

Key Points
The ERK2-DBP substrate interaction domain opposes progression in a JAK2V617F dependent model of myeloproliferative neoplasm.
The ERK-D domain may represent an effective therapeutic target for pharmacologic intervention.

Abstract
Although Ras/mitogen-activated protein kinase (MAPK) signaling is activated in most human cancers, attempts to target this pathway using kinase-active site inhibitors have not typically led to durable clinical benefit. To address this shortcoming, we sought to test the feasibility of an alternative targeting strategy, focused on the ERK2 substrate binding domains, D and DEF binding pocket (DBP). Disabling the ERK2-DBP domain in mice caused baseline erythrocytosis. Consequently, we investigated the role of the ERK2-D and -DBP domains in disease, using a JAK2-dependent model of polycythemia vera (PV). Of note, inactivation of the ERK2-DBP domain promoted the progression of disease from PV to myelofibrosis, suggesting that the ERK2-DBP domain normally opposes progression. ERK2-DBP inactivation also prevented oncogenic JAK2 kinase (JAK2V617F) from promoting oncogene-induced senescence in vitro. The ERK2-DBP mutation attenuated JAK2-mediated oncogene-induced senescence by preventing the physical interaction of ERK2 with the transcription factor Egr1. Because inactivation of the ERK2-DBP created a functional ERK2 kinase limited to binding substrates through its D domain, these data suggested that the D domain substrates were responsible for promoting oncogene-induced progenitor growth and tumor progression and that pharmacologic targeting of the ERK2-D domain may attenuate cancer cell growth. Indeed, pharmacologic agents targeting the ERK2-D domain were effective in attenuating the growth of JAK2-dependent myeloproliferative neoplasm cell lines. Taken together, these data indicate that the ERK-D and -DBP domains can play distinct roles in the progression of neoplasms and that the D domain has the potential to be a potent therapeutic target in Ras/MAPK-dependent cancers.
Introduction
Ras/MAPK signaling is activated in more than 85% of human cancers.1,2 The terminal effectors of the Ras/Raf/MEK sequential cascade, ERK1/2, play a central role in regulating both transformation and numerous normal biological processes, by phosphorylating downstream targets in the cytoplasm and nucleus.3-5 Interestingly, despite the high homology shared by ERK1 and ERK2,6 ERK2 has functional capabilities that are distinct from those of ERK1 in several pathophysiological contexts, including embryonic development, metabolic stress responses, epithelial-to-mesenchymal transformation, tumor growth, and cancer drug addiction.7-11 Despite the fundamental importance of ERK2 in the pathogenesis of cancer, our understanding of the collection of ERK2 targets that support this process remains incomplete. ERK2 interacts with substrates through 2 domains on opposing faces of the protein: the common docking (D) domain and DEF binding pocket (DBP) domain.12 The D domain targets ∼80% of canonical ERK substrates, and the remainder are targeted by the DBP domain, which recognizes substrates through the presence of a DEF (docking site for ERK) motif.12,13 We previously explored the functions of the substrate interaction domains of ERK2 in T-cell development, finding that the ERK2-DBP domain plays a critical role in the early development of γδ T cells, but is dispensable for early development of the alternate αβ T-cell lineage.14 The role of the ERK2-D and -DBP domains in other ERK2-dependent processes, specifically cancer development and progression, has not been explored15,16; however, their importance in cancer is hinted at by the presence of recurrent ERK2 somatic mutations in lymphomas and leukemias.17-19
Myeloproliferative neoplasms (MPNs) are a collection of chronic myeloid malignancies characterized by clonal expansion of ≥1 myeloerythroid lineages; MPN present clinically as an overproduction of erythrocytes (polycythemia vera [PV]), increased thrombocytes (essential thrombocythemia), excessive white blood cells with progressive bone marrow (BM) fibrosis (primary myelofibrosis), and ultimate transformation into highly aggressive and rapidly lethal acute myeloid leukemia.20-23 Acquired JAK2V617F gain-of-function mutations that produce constitutive activation of JAK/STAT signaling are evident in >95% of patients with PV.22-25 JAK2V617F+ MPNs provide a unique ERK-dependent model for studying novel molecular mechanisms underlying disease progression from chronic PV/essential thrombocythemia to advanced primary myelofibrosis/acute leukemia,26-28 in that MAPK/ERK signaling is both an important driver and a therapeutic target for decreasing the excessive proliferation, survival, and fibrosis caused by JAK2V67F+ MPN cells.29
In our study, disabling the DBP domain of ERK2 promoted baseline erythrocytosis and progression of PV from erythrocytosis to myelofibrosis (MF) in a JAK2V617F-driven model of MPN. Furthermore, the enhanced disease progression displayed by ERK2-DBP–mutant progenitors in vivo was paralleled by their failure to undergo JAK2V617F-mediated oncogene-induced senescence (OIS) in vitro. ERK2-DBP inactivation abrogates OIS by preventing the physical association of ERK2 with Egr1, a well-established tumor suppressor in myeloid malignancies.30-33 Because DBP-mutant ERK2 can only target substrates through its D domain, these data suggest that ERK2 promotes disease progression through the D domain and could serve as a therapeutic target. Indeed, pharmacologic agents targeting the ERK2-D domain reduce both JAKV617F-driven progenitor activity of primary HSPCs and the proliferation and survival of human MPN cells.
Methods
Mice
All mice were maintained in the Association for Assessment and Accreditation of Laboratory Animal Care–accredited animal facility at Fox Chase Cancer Center of Temple University. Erk1−/−Erk2fl/fl,34Erk2Y261A/Y261A(DBP),14Vav-Cre,35 and Egr1−/−31 mice have been described. Rag2−/−;Il2rg−/− immunodeficient mice were obtained from Envigo, Inc. All animal studies were approved by the Institutional Animal Care and Use Committee of Fox Chase Cancer Center.
Constructs and retroviral transduction
pMSCV-IRES-GFP (pMIG) and pMIG-JAK2V617F were provided by Michael Deininger (Huntsman Cancer Institute). pMICherry-Egr1 was generated by subcloning Egr1 from pMSCV-mEgr1/Flag-IRES-GFP.36 The pMICherry-Egr1Y253A/Flag construct was generated by site-directed mutagenesis (Geneart Kit; Invitrogen). The retroviral supernatant was generated by cotransfection of Phoenix-293 T cells with pCL-Eco using jetPRIME (Polyplus Transfection, New York, NY). Cell transduction was performed by spin infection using polybrene (8 μg/mL; Sigma).
Adoptive-transfer PV model
BM HSPCs were enriched by magnetic bead depletion (Qiagen) using anti-CD3 (17A2), anti-B220 (RA3-6B2), anti–Gr-1 (RB6-8C5), anti-CD11b (M1/70), and anti-Ter119 (Ter119) antibodies. HSPCs were cultured in Iscove modified Dulbecco medium containing 10% fetal bovine serum and interleukin 3 (IL-3), IL-6, and stem cell factor cytokines for 2 days before spin infection with the pMIG-JAK2V617F retrovirus. Finally, 4 × 105 infected (GFP+) cells and 8 × 105 noninfected cells were injected IV into sublethally irradiated (220 rad) Rag2−/−Il2rg−/− mice. Disease progression was monitored by biweekly retro-orbital bleeding.
