2 inhibition.
Acknowledgments
The authors thank the Lighthouse Core Facility for cell sorting; Heide Dierbach and Katja Graewe for tissue processing and staining; and the Metabolomics Core Facility at the Max Planck Institute of Immunobiology and Epigenetics, Freiburg, for chromatography-mass spectrometry measurements.
This study is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), SFB-1479 – Project ID: 441891347, SFB TRR167 (to R.Z., M.B., and M.P.), SFB-1453 – Project-ID 431984000 (to M.B.), and SFB1160 (to M.B., M.R., and R.Z.); ERC Consolidator grant (681012 GvHDCure to R.Z.); Germany’s Excellence Strategy (CIBSS – EXC-2189 – Project ID 390939984 to R.Z. and N.K.); and INTERREG V European regional development fund (European Union) program (project 3.2 TRIDIAG to R.Z.). P.A. is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - project number 492259164, and by the Deutsches Konsortium für Translationale Krebsforschung (DKTK, German Cancer Consortium) – project number FR01-375. J.R. is supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (2019R1A6A3A03032009). M.B. is supported by the German Federal Ministry of Education and Research (BMBF) within the framework of the e:Med research and funding concept (coNfirm, FKZ 01ZX1708F) and within the Medical Informatics Funding Scheme (MIRACUM, FKZ 01ZZ1606A-H). B.R.B. received funding from the National Institutes of Health R01 HL56067 and R37 AI34495. B.B. received funding through the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program grant agreement No. 882424 (ERC-AdG:IMPACT), the Swiss National Science foundation CRSII5_183478 and 310030_188450, and the University Priority Project translational cancer research.
Authorship
Contribution: J.N.H.G.H., D.S., T.L., J.R., and E.-P.D. performed the majority of the experiments, helped to develop the overall concept, analyzed data, and helped to write the manuscript; N.G.N. and S.C.-P. acquired and analyzed CyTOF data; V.F. performed p53 Western blots; C.T. and M.P. analyzed human data from the Cancer Genome Atlas Research Network initiative and BEAT AML initiative; T.W. helped to analyze data; M.-C.R. performed experiments using AML models; M.F. helped with qPCR; C.B. helped with qPCR and flow-cytometry; N.K. helped to analyze data and write the manuscript; J.M.V. and M.L. helped to perform in vivo experiments; K.S. and F.K. analyzed patients samples; H.E. and L.M.B. helped to characterize the leukemia models; T.R. helped with sea horse experiments; M.M. and S.H. provided human AML relapse samples and clinical data; A.L.I. helped with the murine leukemia models and provided essential reagents; F.M. helped with western blot experiments; G.A. analyzed microarray data; S.D. helped to perform experiments; D.P. helped to analyze microarray data; J.S. and M.R. helped to perform TRAIL-R1 and R2 knockout experiments; C.M. provided p53 tg OCI-AML3 cells; J.D. and H.D.M. helped to develop the studies and to analyze data; M.B. analyzed microarray data; B.R.B. helped to analyze data and write the manuscript; P.A. performed experiments, analyzed data, and helped to write the manuscript; L.V. analyzed data from the Cancer Genome Atlas Research Network initiative and BEAT AML initiative; E.L.P. helped to develop the concept and analyzed data on T-cell metabolism; J.M.B. and N.C.-W. performed metabolomics analysis and analyzed data; B.B. helped to develop the concept, helped with CyTOF-based analysis, and helped to write the manuscript; and R.Z. developed the overall concept, supervised the experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: H.D.M. holds a patent application for the use of MDM2 inhibition to prevent or treat leukemia relapse after allo-HCT (application number: PCT/EP2021/075896). H.D.M. is an employee of Novartis. K.S. received honoraria from Novartis and Blueprint Medicines. B.R.B. receives remuneration as an advisor to Magenta Therapeutics and BlueRock Therapeutics; research funding from BlueRock Therapeutics, Rheos Medicines, Equilibre Biopharmaceuticals, and Carisma Therapeutics, Inc.; and is a cofounder of Tmunity Therapeutics. R.Z. received honoraria from Novartis, Incyte, and Mallinckrodt outside of the submitted work. The remaining authors declare no competing financial interests.
Correspondence: Robert Zeiser, Department of Hematology, Oncology, and Stem Cell Transplantation, University Medical Center Freiburg, Hugstetter Str. 53, D-79106 Freiburg, Germany; e-mail: robert.zeiser@uniklinik-freiburg.de.
All data are available from the authors upon request. Microarray data are deposited in the Gene Expression Omnibus (GEO) database repository under the GEO accession GSE158103. All other materials and methods are described in the supplemental Methods.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal