

Key Points
The RNase H–like superfamily member PIWIL4 is required for leukemic stem cell function but dispensable for human hematopoietic stem cells.
PIWIL4 prevents R-loop accumulation, DNA damage, replication stress, and activation of the ATR pathway in AML cells.

Abstract
RNA-binding proteins (RBPs) form a large and diverse class of factors, many members of which are overexpressed in hematologic malignancies. RBPs participate in various processes of messenger RNA (mRNA) metabolism and prevent harmful DNA:RNA hybrids or R-loops. Here, we report that PIWIL4, a germ stem cell–associated RBP belonging to the RNase H–like superfamily, is overexpressed in patients with acute myeloid leukemia (AML) and is essential for leukemic stem cell function and AML growth, but dispensable for healthy human hematopoietic stem cells. In AML cells, PIWIL4 binds to a small number of known piwi-interacting RNA. Instead, it largely interacts with mRNA annotated to protein-coding genic regions and enhancers that are enriched for genes associated with cancer and human myeloid progenitor gene signatures. PIWIL4 depletion in AML cells downregulates the human myeloid progenitor signature and leukemia stem cell (LSC)-associated genes and upregulates DNA damage signaling. We demonstrate that PIWIL4 is an R-loop resolving enzyme that prevents R-loop accumulation on a subset of AML and LSC-associated genes and maintains their expression. It also prevents DNA damage, replication stress, and activation of the ATR pathway in AML cells. PIWIL4 depletion potentiates sensitivity to pharmacological inhibition of the ATR pathway and creates a pharmacologically actionable dependency in AML cells.
Introduction
In cancer, large-scale gene expression studies and a census of cancer dependencies have illuminated the landscape of nononcogenic dependencies in cancer.1,2 Among these, RNA-binding proteins (RBPs) form a large and diverse class of factors, many members of which are overexpressed adjacent to healthy tissues and nononcogenic dependencies in diverse tumors, including hematologic malignancies.3-6 RBPs regulate gene expression by participating in messenger RNA metabolism, including transcription, capping, splicing, stability, export, and translation.7 Recent studies also demonstrate the role of RBPs in preventing harmful DNA:RNA hybrids or R-loops and in the DNA damage response.8 However, the promise of targeting RBPs therapeutically could be limited by their dispensability for cancer stem cells or the indiscriminate cytotoxicity of their inhibitors for cancer and healthy stem cells.
Acute myeloid leukemia (AML) is an aggressive hematologic malignancy propagated by a population of leukemia stem cells (LSCs).9 Patients with AML have a poor survival rate, highlighting the need for improved therapeutic interventions.10 The PIWI-like (PIWIL) proteins are stem cell–associated, RNA-binding, and cleaving enzymes evolutionarily related to the RNase H–like (RNHL) superfamily.11,12 They are crucial for the survival of the self-renewing population of germ stem cells,12-14 acutely proliferative intestinal stem cells in lower organisms,15 and epigenetic silencing of repetitive elements.16-19 Human PIWIL proteins are upregulated and play an oncogenic role in multiple cancers.20-24 However, studies in murine models suggest that PIWIL proteins could be dispensable for healthy hematopoietic stem cells (HSCs), making them a potentially important target for therapeutic intervention in AML. Here, we identify the PIWI family member PIWIL4 to be aberrantly expressed in most patients with AML and functionally validated LSCs. PIWIL4 is required for the sustenance of AML blasts and LSCs but is dispensable for healthy human hematopoietic stem progenitor (HSPC) function in vivo. We describe a crucial and unanticipated role for PIWIL4 as a functional R-loop resolving enzyme in AML. PIWIL4 prevents R-loop accumulation, DNA damage, replication stress, and stalling of transcription at a subset of LSC-relevant genes in AML cells. Depletion of PIWIL4 in AML cells potentiates sensitivity to ataxia telangiectasia and Rad3 related (ATR) kinase inhibition and opens an avenue for targeting de novo AML via R-loop induction in tandem with ATR inhibition.
Methods
Patients
This study was conducted in accordance with the Declaration of Helsinki for the protection of human participants and the institutional review board of the University Hospital Ulm and Klinikum Grosshadern, Munich. Bone marrow (BM) aspirates and peripheral blood were collected from patients with written informed consent who satisfied diagnostic and immunophenotypic criteria for AML.
Isolation and validation of leukemic stem cell subpopulations and transcriptome analysis
Primary de novo AML cells (supplemental Table 1, available on the Blood website) were sorted for HSC markers (lymphoid-primed multipotential progenitor LMPP: CD34+CD38−CD45RA+CD90− and granulocyte-macrophage progenitor GMP: CD34+CD38+CD123+CD110−CD45RA+) as well as CD34− cells by fluorescence-activated cell sorting (FACS). Sorted cells from different fractions were adjusted to identical numbers before IV transplantation into 1 to 3 sublethally irradiated NOD scid gamma (NSG) mice per subpopulation for each patient. The cell fractions that resulted in leukemic engraftment (>1%) and the phenotype of the NSG mice were defined as functionally validated LSCs. In parallel, all fractions were used for RNA sequencing (RNA-seq).
Please refer to the supplemental Data for further methods.
Results
PIWIL4 is overexpressed in AML bulk and LSCs
Gene expression can be used as a basis for predicting most of the cancer dependencies, and small-molecule cancer dependencies are most commonly predicted by gene expression.1,25 Several gene expression-based observations suggested that PIWIL4 was a potential vulnerability and therapeutic target in AML cells: among all cancers, PIWIL4 was the highest expressed in AML (Figure 1A) and the only PIWI family member that consistently exhibited several-fold overexpression in most of the patients with AML compared with healthy counterparts (Figure 1B-D; supplemental Figure 1A-B, data not shown; supplemental Table 1).2,28 It was higher expressed in patients with de novo AML and secondary AML arising from myelodysplastic syndrome (MDS) vs those with MDS (Figure 1E). Among patients with AML, those harboring the MLL-AF9 translocation consistently exhibited the highest expression (supplemental Figure 1C-D). Importantly, PIWIL4 also exhibited overexpression compared with healthy counterparts in functionally validated, AML LSC–harboring, lymphoid-myeloid–primed multipotent progenitors (L-LMPPs) and in the CD34+ CD8− subpopulation derived from cytogenetically normal patients with AML (Figure 1F; supplemental Figure 1E).29 In contrast, although healthy hematopoietic tissue harbored high expression of PIWIL4 compared with other healthy tissues, studies in the murine model indicated that PIWIL4 had no discernable role in HSCs or healthy hematopoiesis.30,31 Based on these data, we reasoned that an investigation was warranted into whether the aberrant expression of PIWIL4 was of functional significance for AML cells.

PIWIL4 is overexpressed in most patients with AML and AML LSCs. (A) PIWIL4 messenger RNA (mRNA) expression in types of cancer from The Cancer Genome Atlas (TCGA) data set. The boxplot indicates median expression and range of expression.26 (B) qRT-PCR mRNA expression of PIWIL4 in patients with AML, bone marrow mononuclear cells (BMNCs) and CD34+ BM and cord blood CD34+ (CB CD34+) HSPCs. Dots indicate biological replicates; horizontal bars represent median expression. (C) mRNA expression of PIWIL4 in patients with AML vs healthy CD34+ BM HSPCs from the Vizome RNA-seq data set.27 (D) Upper panel: PIWIL4 protein levels vs β-actin as loading control in independent biological replicates of cytogenetically normal (CN) patients with AML and healthy bulk CB. Lower panel: densitometry analysis of PIWIL4 protein expression relative to β-actin protein levels, analyzed using ImageJ. (E) RNA-seq mRNA expression PIWIL4 in de novo AML, secondary AML (sAML), and MDS. Dots indicate individual patients and the boxplot indicates median expression and range of expression. (F) PIWIL4 expression in lymphoid-myeloid primed multipotent progenitors (H-LMPPs vs L-LMPPs) derived from functionally validated, healthy individuals vs patients with leukemia patient. (G) Localization of PIWIL4 protein in AML cell line, NB-4. Nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI) and nucleolus with fibrillarin. The size bar indicates 20 μm distance. (H) Fold increase in Piwil4 mRNA expression in murine HSPCs transduced with AML-specific oncogenes (MLL-AF9, Cdx2, and AML1-ETO9A) vs empty vector control, 72 hours after transduction. “n” indicates number of independent experiments.
PIWIL4 is overexpressed in most patients with AML and AML LSCs. (A) PIWIL4 messenger RNA (mRNA) expression in types of cancer from The Cancer Genome Atlas (TCGA) data set. The boxplot indicates median expression and range of expression.26 (B) qRT-PCR mRNA expression of PIWIL4 in patients with AML, bone marrow mononuclear cells (BMNCs) and CD34+ BM and cord blood CD34+ (CB CD34+) HSPCs. Dots indicate biological replicates; horizontal bars represent median expression. (C) mRNA expression of PIWIL4 in patients with AML vs healthy CD34+ BM HSPCs from the Vizome RNA-seq data set.27 (D) Upper panel: PIWIL4 protein levels vs β-actin as loading control in independent biological replicates of cytogenetically normal (CN) patients with AML and healthy bulk CB. Lower panel: densitometry analysis of PIWIL4 protein expression relative to β-actin protein levels, analyzed using ImageJ. (E) RNA-seq mRNA expression PIWIL4 in de novo AML, secondary AML (sAML), and MDS. Dots indicate individual patients and the boxplot indicates median expression and range of expression. (F) PIWIL4 expression in lymphoid-myeloid primed multipotent progenitors (H-LMPPs vs L-LMPPs) derived from functionally validated, healthy individuals vs patients with leukemia patient. (G) Localization of PIWIL4 protein in AML cell line, NB-4. Nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI) and nucleolus with fibrillarin. The size bar indicates 20 μm distance. (H) Fold increase in Piwil4 mRNA expression in murine HSPCs transduced with AML-specific oncogenes (MLL-AF9, Cdx2, and AML1-ETO9A) vs empty vector control, 72 hours after transduction. “n” indicates number of independent experiments.
PIWI proteins are present in the cytoplasm and nucleus of cells, whereas their exclusive nuclear localization has been linked to stem cell function and cancer.15,21 Subcellular localization studies placed PIWIL4 exclusively in the nucleus of AML cells (Figure 1G). Furthermore, overexpression of AML-specific oncogenes in healthy murine HSPCs prompted a six- to eightfold upregulation of Piwil4 expression within 72 hours of the end of transduction (Figure 1H; supplemental Figure 1E). Similarly, upregulation of PIWIL4 was previously reported in human CD34+ HSPCs immortalized with MLL-AF9 compared with HSPCs transduced with AML1-ETO and inv16, translocations that do not immortalize HSPCs (supplemental Figure 1F).32 Published chromatin immunoprecipitation data sets revealed that AML-specific oncoproteins MLL-AF9 and AML1-ETO9A did not bind on the promoter of Piwil4 (data not shown).33-35 Nevertheless, published data sets illustrate that oncogenic transcription factors, such as MYC proto-oncogene and its binding partner MYC associated factor X (MAX), bind on the promoter region of PIWIL4 in myeloid leukemia cells (supplemental Figure 1G).36,37 The knockdown (KD) of MYC in AML cells resulted in a >50% reduction in PIWIL4 expression, suggesting that oncogenes could mediate the overexpression of PIWIL4 in AML cells via transcription factors such as MYC (supplemental Figure 1H).
AML LSCs and their progeny are dependent on PIWIL4 expression
To examine the significance of PIWIL4 overexpression in AML bulk and LSCs, we depleted PIWIL4 via short hairpin RNA (shRNA)-mediated KD or gRNA-mediated knockout (KO). Using 2 independent shRNA and single guide RNA (sgRNA), KD or KO of PIWIL4 induced a dramatic reduction in colony-forming ability and proliferation in liquid culture in vitro in AML cell lines representing a variety of molecular subtypes (Figure 2A-B; supplemental Figure 2A-G). Similarly, the KD of PIWIL4 in samples from patients with AML induced a significant reduction in colony formation (supplemental Figure 2H). However, KD of PIWIL4 in RAJI, a Burkitt lymphoma cell line with undetectable PIWIL4 expression, yielded no impact on colony-forming potential, confirming that the shRNA had no discernable off-target effects (supplemental Figure 2I). Importantly, the depletion of PIWIL4 adversely affected AML LSC function as demonstrated by the reduction of engraftment of PIWIL4-depleted primary cells from patients with AML in xenografts 16 weeks after transplantation (Figure 2C), the reduced leukemic engraftment potential of the PIWIL4 KO AML cell line, OCI-AML3 (Figure 2D), and the delayed onset of leukemia in xenografts transplanted with PIWIL4-depleted MV4-11 and THP-1 cells (Figure 2E). Quantitative real-time polymerase chain reaction (qRT-PCR) analysis of the BM of mice that succumbed to leukemia revealed that shRNA-transduced THP-1 cells no longer exhibited PIWIL4 KD, suggesting a selection pressure for high PIWIL4 expression (supplemental Figure 2J). Finally, in limiting dilution transplantation assays of the ckit+ LSC-harboring population of MLL-AF9 primary leukemic AML cells,38 the estimated frequency of LSCs was calculated as 1 per 834 cells in the control arm vs 1 per 32 526 cells in the Piwil4-depleted arm. This reflected a 39-fold reduction in LSCs upon Piwil4 depletion (Figure 2F). Collectively, these data demonstrated that PIWIL4 is required for AML LSCs, their progeny, and AML progression.

Aberrant expression of PIWIL4 is required for sustenance of AML growth. (A) Percentage difference in clonogenicity in CFU assay of PIWIL4-depleted vs control AML cell lines. Bars indicate mean and standard error of the mean (SEM). “n” indicates number of independent experiments. (B) Number of colonies in CFU assay of AML cell lines transduced with sgScr vs sgRNAs against PIWIL4 (sgRNA-A and B). Bars indicate mean colony number and SEM. “n” indicates number of independent experiments. (C) Flow cytometry analysis of human engraftment in BM of NSG mice that underwent transplantation with cells from patients with AML transduced with shRNA or scrambled control. Engraftment was determined by CD45 and GFP double positivity. Dots indicate engraftment levels in individual mice. (D) Flow cytometry analysis of human engraftment in BM of NSG mice that underwent transplantation with OCI-AML3 cells transduced with sgPIWIL4 or sgSCR control. Engraftment was determined by positivity for CD45. Dots indicate engraftment levels in individual mice. (E) Kaplan Meier plot showing the survival of NSG mice that underwent transplantation with PIWIL4-depleted AML cell lines. (F) Kaplan Meier plot of mice that underwent transplantation with cell number dilutions (105, 104, and 103) of ckit+ MLL-AF9+ cells from primary leukemic mice transduced with shRNA against Piwil4 or scrambled control. “n” indicates the number of individual mice that underwent transplantation. The table shows calculated LSC frequencies and the right panel shows hematoxilin and eosin-stained histopathology and myeloperoxidase (MPO) immunohistochemistry of representative mice. (G) Apoptosis assay of PIWIL4-depleted cells from patients with primary AML. “n” indicates the number of biological replicates.
Aberrant expression of PIWIL4 is required for sustenance of AML growth. (A) Percentage difference in clonogenicity in CFU assay of PIWIL4-depleted vs control AML cell lines. Bars indicate mean and standard error of the mean (SEM). “n” indicates number of independent experiments. (B) Number of colonies in CFU assay of AML cell lines transduced with sgScr vs sgRNAs against PIWIL4 (sgRNA-A and B). Bars indicate mean colony number and SEM. “n” indicates number of independent experiments. (C) Flow cytometry analysis of human engraftment in BM of NSG mice that underwent transplantation with cells from patients with AML transduced with shRNA or scrambled control. Engraftment was determined by CD45 and GFP double positivity. Dots indicate engraftment levels in individual mice. (D) Flow cytometry analysis of human engraftment in BM of NSG mice that underwent transplantation with OCI-AML3 cells transduced with sgPIWIL4 or sgSCR control. Engraftment was determined by positivity for CD45. Dots indicate engraftment levels in individual mice. (E) Kaplan Meier plot showing the survival of NSG mice that underwent transplantation with PIWIL4-depleted AML cell lines. (F) Kaplan Meier plot of mice that underwent transplantation with cell number dilutions (105, 104, and 103) of ckit+ MLL-AF9+ cells from primary leukemic mice transduced with shRNA against Piwil4 or scrambled control. “n” indicates the number of individual mice that underwent transplantation. The table shows calculated LSC frequencies and the right panel shows hematoxilin and eosin-stained histopathology and myeloperoxidase (MPO) immunohistochemistry of representative mice. (G) Apoptosis assay of PIWIL4-depleted cells from patients with primary AML. “n” indicates the number of biological replicates.
Bromodeoxyuridine cell cycle analysis revealed that PIWIL4-depleted AML cells exhibited an increase in the sub-G1 phase and the G2M phase (supplemental Figure 2K). The increase in G2M indicated the accumulation of DNA damage, and the increase in sub-G1 phase implied that PIWIL4-depleted cells were either apoptotic or senescent. The senescence-associated β-galactosidase assay yielded no significant differences (data not shown). However, annexin-V apoptosis assays revealed a significant increase in apoptotic and dead cells (Figure 2G; supplemental Figure 2L-N).
PIWIL4 is dispensable for the long-term repopulation capacity of healthy HSPCs
In comparison to AML cells, the expression of PIWIL4 was relatively lower in healthy human HSPCs. However, compared with other human tissues, RNA-seq data showed that the expression levels of PIWIL4 are considerably high in human hematopoietic cells, only surpassed by expression levels in the testis (supplemental Figure 3A).31 The western blot analysis of healthy cord blood yielded PIWIL4 protein levels that, albeit significantly lower than those in patients with AML, were detectable. Furthermore, CT values obtained from qRT-PCR analysis of CD34+ BM and cord blood–derived HSPCs were suggestive of moderate levels of transcription. Collectively, these data suggested that an shRNA-mediated KD was feasible and appropriate (Figure 1D; supplemental Figure 3B).
PIWIL4 depletion had no discernable impact on the colony-forming ability or normal differentiation in vitro, as evidenced by the distribution of the multipotent progenitor (CFU-GEMM: colony-forming unit granulocyte, erythrocyte macrophage, monocyte), myeloid (CFU-GM: granulocyte monocyte), and erythroid (BFU-E: blast forming unit erythroid; CFU-E–erythroid) colonies and their morphology (Figure 3A-B; supplemental Figure 3C). In contrast to AML cells, PIWIL4 depletion in human HSPCs did not affect human engraftment levels in NSG mice 12 weeks after transplantation (Figure 3C; supplemental Figure 3D). In addition, depleted human HSPCs did not exhibit skewed hematopoietic lineage specification in mice that underwent transplantation, with no significant differences in distribution between stem-progenitor–enriched (CD34+), myeloid (CD33+), or B-cell (CD19+) cellular compartments compared with the scrambled control arm (Figure 3D; supplemental Figure 3E). The KD of PIWIL4 was maintained after engraftment in the GFP+ BM cells of NSG mice, showing that the observed hematopoiesis arose from PIWIL4-depleted cells (supplemental Figure 3F). Consistent with these findings, the depletion of PIWIL4 in healthy CD34+ cells also did not cause apoptosis in liquid culture in the annexin-V assay (Figure 3E). In sum, these data highlight a differential vulnerability toward PIWIL4 depletion between normal and leukemic stem and progenitor cells.