Peripheral blood and histological tissue analysis
Complete blood counts (CBCs) were measured with the VetScan (Abaxis, Union City, CA) according to the manufacturer’s instructions. Blood smears were stained with Wright-Giemsa for morphological analysis. For histopathology, mouse tissues were fixed in neutral buffered formalin, dehydrated, embedded in paraffin, and stained with hematoxylin and eosin. Gömöri’s silver impregnation technique was used for reticulin staining. For collagen staining, femurs, prepared as above for histopathology, were hydrated in distilled water, soaked in preheated (58°C) Bouin solution for 15 minutes, and stained sequentially with modified Weigert’s iron hematoxylin, acid-alcohol solution, Biebrich scarlet-acid fuchsin solution, 1% phosphomolybdic acid, aniline blue, and acetic acid. Stained tissue was dehydrated in graded alcohol and cleared in xylene before viewing.
Flow cytometry
Single-cell suspensions were stained with anti–Gr-1, anti-CD11b, anti-Ter119, anti-CD41, anti-CD19, anti-CD44, and anti-Thy1.2. All antibodies were purchased from eBioscience, BD Biosciences, or Biolegend. Dead cells were excluded by using propidium iodide or 4′,6-diamidino-2-phenylindole (Biolegend). Data were acquired on an LSR-II Flow Cytometer and analyzed with Flowjo software (Treestar, Ashland, OR).
Colony assays
Retrovirally transduced HSPCs were sorted by fluorescence-activated cell sorting and plated in triplicate in cytokine-supplemented methylcellulose medium (M3434, M3534, or M3436; Stemcell Technologies) using 3000 or 1000 cells per well in 12-well plates. Colonies were scored at day 7 in culture. Images were captured with an SMZ1500 microscope equipped with a DS-Fi1 digital camera and Ar imaging software (all from Nikon).
Senescence-associated β-galactosidase staining
Transduced HSPCs were cultured as for the colony assays for 3 days. Senescence was determined by senescence-associated β-galactosidase (SA-β-gal) staining with the histochemical staining kit (CS0030; Sigma), as previously reported.37,38 Blue-green–stained senescent cells were photographed and scored, using a ×40 objective under a bright-field microscope (Eclipse TE300; Nikon) equipped with the Ar imaging software. For fluorescence-based analysis of senescence, HSPCs were stimulated as for the colony assays and transduced with either pMIG or pMIG-JAK2-V617F retrovirus. After 4 days, transduced cells were treated with bafilomycin A1 (500 nM) for 30 minutes, then C12RG (33 μM) for an additional 30 minutes. Phenyl-ethyl β-d-thiogalactopyranoside (1 mM) was added to stop the reaction, after which the cells were stained with fluorescent conjugated antibodies (cKit, Sca-1, CD16/32, CD34, CD150, CD48, CD105, and CD41) and analyzed by flow cytometry.
Real-time polymerase chain reaction
Total RNA was extracted by using Trizol (Invitrogen) and glycogen (Ambion). RNA was reverse transcribed with random hexamers and the Super Script II Kit (Invitrogen), after which RNA targets were quantified with stock TaqMan primer/probe sets (Applied Biosystems [ABI]) or SYBR Green (Qiagen) on an ABI Prism 7700 real-time polymerase chain reaction system. All measurements were normalized to β-actin.
Cell lines, drug treatment, and proliferation assay
SCID.adh murine lymphoma cells39 were pretreated with MEK1/2 inhibitor U0126 (Sigma) or ERK-D inhibitor (76) for 30 minutes, followed by phorbol myristate acetate (PMA; 50 ng/mL) stimulation for 2 hours for analysis of signaling pathways. The following JAK2V617F-expressing cell lines were maintained in RPMI-1640 medium supplemented with 20% fetal bovine serum: SET-2, UKE-1, Ku812, and BAF/3-JAK2VF. The effect of ERK inhibition of cell growth and viability was analyzed with WST-8 (Cell Counting Kit 8; Sigma). The absorbance was read at 450 nm with a TECAN plate reader (Infinite M200; TECAN). The relative change in viability was calculated using values normalized to baseline at day 0.
Coimmunoprecipitation and western blot analysis
Primary mouse HSPCs or SCID.adh cells were lysed in mammalian protein extraction reagent (M-PER; Pierce). Immunoblot analysis was performed as previously described.14 The following antibodies were used: anti–phospho-STAT3 (9131; Cell Signaling), anti–total-STAT3 (9139), anti–phospho-ERK1/2 (9106S), anti-ERK2 (9102), anti–phospho-RSK (12032), anti–total-RSK (8408), and anti-FLAG (2368; all from Cell Signaling); anti-Egr1 (A303-390A; Bethyl Laboratories); and anti–β-actin (sc-47778; Santa Cruz Biotechnology). SCID.adh cells were transduced with pMICherry, pMICherry-Egr1(WT)/FLAG, or pMICherry-Egr1Y253A/FLAG), pretreated with PMA (50 ng/mL) for 30 minutes, then lysed with lysis buffer (50 mM Tris [pH 7.5], 120 mM NaCl, 1 mM EDTA, 5% glycerol, 1% Triton X-100, and 0.15% sodium deoxycholate, containing 1 mM phenylmethylsulfonyl fluoride and protease/phosphatase inhibitors) at 4°C. The protein extracts were immunoprecipitated with anti-FLAG M2 magnetic beads (M8823; Sigma) and subjected to immunoblot analysis.
In vivo assessment of 76 efficacy
SET-2 cells (1 × 107) were subcutaneously engrafted on the flank of Il2rg−/−;Rag2−/− mice in 1:1 medium (Cultrex type II gel; R&D Systems). When the tumor reached 40 mm3, either dimethyl sulfoxide or compound 76 was administered intraperitoneally every other day at 10 mg/kg. Tumor volume was measured with calipers and calculated with the formula longest side × width × height/2. Single-cell suspensions were evaluated by flow cytometry with antibodies to HLA-DR.
3D structure modeling and bioinformatics
The crystal structure of ERK2 protein complexed with an MAPK docking peptide (MKNK1) was acquired from the Protein Data Bank (http://www.rcsb.org/structure/2Y9Q). Multiple alignments of Egr1 amino acid sequences were obtained with Clustal Ω (https://www.ebi.ac.uk/Tools/msa/clustalo/).
Statistical analysis
Statistical analysis was performed in Prism (GraphPad) by 1-way analysis of variance or Student t test. Statistical significance was set at P < .05. All experiments were repeated ≥3 times.
Results
ERK2-DBP inactivation promotes progression of JAK2V617F-induced PV
ERK2-Y261A (Erk2Y261A) mutant mice bear an Erk2 allele in which the ERK2-DBP domain is inactivated, but ERK2 activity and D-domain function are preserved.14 The role of the ERK2-DBP domain in early hematopoiesis has not been explored. To address this deficit, we intercrossed Erk1+/−Erk2+/Y261A mice, which revealed that Erk2Y261A/Y261A mice exhibited a survival defect, suggesting that the ERK2-DBP domain plays an important role during embryogenesis (supplemental Figure 1A-B, available on the Blood Web site). Surprisingly, among the surviving Erk1−/−Erk2Y261A/Y261A mice, we observed considerable splenomegaly (Figure 1A) and erythrocytosis, characterized by marked expansion of CD44+/Ter119+ early erythroblasts and Ter119+CD44+/FSChi immature erythroid progenitors (Figure 1B).40
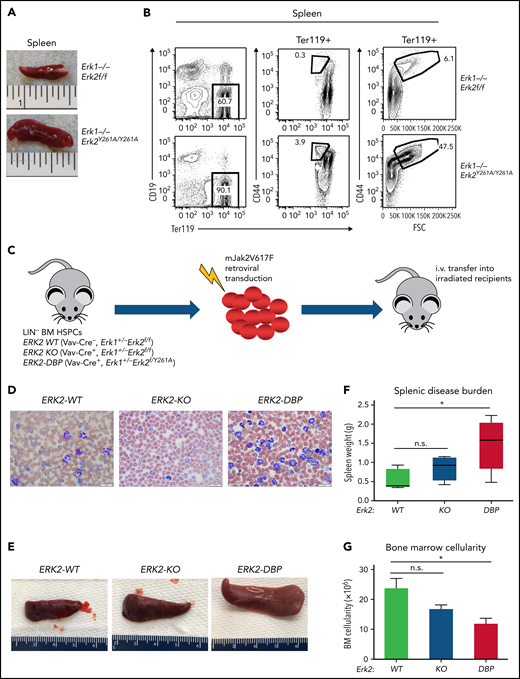
Effect of disabling the ERK2-DBP domain on erythropoiesis and JAK2V617F-driven PV progression. (A) Photograph of representative spleens from Erk2-WT and ERK2Y261A/Y261A mice. (B) Flow cytometry analysis on the effect of disabling the ERK2-DBP domain on expansion of red blood cell precursors (CD44+/Ter119+) in mice of the indicated genotypes. (C) Adoptive transfer model of PV. Lin− HSPCs were harvested from the BM of ERK2-WT, -KO or -Y261A (DBP) mice; transduced with JAK2V617F-expressing retrovirus; and IV injected into sublethally irradiated immunodeficient recipient mice (n = 7 per group). (D) Peripheral blood smears from ERK2-WT, -KO, and –DBP-Y261A HSPC recipient mice at 12 weeks after transfer. Scale bars, 20 µm, Wright-Giemsa stain. (E-F) Spleen size and weight of mice transferred with JAK2V617F-transduced ERK-WT–, -KO–, and -DBP–mutant HSPCs. Results are expressed as the mean ± standard deviation (SD). *P < .05. (G) BM cell counts of JAK2V617F-transduced ERK-WT–, -KO–, or -DBP–mutant mice. The data are expressed as the mean ± SD. *P < .05; n.s., not significant.
Effect of disabling the ERK2-DBP domain on erythropoiesis and JAK2V617F-driven PV progression. (A) Photograph of representative spleens from Erk2-WT and ERK2Y261A/Y261A mice. (B) Flow cytometry analysis on the effect of disabling the ERK2-DBP domain on expansion of red blood cell precursors (CD44+/Ter119+) in mice of the indicated genotypes. (C) Adoptive transfer model of PV. Lin− HSPCs were harvested from the BM of ERK2-WT, -KO or -Y261A (DBP) mice; transduced with JAK2V617F-expressing retrovirus; and IV injected into sublethally irradiated immunodeficient recipient mice (n = 7 per group). (D) Peripheral blood smears from ERK2-WT, -KO, and –DBP-Y261A HSPC recipient mice at 12 weeks after transfer. Scale bars, 20 µm, Wright-Giemsa stain. (E-F) Spleen size and weight of mice transferred with JAK2V617F-transduced ERK-WT–, -KO–, and -DBP–mutant HSPCs. Results are expressed as the mean ± standard deviation (SD). *P < .05. (G) BM cell counts of JAK2V617F-transduced ERK-WT–, -KO–, or -DBP–mutant mice. The data are expressed as the mean ± SD. *P < .05; n.s., not significant.
The overproduction of erythroid precursors in ERK2-DBP–mutant mice resembled the erythrocytosis observed in PV, raising the question of whether the ERK2-DBP influences PV pathogenesis. PV is characterized by activating mutations in JAK2 kinase (JAK2V617F),41 which promote disease pathogenesis in an ERK-dependent manner.42 Thus, we used a Jak2V2617F-driven adoptive transfer model to assess the ERK2-DBP domain function in PV pathogenesis. To avoid survival issues and focus on the hematopoietic compartment, we conditionally activated the Erk2Y261A allele in hematopoietic progenitors by excision of the Erk2Fl allele in hemizygous Erk2Y261A/Fl mice, by using Vav-Cre.35 Ectopic expression of JAK2V617F in murine HSPCs produces a myeloproliferative disease that mimics human PV.43-46 To assess the effect of the loss of ERK2-DBP function on PV pathogenesis in vivo, we transduced mJAK2V617F into BM HSPCs derived from ERK2-WT (VavCr−, Erk1+/−Erk2fl/fl), ERK2-KO (VavCre+, Erk1+/−Erk2fl/fl), and ERK2-DBP–mutant (VavCre+, Erk1+/−Erk2fl/Y261A) mice, followed by transplantation into irradiated, immunodeficient recipient mice (Rag2−/−Il2rg−/−; Figure 1C). All transduced donor HSPCs had an intact Erk1 allele and exhibited equivalent engraftment (supplemental Figure 1C). Recipients of Jak2V617F-expressing wild-type (WT) HSPCs developed disease resembling human PV, with elevated red blood cell counts, hematocrit, and hemoglobin levels (supplemental Figure 2A-C). ERK2-KO HSPC recipients exhibited transient erythrocytosis (supplemental Figure 2A-C), suggesting that ERK2 is essential for sustaining erythrocytosis. ERK2-DBP–mutant recipients exhibited mild but sustained erythrocytosis (supplemental Figure 2A-C). Interestingly, ERK2-DBP–mutant HSPC recipients also exhibited a marked increase in circulating GFP+ leukocytes with a predominance of mature neutrophils and monocytes (Figure 1D; supplemental Figure 2D-H), suggestive of disease progression in ERK2-DBP–mutant HSPC recipients. Consistent with previous studies,43-46 thrombocytosis was not observed in any recipient mice (supplemental Figure 2I). At 12 weeks after transplantation, ERK2-DBP HSPC recipients exhibited massive splenomegaly (Figure 1E-F; supplemental Figure 2J) and decreased BM cellularity (Figure 1F-G). Together, these data suggest that that the ERK2-DBP domain normally acts to oppose PV progression, perhaps to MF.