PIWIL4 is dispensable for normal human hematopoiesis in vivo. (A) CFU assay showing number of CFU-GEMM, myeloid (CFU-GM), and the erythroid (BFU-E blast forming unit erythroid, CFU-E) colonies at day 14, formed by CB-derived CD34+ HSPCs transduced with scrambled control or shPIWIL4. Bars indicate mean colony number and SEM. “n” indicates number of independent experiments. (B) Representative image of the morphology of colonies observed in CFU assay of PIWIL4-depleted healthy human HSPCs vs control. (C) Total percentage of human engraftment in BM of NSG mice at 12 weeks after transplantation with scrambled or shRNA-transduced healthy CD34+ HSPCs. Engraftment was determined by GFP positivity. Dots indicate engraftment levels in individual mice. (D) Flow cytometry analysis of immunophenotypic subpopulations, stem cell–enriched (CD34+), myeloid (CD33+), or B-cell (CD19+), in BM of NSG mice that underwent transplantation with scrambled or shRNA-transduced healthy CD34+ HSPCs. (E) Apoptosis assay of PIWIL4-depleted CD34+ HSPCs vs scrambled control. “n” indicates the number of biological replicates.
PIWIL4 is dispensable for normal human hematopoiesis in vivo. (A) CFU assay showing number of CFU-GEMM, myeloid (CFU-GM), and the erythroid (BFU-E blast forming unit erythroid, CFU-E) colonies at day 14, formed by CB-derived CD34+ HSPCs transduced with scrambled control or shPIWIL4. Bars indicate mean colony number and SEM. “n” indicates number of independent experiments. (B) Representative image of the morphology of colonies observed in CFU assay of PIWIL4-depleted healthy human HSPCs vs control. (C) Total percentage of human engraftment in BM of NSG mice at 12 weeks after transplantation with scrambled or shRNA-transduced healthy CD34+ HSPCs. Engraftment was determined by GFP positivity. Dots indicate engraftment levels in individual mice. (D) Flow cytometry analysis of immunophenotypic subpopulations, stem cell–enriched (CD34+), myeloid (CD33+), or B-cell (CD19+), in BM of NSG mice that underwent transplantation with scrambled or shRNA-transduced healthy CD34+ HSPCs. (E) Apoptosis assay of PIWIL4-depleted CD34+ HSPCs vs scrambled control. “n” indicates the number of biological replicates.
PIWIL4 binds to cancer-associated protein-coding RNA in AML cells and its depletion induces the deregulation of LSC and DNA repair–associated pathways
The function of PIWI proteins in germ cells is based on their ability to bind piwi-interacting RNA (piRNA). To identify the genome-wide RNA targets of PIWIL4 and understand whether PIWIL4 associates with piRNA in AML cells, we performed photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) on THP-1 cells (n = 2). We attained 8967 sequence clusters that corresponded to over 4500 annotated transcripts [false discovery rate (FDR) of <0.05 and log(FC) >1]. Our data revealed that known piRNA constituted only 6% of PIWIL4-bound RNA. Strikingly, nearly half of the PIWIL4-associated RNA mapped to protein-coding genic regions (48%) and 17% mapped to enhancers (Figure 4A; supplemental Figure 4A; supplemental Table 3). Motif analysis of binding sites showed enrichment of guanine-rich sequences (Figure 4B). In patients with primary AML, LSC populations immunophenotypically and transcriptionally resemble normal myeloid progenitors.29 The pathway and cell radar analysis of the RNA targets of PIWIL4 annotated to protein-coding genic regions showed enrichment for cancer pathways and association with gene signatures of human myeloid progenitors, common myeloid progenitors (CMP), GMP, and myelocytes (Figure 4C; supplemental Figure 4B).

PIWIL4 binds to cancer-associated protein coding RNA in AML cells and its depletion induces deregulation of LSC and DNA repair–associated pathways. (A) Genomic distribution of PIWIL4 bound RNA in THP-1 cells from 2 independent PAR-CLIP experiments. (B) The top 2 significantly enriched motifs of PIWIL4-binding sites in THP-1 cells. (C) MSigDB pathway analysis of RNA targets of PIWIL4 annotated to protein-coding genic regions. (D) Heatmap of differentially expressed genes in shPIWIL4 vs scrambled transduced THP-1 cells, depicting expression of LSC-associated genes identified in HSC/CMP/GMP lineage LSCs from MLL-AF9, MOZ-TIF2, and MN1 models. (E) Cell radar analysis of differentially expressed genes. (F) gene set enrichment analysis (GSEA) of RNA-seq of PIWIL4-depleted THP-1 cells.39 (G) γ-H2AX foci in Piwil4-depleted ckit+ MLL-AF9+ cells. The horizontal size bar represents a distance of 20 μM. The right panel shows the mean percentage of cells with more than 5 foci combined from 3 independent experiments (H) Neutral comet assay depicting damaged DNA in PIWIL4-depleted cells from patients with primary AML. The size bar depicts 100 μm distance. The right panel shows a summary of the tail moment of single cells combined from 3 biological replicates. (I) Left panel: representative western blot for PIWIL4, p-ATR, p-RPA2, and γ-H2AX and β-actin as a control on PIWIL4-depleted THP-1 cells and depleted cells rescued by ectopic expression of PIWIL4. Right panel: densitometry data analyzed using ImageJ of PIWIL4, p-ATR, p-RPA2, and γ-H2AX protein levels normalized to β-actin protein levels in scrambled vs shPIWIL4-A-transduced THP-1 cells with (+) or without (−) the ectopic expression of PIWIL4. The data is a summary of 3 independent experimental replicates. (J) Native bromodeoxyuridine (BrdU) staining of PIWIL4-depleted AML cells. The left panel shows a representative figure of anti–BrdU-488–stained THP-1 cells. The size bar represents a distance of 20 μM. The right panel shows mean fluorescent intensity of anti–BrdU-488–stained cells.