ERK2-DBP inactivation promotes the development of MF in a murine PV model
To determine whether the ERK2-DBP mutation promotes progression to MF, we performed histopathological and reticulin staining analyses. At 12 weeks after transplantation, recipients of WT HSPCs displayed hypercellularity with expansion of erythroid precursors and increased megakaryocytes, reminiscent of early-stage, PV-like disease (Figure 2A-C). These features were less pronounced in the recipients of ERK2-KO HSPCs (Figure 2A-C). In contrast, ERK2-DBP–mutant HSPC recipients exhibited osteosclerosis and granulocytic hyperplasia in the BM, together with expansion and clustering of atypical megakaryocytes and distorted architecture in the spleen (Figure 2A-C). Reticulin and collagen staining confirmed more extensive fibrosis with interstitial infiltration in both the BM and spleen of ERK2-DBP–mutant HSPC recipients (Figure 2B-B′,D; supplemental Figure 3). Recipients of ERK2-DBP–mutant HSPCs also exhibited far greater lobular infiltration of neutrophils, megakaryocytes, and erythroid progenitors into the liver (supplemental Figure 3A). Consistent with the morphologic assessment, flow cytometry on GFP+ cells revealed that the frequency of myeloid cells (GFP+/Gr1+ and GFP+/Mac1+) in the BM of ERK2-DBP–mutant recipient mice increased significantly (Figure 3A-B). In contrast, neither erythroid cells (GFP+/Ter119+) nor megakaryocytes (GFP+/CD41+) in BM and spleen were altered in ERK2-DBP–mutant HSPC recipients (Figure 3C-D). Thus, inactivation of the ERK2-DBP domain potentiated JAK2V617F-driven disease, including extensive extramedullary hematopoiesis and progression to MF.

Morphologic evidence of MF in the BM and spleen of mice receiving ERK2-DBP–mutant HSPCs. (A-B) Hematoxylin and eosin and reticulin staining of representative BM sections from mice that received ERK2-WT–, -KO– and -DBP–mutant HSPCs, at 12 weeks after BM transplant. (B′) Enlarged images delimited by the blue inset in panel B. Scale bars, 20 µm. (C-D) Representative images of sections of spleen from the mice above, stained with hematoxylin and eosin (C) or reticulin (D) 12 weeks after BMT. Scale bars, 50 µm.
Morphologic evidence of MF in the BM and spleen of mice receiving ERK2-DBP–mutant HSPCs. (A-B) Hematoxylin and eosin and reticulin staining of representative BM sections from mice that received ERK2-WT–, -KO– and -DBP–mutant HSPCs, at 12 weeks after BM transplant. (B′) Enlarged images delimited by the blue inset in panel B. Scale bars, 20 µm. (C-D) Representative images of sections of spleen from the mice above, stained with hematoxylin and eosin (C) or reticulin (D) 12 weeks after BMT. Scale bars, 50 µm.
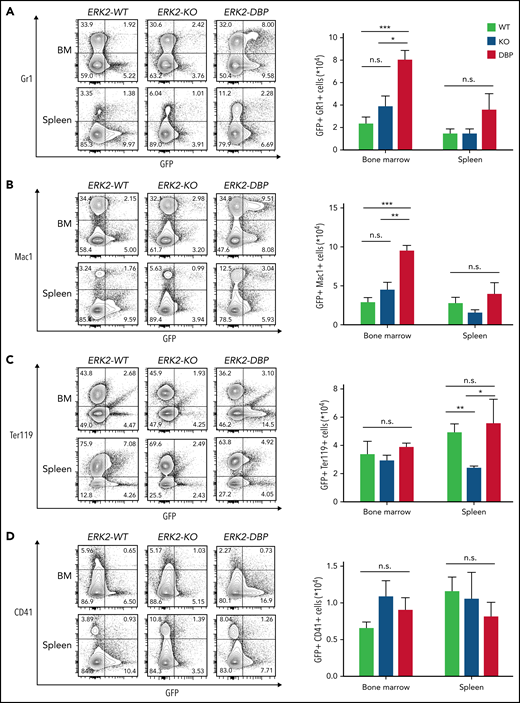
Preferential accumulation of myeloid progenitors in the BM of mice that received ERK2-DBP–mutant HSPCs. (A-B) Percentage and absolute number of GFP+/Gr1+ or GFP+/Mac1+ myeloid cells per 1 × 106 cells in the BM and spleen of mice receiving ERK2-WT–, -KO–, and -DBP–mutant HSPCs. (C) Percentage and absolute number of GFP+/Ter119+ erythroid cells per 1 × 106 cells in BM and spleen in the mice in panels A and B. (D) Percentage and absolute number of GFP+/CD41+ platelets per 1 × 106 cells in BM and spleen in the same mice as in panels A and B. Data are expressed as the mean ± standard error of the mean (n = 5). *P < .05; **P < .01; ***P < .001.
Preferential accumulation of myeloid progenitors in the BM of mice that received ERK2-DBP–mutant HSPCs. (A-B) Percentage and absolute number of GFP+/Gr1+ or GFP+/Mac1+ myeloid cells per 1 × 106 cells in the BM and spleen of mice receiving ERK2-WT–, -KO–, and -DBP–mutant HSPCs. (C) Percentage and absolute number of GFP+/Ter119+ erythroid cells per 1 × 106 cells in BM and spleen in the mice in panels A and B. (D) Percentage and absolute number of GFP+/CD41+ platelets per 1 × 106 cells in BM and spleen in the same mice as in panels A and B. Data are expressed as the mean ± standard error of the mean (n = 5). *P < .05; **P < .01; ***P < .001.
The ERK2-DBP domain regulates JAK2V617F-mediated OIS
To determine how inactivation of the ERK2-DBP domain promotes JAK2V617F-induced disease progression, we assessed the clonogenic potential of JAK2V617F-expressing HSPCs in vitro in methylcellulose supporting distinct progenitor populations (myeloid/erythroid/megakaryocyte, M3434; and granulocyte-macrophage, M3536). HSPCs from ERK2-WT–, -KO–, and -DBP–mutant mice exhibited no difference in baseline colony formation (Figure 4A-B); however, they responded very differently to JAK2V617F transduction, which markedly reduced colony formation by HSPCs from both ERK2-WT and -KO mice (Figure 4A-B). Importantly, JAK2V617F transduction did not reduce colony formation by HSPCs from ERK2-DBP–mutant mice (Figure 4A-B). These differences in colony formation were not evident in methylcellulose cultures supporting erythroid progenitors (supplemental Figure 4A).
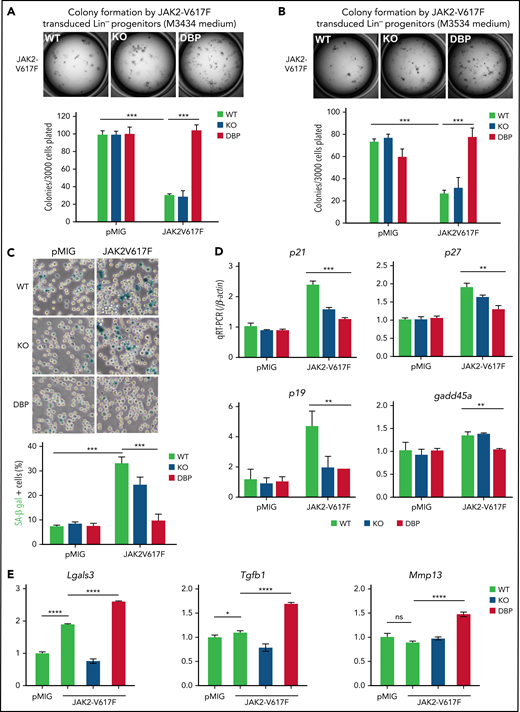
JAK2V617F-induced senescence was alleviated by inactivation of the ERK2-DBP domain. (A-B) Control and JAK2V617F-transduced HSPCs (3 × 103) derived from ERK2-WT–, -KO–, or -DBP–mutant mice were sorted and plated on M3434 (A), or M3534 methylcellulose medium (B). The number of colonies was scored 7 days after plating. The transduction of the pMIG empty vector was used as the control. Data are expressed as the mean ± standard deviation (SD). ***P < .001. (C) Control and JAK2V617F-transduced HSPCs from mice with the same genotype as in panels A and B were sorted and then cultured with IMDM supplemented with IL-3, IL-6, and stem-cell factor cytokines for 3 days. Representative images of SA-β-gal staining are depicted on the left. Percentages of SA-β-gal+ cells were determined by counting 100 cells in randomly selected fields from triplicate cultures. Data are expressed as the mean ± SD.***P < .001. (D-E) pMIG and JAK2V617F-transduced HSPCs from the mice above were cultured for 3 days after transduction, and then GFP+ cells were sorted into Trizol. The relative expression of senescence-associated and profibrotic genes was measured by real-time quantitative polymerase chain reaction. Data are expressed as the mean ± SD. All results represent ≥3 independent experiments. **P < .01; ***P < .001; ****P < .0001. IMDM, Iscove modified Dulbecco medium.
JAK2V617F-induced senescence was alleviated by inactivation of the ERK2-DBP domain. (A-B) Control and JAK2V617F-transduced HSPCs (3 × 103) derived from ERK2-WT–, -KO–, or -DBP–mutant mice were sorted and plated on M3434 (A), or M3534 methylcellulose medium (B). The number of colonies was scored 7 days after plating. The transduction of the pMIG empty vector was used as the control. Data are expressed as the mean ± standard deviation (SD). ***P < .001. (C) Control and JAK2V617F-transduced HSPCs from mice with the same genotype as in panels A and B were sorted and then cultured with IMDM supplemented with IL-3, IL-6, and stem-cell factor cytokines for 3 days. Representative images of SA-β-gal staining are depicted on the left. Percentages of SA-β-gal+ cells were determined by counting 100 cells in randomly selected fields from triplicate cultures. Data are expressed as the mean ± SD.***P < .001. (D-E) pMIG and JAK2V617F-transduced HSPCs from the mice above were cultured for 3 days after transduction, and then GFP+ cells were sorted into Trizol. The relative expression of senescence-associated and profibrotic genes was measured by real-time quantitative polymerase chain reaction. Data are expressed as the mean ± SD. All results represent ≥3 independent experiments. **P < .01; ***P < .001; ****P < .0001. IMDM, Iscove modified Dulbecco medium.
Because the capacity of activated JAK2 and ERK signaling to induce quiescence and senescence in HSPCs is well established,47-51 we used staining for SA-β-gal to inquire whether enforced JAK2V617F expression reduces colony formation by inducing senescence.37 JAK2V617F markedly increased SA-β-gal activity in ERK2-WT HSPCs (Figure 4C); however, the increase was strikingly attenuated by inactivation of the ERK2-DBP domain (Figure 4C), indicating that the ERK2-DBP domain acts to promote OIS. In support, the induction of other markers of senescence (p21CIP, p27KIP, p19ARF, and gadd45a)52 was also attenuated by inactivation of the ERK2-DBP domain (Figure 4D). The reduction in OIS was not caused by impaired JAK2V617F signaling by the ERK2-DBP–mutant, because STAT3 activation was unaffected (supplemental Figure 4B). Expression of other pro- or antiapoptotic genes and proliferation markers was not altered by JAK2V617F expression, irrespective of the status of ERK2 (supplemental Figure 4C-K). The attenuation of OIS induction in ERK2-DBP–mutant HSPCs was also evident when assessed by flow cytometry using a fluorescent SA-β-gal substrate, C12RG, which revealed that most HSPC subpopulations were equally susceptible to OIS (supplemental Figure 5). Expression of profibrotic genes was increased in ERK2-DBP–mutant HSPCs (Figure 4E), consistent with the observed increase in MF in ERK2-DBP recipients (Figures 1 and 2). These data strongly implicate the ERK2-DBP domain in suppressing progenitor activity by promoting JAK2V617F-mediated OIS, given that we observed no significant changes in apoptosis or proliferation (supplemental Figure 5E).
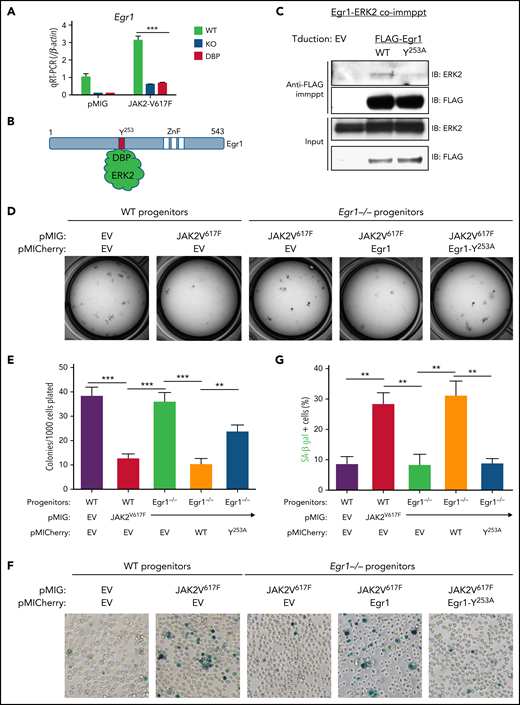
ERK-DBP induction of senescence depends upon interaction with Egr1. (A) Real-time quantitative polymerase chain reaction analysis of Egr1 expression in pMIG or pMIG-JAK2V617F–transduced HSPCs from ERK2-WT–, -KO–, and -DBP–mutant mice that were cultured for 3 days. (B) The physical interaction between ERK2-DBP domain and the DEF motif of Egr1. (C) Physical association of ERK2 and Egr1 after PMA stimuli. Anti-FLAG anti-ERK immunoblots were performed on input and anti-FLAG immunoprecipitates (immppt) from protein extracts of SCID.adh cells transduced with pMICherry empty vector (EV), FLAG-Egr1 (WT), and FLAG-Egr1Y253A mutant. (D-E) Colony-forming assays were performed on WT and Egr1−/− HSPCs transduced with the indicated constructs (pMIG, EV; pMIG-JAK2V617F; pMICherry, EV; pMICherry-Egr1; pMICherry-Egr1Y253A). Sorted cells (1 × 103) were plated on M3434 medium and scored after 7 days in culture. (D) Representative images of cultures and a graphic depiction of the mean ± standard deviation (SD) colonies are shown. **P < .01; ***P < .001. (F-G) Representative images of SA-β-gal staining and the percentages of SA-β-gal+ cells were determined in cultures equivalent to those in panel D. All results are from ≥3 independent experiments. Data are expressed as the mean ± SD. **P < .01.