PIWIL4 binds to cancer-associated protein coding RNA in AML cells and its depletion induces deregulation of LSC and DNA repair–associated pathways. (A) Genomic distribution of PIWIL4 bound RNA in THP-1 cells from 2 independent PAR-CLIP experiments. (B) The top 2 significantly enriched motifs of PIWIL4-binding sites in THP-1 cells. (C) MSigDB pathway analysis of RNA targets of PIWIL4 annotated to protein-coding genic regions. (D) Heatmap of differentially expressed genes in shPIWIL4 vs scrambled transduced THP-1 cells, depicting expression of LSC-associated genes identified in HSC/CMP/GMP lineage LSCs from MLL-AF9, MOZ-TIF2, and MN1 models. (E) Cell radar analysis of differentially expressed genes. (F) gene set enrichment analysis (GSEA) of RNA-seq of PIWIL4-depleted THP-1 cells.39 (G) γ-H2AX foci in Piwil4-depleted ckit+ MLL-AF9+ cells. The horizontal size bar represents a distance of 20 μM. The right panel shows the mean percentage of cells with more than 5 foci combined from 3 independent experiments (H) Neutral comet assay depicting damaged DNA in PIWIL4-depleted cells from patients with primary AML. The size bar depicts 100 μm distance. The right panel shows a summary of the tail moment of single cells combined from 3 biological replicates. (I) Left panel: representative western blot for PIWIL4, p-ATR, p-RPA2, and γ-H2AX and β-actin as a control on PIWIL4-depleted THP-1 cells and depleted cells rescued by ectopic expression of PIWIL4. Right panel: densitometry data analyzed using ImageJ of PIWIL4, p-ATR, p-RPA2, and γ-H2AX protein levels normalized to β-actin protein levels in scrambled vs shPIWIL4-A-transduced THP-1 cells with (+) or without (−) the ectopic expression of PIWIL4. The data is a summary of 3 independent experimental replicates. (J) Native bromodeoxyuridine (BrdU) staining of PIWIL4-depleted AML cells. The left panel shows a representative figure of anti–BrdU-488–stained THP-1 cells. The size bar represents a distance of 20 μM. The right panel shows mean fluorescent intensity of anti–BrdU-488–stained cells.
Next, to better understand the molecular impact of PIWIL4 depletion and, specifically, whether it affected the expression of PIWIL4 RNA targets in AML cells, we performed RNA-seq in THP-1 cells. We observed 5305 differentially expressed genes (FDR < 0.05, P < .05) (supplemental Figure 4A; supplemental Table 2). PIWIL4 KD induced the downregulation of LSC-associated genes originating from AML HSC, CMP, and GMP gene signatures of MN1-, MOZ-TIF2–, and MLL-AF9–induced AML models (Figure 4D; supplemental Table 3).40-42 These also included genes from the 17-gene LSC signature that associates gene expression in LSCs with inferior outcomes in patients with AML (Figure 4A; supplemental Table 3).43 Cell radar analysis indicated that PIWIL4 depletion led to the downregulation of genes associated with human HSC, CMP, GMP, and myelocyte gene signatures, whereas upregulated genes were mainly associated with the megakaryocyte-erythroid progenitor gene signature (Figure 4E). In gene set enrichment analysis, LSC maintenance associated hypoxia and tumor necrosis factor α (TNF-α) via NF-κB signaling pathways, and LSC signature genes were significantly downregulated in THP-1 cells (Figure 4F).44-46 Of note, in patients with AML, PIWIL4 expression positively correlated with hypoxia and TNF-α via NF-κB signaling pathways and additionally with the expression of MYC targets and LSC-associated metabolic pathways (supplemental Figure 4D).
The overlap of PAR-CLIP data with RNA-seq revealed that PIWIL4 targets exhibited higher median expression than nontargets in THP-1 cells (supplemental Figure 4E). In our RNA-seq data, 30% of PIWIL4 targets (1193) were differentially expressed, of which nearly two-thirds (754) were downregulated. As before, downregulated genes largely belonged to hypoxia and TNF-α via NF-κB signaling pathways and consisted of multiple AML and LSC-associated genes (data not shown, supplemental Table 3). Because of the binding of PIWIL4 to sequences that mapped to introns, we also analyzed splicing events. The analysis of alternative splicing (AS) events in PIWIL4-depleted THP-1 cells revealed 4614 AS splicing events, the highest of which were intron retention events (supplemental Figure 4F; supplemental Table 3). The genes that underwent AS events were associated with the GMP lineage signature (data not shown), were downstream targets of MYC, and were associated with cell cycle, DNA repair, and apoptosis pathways (supplemental Figure 4G). However, only 20% of transcripts that retained introns showed downregulation in RNA-seq, suggesting that only select transcripts showed decreased expression due to nonsense-mediated decay. These select transcripts belonged to TNF signaling, hypoxia, and cholesterol homeostasis pathways (MSigDB, P < .01, FDR < 0.01).