ERK-DBP induction of senescence depends upon interaction with Egr1. (A) Real-time quantitative polymerase chain reaction analysis of Egr1 expression in pMIG or pMIG-JAK2V617F–transduced HSPCs from ERK2-WT–, -KO–, and -DBP–mutant mice that were cultured for 3 days. (B) The physical interaction between ERK2-DBP domain and the DEF motif of Egr1. (C) Physical association of ERK2 and Egr1 after PMA stimuli. Anti-FLAG anti-ERK immunoblots were performed on input and anti-FLAG immunoprecipitates (immppt) from protein extracts of SCID.adh cells transduced with pMICherry empty vector (EV), FLAG-Egr1 (WT), and FLAG-Egr1Y253A mutant. (D-E) Colony-forming assays were performed on WT and Egr1−/− HSPCs transduced with the indicated constructs (pMIG, EV; pMIG-JAK2V617F; pMICherry, EV; pMICherry-Egr1; pMICherry-Egr1Y253A). Sorted cells (1 × 103) were plated on M3434 medium and scored after 7 days in culture. (D) Representative images of cultures and a graphic depiction of the mean ± standard deviation (SD) colonies are shown. **P < .01; ***P < .001. (F-G) Representative images of SA-β-gal staining and the percentages of SA-β-gal+ cells were determined in cultures equivalent to those in panel D. All results are from ≥3 independent experiments. Data are expressed as the mean ± SD. **P < .01.
ERK2-DBP regulation of senescence depends on its association with EGR1
To determine how the ERK-DBP domain regulates senescence, we focused on the transcription factor Early growth response 1 (Egr1), a well-established tumor suppressor for myeloid neoplasms that contains a DEF motif, through which the ERK2-DBP domain recognizes substrates.30,31,33,53-56 JAK2V617F transduction of WT HSPCs markedly upregulated Egr1 mRNA, which was attenuated by inactivation of the ERK2-DBP domain (Figure 5A). Egr1 has a highly conserved F/Y-X-F/Y-P DEF motif12 (YFLP; Figure 5B; supplemental Figure 6A-B). Egr1 was physically associated with activated ERK2 in PMA-stimulated thymic lymphoma cells, and the association was impaired by a Y253A mutation in the Egr1 DEF motif (Figure 5C). To test the role of Egr1 in regulating colony formation and senescence, we performed loss-of-function and replacement experiments with intact Egr1, and Egr1 that does not associate with ERK2 (Egr1Y253A;Figure 5D-G). Egr1-deficiency abrogated the ability of JAK2-V617F-signaling both to suppress colony formation and to promote senescence, as measured by SA-β-gal staining (Figure 5D-G). Importantly, reintroduction of Egr1 restored responsiveness to JAK2-V617F-mediated suppression of colony formation and induction of senescence (Figure 5D-G); however, JAK2-V617F–mediated suppression of colony formation and senescence induction were not restored by Egr1 with an inactivated DEF motif (Egr1-Y253A; Figure 5D-G). The expression levels of Egr1 and Egr1-Y253A mutants were equivalent (supplemental Figure 6C). Thus, the ability of ERK2 to mediate OIS and suppress colony formation is dependent upon physical association of Egr1.
Pharmacologic targeting of the ERK-D domain attenuates MPN growth
Our data suggest that the ERK2-DBP domain antagonizes progression of JAK2-V617F–driven neoplasms in vivo and promotes OIS induction in vitro. Because DBP-mutant ERK2 can target substrates only through the D domain, these findings implicate the ERK2-D domain in promoting disease progression in the ERK-DBP–mutant recipients (Figure 1).12,14 The ERK2-D domain mediates phosphorylation of most (80%) ERK substrates, including MKNK1 and p90RSK kinases, which promote cellular proliferation and survival.13 Consequently, we sought to test whether the pharmacologic inhibition of ERK-D domain substrate interactions had therapeutic potential (Figure 6A). We generated ERK inhibitors that block phosphorylation of D-domain–containing substrates.57 In support, in PMA-stimulated thymic lymphoma cells, MEK inhibition (U0126) blocks ERK phosphorylation, phosphorylation of the D-domain substrate, RSK, and induction of the DBP substrate Egr1 (Figure 6B). In contrast, the ERK-D–domain inhibitor #7657 spared ERK phosphorylation, Egr1 induction, and Egr1-ERK association, but impaired RSK phosphorylation, attesting to its selectivity for the D domain57 (Figure 6B; supplemental Figure 6D). Importantly, selective inhibition of D domain function attenuated colony formation by JAK2V617-expressing HSPCs (Figure 6C-D). Moreover, the D-domain inhibitor impaired the growth of the ERK-dependent human MPN cell line SET-2,29 in vitro and in vivo (Figure 6E-F).
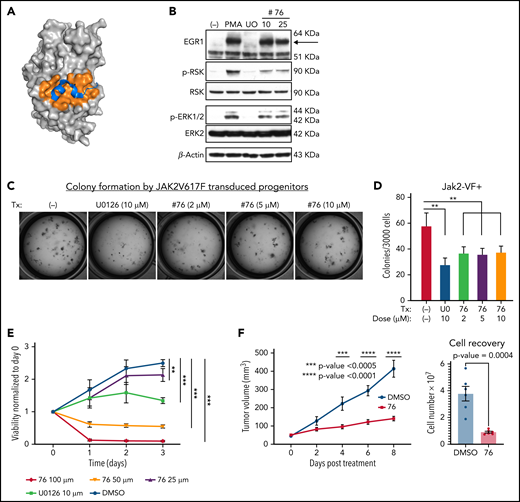
ERK-D domain targeting inhibited cellular proliferation of JAK2V617F-dependent cells. (A) Space filling model of ERK2 and the D docking domain (orange pocket region) complexed with a MAPK docking peptide (MKNK1, blue colored structure). (B) Selective inhibition of ERK2-mediated phosphorylation of RSK by D-domain inhibitor (#76). SCID.adh cells were pretreated with U0126 or #76 inhibitor and then stimulated with PMA. Immunoblot analysis of protein extracts was performed, using the following antibodies: anti-EGR1, anti–phospho-RSK, anti-RSK, anti–phospho-ERK, anti- ERK, and anti–β-actin, which served as a loading control. (C-D) Colony forming analysis was performed on JAK2V617F-transduced HSPCs treated with vehicle control, U0126 or #76. Cells (3 × 103) were plated on M3434 medium and cultured for 7 days. The number of colonies was scored and compared. Data are expressed as the mean ± standard deviation (SD). **P < .01. (E) Human SET-2 cells expressing JAK2V617F were treated with vehicle control, U0126 (10 µM), or #76 at the indicated doses. Cell viability and proliferation were measured each day for 3 days and normalized to untreated cells at day 0 and are expressed as the mean ± SD. **P < .01; ***P < .001. (F) Human SET-2 cells were implanted subcutaneously into Il2rg−/−Rag2−/− mice and left to reach a size of 40 mm3 before treatment every other day by intraperitoneal injection with vehicle control (dimethyl sulfoxide) or #76 at 10 mg/kg. Tumor size and cell recovery upon tumor disaggregation are expressed as the mean ± SD (n = 6 per condition). Results in panels B-E were derived from at least 3 independent experiments.
ERK-D domain targeting inhibited cellular proliferation of JAK2V617F-dependent cells. (A) Space filling model of ERK2 and the D docking domain (orange pocket region) complexed with a MAPK docking peptide (MKNK1, blue colored structure). (B) Selective inhibition of ERK2-mediated phosphorylation of RSK by D-domain inhibitor (#76). SCID.adh cells were pretreated with U0126 or #76 inhibitor and then stimulated with PMA. Immunoblot analysis of protein extracts was performed, using the following antibodies: anti-EGR1, anti–phospho-RSK, anti-RSK, anti–phospho-ERK, anti- ERK, and anti–β-actin, which served as a loading control. (C-D) Colony forming analysis was performed on JAK2V617F-transduced HSPCs treated with vehicle control, U0126 or #76. Cells (3 × 103) were plated on M3434 medium and cultured for 7 days. The number of colonies was scored and compared. Data are expressed as the mean ± standard deviation (SD). **P < .01. (E) Human SET-2 cells expressing JAK2V617F were treated with vehicle control, U0126 (10 µM), or #76 at the indicated doses. Cell viability and proliferation were measured each day for 3 days and normalized to untreated cells at day 0 and are expressed as the mean ± SD. **P < .01; ***P < .001. (F) Human SET-2 cells were implanted subcutaneously into Il2rg−/−Rag2−/− mice and left to reach a size of 40 mm3 before treatment every other day by intraperitoneal injection with vehicle control (dimethyl sulfoxide) or #76 at 10 mg/kg. Tumor size and cell recovery upon tumor disaggregation are expressed as the mean ± SD (n = 6 per condition). Results in panels B-E were derived from at least 3 independent experiments.
We have been developing more potent ERK-D-domain inhibitors by altering the 4-ethoxyphenyl moiety of #76 with parabenzyloxy or paraisobutoxy derivatives to generate compounds 30 g and 30 h, respectively.58 These compounds were tested on a panel of human MPN lines bearing the JAK2V617F mutations SET-2,59 UKE-1,60 and Ku812,61 as well as a murine pro-B cell line (BAF/3) ectopically expressing JAK2V617F.24 As with the parental ERK-D–domain inhibitor #76, the 30 g and 30 h analogs spared ERK phosphorylation and Egr1 induction, both of which were completely inhibited by comparable doses of U0126, a MEK inhibitor (Figure 7A). Importantly, 30 g and 30 h, but particularly 30 h, more effectively attenuated the growth of the JAK2-dependent cell lines (Figure 7B).
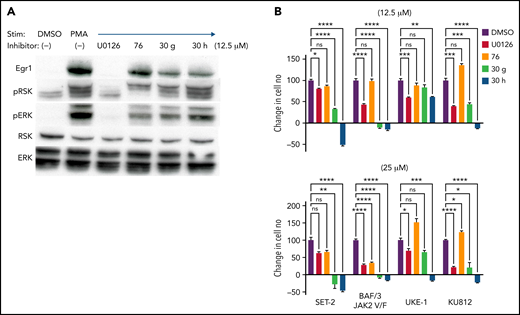
Derivatives of ERK-D domain inhibitor #76 exhibit greater potency in inhibiting the growth of JAK-dependent cells. (A) Selective inhibition of ERK2-mediated phosphorylation of RSK by the D-domain inhibitors #76, 30 g, and 30 h. SCID.adh cells were pretreated with the indicated inhibitors and then stimulated with PMA. Immunoblot analysis of protein extracts was performed with the following antibodies: anti-EGR1, anti–phospho-RSK, anti-RSK, anti–phospho-ERK, anti- ERK, and anti–β-actin, which served as a loading control. (B) Cell lines expressing JAK2V617F (SET-2, UKE-1, KU812, and BAF/3-JAK2) were treated with vehicle control, U0126, #76, 30 g, and 30 h at the indicated doses. All results are from ≥3 independent experiments. Cell viability and proliferation were measured after 1 day of culture and normalized to untreated cells at day 0. Data are expressed as the mean ± standard deviation. *P < .05; **P < .01; ***P < .001; ns, not significant.
Derivatives of ERK-D domain inhibitor #76 exhibit greater potency in inhibiting the growth of JAK-dependent cells. (A) Selective inhibition of ERK2-mediated phosphorylation of RSK by the D-domain inhibitors #76, 30 g, and 30 h. SCID.adh cells were pretreated with the indicated inhibitors and then stimulated with PMA. Immunoblot analysis of protein extracts was performed with the following antibodies: anti-EGR1, anti–phospho-RSK, anti-RSK, anti–phospho-ERK, anti- ERK, and anti–β-actin, which served as a loading control. (B) Cell lines expressing JAK2V617F (SET-2, UKE-1, KU812, and BAF/3-JAK2) were treated with vehicle control, U0126, #76, 30 g, and 30 h at the indicated doses. All results are from ≥3 independent experiments. Cell viability and proliferation were measured after 1 day of culture and normalized to untreated cells at day 0. Data are expressed as the mean ± standard deviation. *P < .05; **P < .01; ***P < .001; ns, not significant.
Discussion
The Ras/MAPK cascade is activated in most human cancers and plays a critical role in cancer pathogenesis,1,2 but the molecular basis by which the terminal kinases ERK1/ERK2 contribute to cancer development remains incompletely understood. We report analysis of the opposing roles of the ERK2-D and DBP domains in pathogenesis of JAK2V617F-driven mouse MPN model. We found that the ERK2-DBP domain opposes the progression of PV to MF in vivo and promotes cellular senescence in vitro. Senescence induction depends on physical interaction of ERK2 with Egr1, providing the first indication that the function of Egr1 in opposing tumor progression may be dependent on its ability to interact with ERK. In contrast, the ERK2-D domain appears to promote tumor progression, in that it remains intact in the ERK2-DBP–mutant and is the only substrate-binding domain through which the ERK2-DBP mutant can promote disease progression. Moreover, pharmacologic inhibition of ERK-D substrate interaction attenuated the growth of JAK2V617F-dependent MPN cell lines in vitro and disease pathogenesis in vivo. These data support a model where the ERK2-D and -DBP domains serve opposing roles in the pathogenesis of JAK2V617F-dependent neoplasms and suggest that targeting ERK-substrate interaction domains, may serve as a more effective alternative to active site-focused inhibitors.