PIWIL4 depletion induces DNA damage, replication stress, and activation of the ATR pathway in AML cells
The upregulated pathways in our RNA-seq data largely consisted of DNA repair pathway genes associated with homologous recombination repair, suggesting that PIWIL4 prevents DNA damage in AML cells (Figure 4F; supplemental Figure 4H). Because of the upregulation of DNA repair pathways, we examined the status of DNA damage in PIWIL4-depleted AML cell lines and patients with primary AML. We observed a clear increase in double-stranded DNA damage through γH2AX staining and neutral comet assays (Figure 4G-H; supplemental Figure 5A-B). The ATM (ataxia-telangiectasia–mutated), ATR (ATM- and Rad3-related), and DNA-PKcs (DNA-dependent protein kinase) kinases are upstream DNA damage response kinases. Western blots showed that PIWIL4 depletion did not activate ATM signaling in AML cells (data not shown). However, we observed activation of the ATR pathway, evidenced by the increased phosphorylation of ATR and replication protein A1 (RPA1), whereas unphosphorylated ATR levels were perceptibly reduced (Figure 4I; supplemental Figure 5C-F). Consistent with our observations using microscopic analyses, we also observed increased levels of γH2AX in western blots. In contrast, the rescue of PIWIL4 KD by forced overexpression of PIWIL4 alleviated the phosphorylated protein levels of ATR, RPA, and H2AX, establishing a causal relationship between PIWIL4 depletion, DNA damage, and activation of the ATR pathway (Figure 4I). As ATR is one of the central DNA replication stress response kinases, we analyzed whether PIWIL4 depletion induced replication stress. Indeed, the native bromodeoxyuridine assay demonstrated increased replication stress in PIWIL4-depleted AML cells (Figure 4J).
PIWIL4 is an R-loop–resolving enzyme in AML cells
PIWIL4 belongs to the RNase H superfamily. RNASEH enzymes are responsible for resolving DNA:RNA hybrid–based structures known as R-loops.47-49 R-loop accumulation can cause activation of the ATR pathway, replication stress, and DNA damage.50-52 Furthermore, guanine-rich sequences are important for the nucleation and elongation of R-loop structures.53 We observed all of the aforementioned phenotypes in PIWIL4-depleted AML cells, and motif analysis of PIWIL4-binding sites showed enrichment of guanine-rich sequences. Therefore, we hypothesized that PIWIL4 could be a functional R-loop–resolving enzyme in AML cells, and its depletion could lead to R-loop accumulation in AML cells.
To test this, we performed intracellular staining using S9.6, a monoclonal antibody against DNA:RNA hybrids, which revealed that PIWIL4 depletion in primary cells from patients with AML and cell lines induced an accumulation of S9.6 signal in the nucleus of AML cell lines and primary patient cells within 72 hours of transduction (Figure 5A-B; supplemental Figure 6A). The accumulation of the S9.6 signal was lost upon treatment with the RNase H enzyme, confirming the presence of R-loops (Figure 5B; supplemental Figure 6A). Secondly, the ectopic overexpression of wild-type PIWIL4 or human RNASEH1 in KD cells also rescued the R-loop accumulation phenotype, underlining the causal relationship between R-loop accumulation and PIWIL4 expression (Figure 5C). These findings were further confirmed with a catalytically inactivated RNASEH fused with a GFP protein, which acts as an R-loop sensor (supplemental Figure 6B).56 Notably, RNASEH1 or PIWIL4 wild-type overexpression but not overexpression of a mutant PIWIL4, PIWIL4-ΔCD, with an intact active site (amino acids 225-450) but lacking the canonical catalytic PIWI domain (amino acids 491-863), allowed AML cells to overcome the reduced colony-forming ability and apoptosis phenotype of PIWIL4-depleted AML cells, demonstrating that the R-loop resolving function of PIWIL4 is critical for AML cell growth and survival (Figure 5D; supplemental Figure 6C). Of note, PIWIL4 depletion in healthy HSPCs did not induce R-loop accumulation (supplemental Figure 6D).
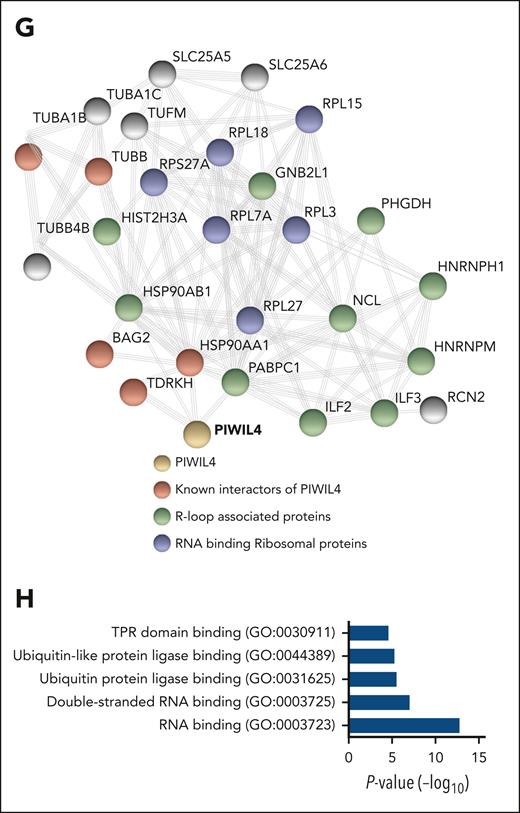
PIWIL4 is a R-loop–resolving enzyme in AML cells. (A) Representative image of nuclear mean fluorescence intensity of S9.6 signal in PIWIL4-depleted sample from patients with AML. The size bar represents 20 μm. The right panel shows summarized nuclear mean fluorescence intensity of individual cells from 3 biological replicates combined. Horizontal lines depict mean intensity of all analyzed cells. (B) Summarized nuclear mean fluorescence intensity of R-loops in individual cells from 3 independent experiments of PIWIL4 depletion in THP-1 and NB-4 AML cell lines. Fixed cells from shRNA-A were treated with RNase H as a control for the presence of R-loops. (C) Nuclear mean fluorescence intensity of R-loops in PIWIL4-depleted THP-1 cells transduced with empty vector vs ectopic overexpression of PIWIL4 or RNASEH1. In the dot plot, each dot represents fluorescence from individual cells combined from 3 independent experiments. (D) Percentage difference in colony formation in CFU assay of PIWIL4 KD AML cell lines, rescued with overexpression of RNASEH1 or wild-type PIWIL4 vs PIWIL4-ΔCD mutant and empty vector control, 72 hours after transduction with overexpression constructs. Bars indicate mean and SEM. “n” indicates number of independent experiments. (E-F) Treatment of (E) DNA:RNA hybrids and (F) R-loops with human wild-type PIWIL4, mutant PIWIL4-ΔCD (0.2, 0.4, 0.8 μM), and RNASEH1 (2.5 μM) for a duration of 1 hour at 37°C. Representative images from 3 independent experiments are shown. The graph represents the quantification of fractions of substrate cleaved in 3 independent experiments (G) STRING network analysis of PIWIL4 protein interactome in 293T HEK cells showing known interactors of PIWIL4,54 R-loop–binding proteins55 and RNA-binding ribosomal proteins (H) GO term pathway analysis of PIWIL4-associated proteins (FDR < 0.05, P < .05).

Structural and functional studies of archaebacterial PIWI and the human PIWI domain of AGO2 strongly suggested that the human PIWI domain was capable of binding and resolving DNA:RNA hybrids.57-59 To test the ability of the human PIWIL4 protein to resolve DNA:RNA hybrids and R-loops in vitro, fluorescent DNA:RNA hybrids and R-loops were treated with either the wild-type PIWIL4 or the mutant protein PIWIL4-ΔCD, lacking the canonical catalytic PIWI domain (amino acids 491-863), or RNASEH1 as a positive control (Figure 5E-F). RNASEH1 treatment confirmed the resolving activity on DNA:RNA hybrids and R-loops.48,60 Importantly, PIWIL4 also demonstrated the ability to resolve DNA:RNA hybrids and R-loops with increasing levels of concentration of protein for a fixed duration of time (1 hour), whereas the mutant PIWIL4-ΔCD showed a marked reduction in catalytic activity on both hybrids and R-loops (Figure 5E-F). In addition, to supplement our findings, we also analyzed the ability of PIWIL4 to resolve R-loops in vivo. For this, we used HEK cells that constitutively express the R-loop–forming portion of the mouse antisense of IGF2R nonprotein coding RNA (AIRN) gene.61,62 Overexpression of wild-type RNASEH1 or PIWIL4 resulted in the resolution of R-loops at the mouse AIRN gene compared with the empty vector, demonstrating that PIWIL4 can resolve R-loops in vivo (supplemental Figure 6E).
To rule out that PIWIL4 partner proteins could be responsible for R-loop resolution, we immunoprecipitated HA-tagged PIWIL4 and performed liquid chromatography/mass spectrometry (LC/MS). In LC/MS of HA-tagged PIWIL4, we identified known interactors such as TDRKH and BAG2, and several R-loop interacting proteins such as premessenger RNA–binding proteins HNRNPH1 and PABPC1 (Figure 5G-H; supplemental Figure 6F; supplemental Table 3).54,55,63,64 Overall, the pathway analysis of PIWIL4-partner proteins showed enrichment for RBPs and downstream targets of MYC (Figure 5H; supplemental Figure 6G). However, no RNase H–like enzymes or RNA/DNA helicases were present in our LC/MS analysis, suggesting that PIWIL4 independently performed the function of hybrid and R-loop resolution (supplemental Table 3).
PIWIL4 binds RNA and prevents R-loop accumulation on a subset of AML and LSC-associated genes
Studies have experimentally demonstrated that, apart from DNA damage, preformed R-loops can also stall transcription.65,66 We reasoned that in addition to R-loop accumulation–induced DNA damage, PIWIL4 depletion could also halt the transcription of genes coding for factors required for LSC function and AML growth. To investigate this