To understand how ERK2 modulates cancer development, we focused on its substrate interaction domains. We found that the ERK2-DBP domain opposed cancer development, in that ERK2-DBP inactivation promoted progression from PV to MF in a JAK2V1617F-driven model. ERK2-DBP inactivation also disabled senescence induction in vitro. Senescence induction by the ERK2-DBP required physical association with Egr1, a well-known tumor suppressor of myeloid neoplasms.30-33 Therefore, our data suggest that Egr1 is a critical ERK2 target through which the ERK-DBP regulates senescence and MPN progression in vivo; however, these molecular links were made in in vitro models, and conclusive proof awaits replication in vivo, including investigation of the link between loss of senescence and disease progression in primary human MPNs, where mutations affecting senescence have been linked to MPN pathogenesis.48,62 It should also be noted that inactivation of the ERK2-DBP domain resulted in reduced senescence in most JAK2V617F-transduced progenitor subsets in vitro. Consequently, it remains unclear how ERK2-DBP inactivation results in the JAK2V617F-mediated selective expansion of particular myeloid subsets in vivo, but may result from OIS-related lineage shifts at later stages of disease pathogenesis in vivo. Moreover, ∼20% of ERK2 substrates are targeted through the DBP domain, and substrates in addition to Egr1 are almost certainly involved.13 Another critical question is whether the ERK2-DBP domain always functions to oppose tumor progression, as it did in our PV model, or if it is context dependent. The ERK2-DBP domain can promote epithelial-to-mesenchymal transition (EMT) and other prometastatic behaviors in epithelial cell lines.9 Likewise, some ERK2-DBP mutations behaved as loss-of-function in an in vitro BRAF-driven, melanoma-based ERK inhibitor resistance assay.63 Conversely, ERK2-D mutations behaved as gain-of-function in the same melanoma cell line.63 These findings appear discordant with our findings that the ERK2-DBP and -D domains are anti- and protumorigenic, respectively. There are 2 explanations. First, targets phosphorylated through the ERK2-D and -DBP domains may have more distinct functions early in transformation (eg, senescence induction) than they do late in tumor progression (eg, metastasis). Second, the substrates targeted through these domains may differ, depending on the tumor cell of origin or the oncogenic drivers. In future efforts, we will use our conditional inactivation models to assess stage- and context-dependent functions of the ERK-DBP and -D domains.
We conclude that the ERK2-D domain is necessary for promoting disease progression in our PV model because the ERK2-DBP mutant that promotes progression can target only substrates via its D domain. Moreover, progression was not observed in ERK2-KO recipients, suggesting that the promotion of progression requires the ERK2-D kinase and D-domain function. Finally, pharmacologic targeting of the ERK-D domain attenuates the growth of several JAK2V617F dependent MPN and lymphoma lines. Definitive tests of the role of the ERK2-D domain in cancer development in vivo must await the construction of genetic models inactivating the D domain. Likewise, the D domain–targeted substrates responsible for disease progression in vivo or MPN cell proliferation or survival in vitro remain unclear. JAK activation in human HSPCs induces the expression of many progrowth ERK2-D substrates, including Zfp36, Fos, Jun, and Mcl1.50,64 Nevertheless, because 80% of ERK2 targets are phosphorylated in a D-domain–dependent manner,13 unbiased functional screening is necessary to arrive at a more complete understanding of the molecular basis for ERK2-D-domain function in cancer pathogenesis. Finally, our findings of a modular role of the substrate binding domains of ERK2 are likely to have important implications in MPNs beyond those driven by JAK2V617F. For example, previous analysis of MPN driven by W515 mutations of the thrombopoietin receptor (TpoR) revealed that mutations that enhance or diminish ERK signaling by TpoR promote or attenuate MPN progression, respectively.65
Because the Ras/MAPK cascade is activated in most human cancers, development of effective pharmacologic interventions to attenuate its activity remains the holy grail; however, much of this effort has focused on targeting the active sites of signaling effectors and has failed to produce lasting clinical benefit. JAK2V617F-driven MPNs remain incurable hematological disorders. JAK inhibitors, such as ruxolitinib, alleviate some constitutional symptoms, but fail to eliminate the core pathologic features or block progression.66 Combination treatment with both JAK (ruxolitinib) and MEK inhibitors (binimetinib) provide greater therapeutic efficacy than either single agent,29 suggesting that combination JAK/MAPK therapy has potential for managing MPNs. Of the targets in the Ras/Raf/MAPK cascade, ERK is a critical hub, as the benefit of Ras/BRAF/MEK inhibitors is always compromised by the acquisition of ERK-dependent drug resistance.11,67-69 Therefore, targeting ERK2 is entirely justified; however, given our finding of opposing roles of the ERK2-D and -DBP domains in JAK2-driven neoplasms, we suggest that targeting the substrate-interacting domains of ERK may be more effective than targeting its active site.
Acknowledgments
The authors thank Michael Deininger for providing the retroviral vector–expressing mouse JAK2V617F, and the core facilities Cell Culture, Cell Sorting, Imaging, Histopathology, Transgenic Mouse, and Laboratory Animal at the Fox Chase Cancer Center for assistance.
This work was supported by National Institutes of Health, National Institute of Allergy and Infectious Diseases Extramural Activities grant R37AI110985 (D.L.W.) and National Cancer Institute (NCI) grant P30CA006927 (D.L.W.), and by the Commonwealth of Pennsylvania and the Bishop Fund (D.L.W.). R.L.L. was supported by NCI grant P01CA10867111.
Authorship
Contribution: Y.Z., B.T., S.P.F., E.M., D.A.L., J.S., R.L.L., and S.F. contributed to the performance of experiments; K.Q.C., E.D.A., Y.G., and R.D. analyzed the data; Y.Z. and D.L.W. wrote the manuscript; and D.K., S.M.S., P.S., Y.Z., and D.L.W. designed and supervised the work.
Conflict-of-interest disclosure: R.L.L. is on the supervisory board of Qiagen and is a scientific advisor to Imago, Mission Bio, Zentalis, Ajax, Auron, Prelude, C4 Therapeutics, and Isoplexis. He receives research support from and consulted for Celgene and Roche and has consulted for Incyte, Janssen, Astellas, Morphosys, and Novartis. He has received honoraria from Astra Zeneca, Roche, Lilly, and Amgen for invited lectures and from Gilead for grant reviews. The remaining authors declare no competing financial interests.
The current affiliation for Y.Z. is Department of Pathology, Penn State Milton S. Hershey Medical Center, Hershey, PA.
Correspondence: David L. Wiest, Blood Cell Development and Function Program, Fox Chase Cancer Center, 333 Cottman Ave, Philadelphia, PA 19111; e-mail: david.wiest@fccc.edu.
Original data are available in response to an e-mail request to the corresponding author.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal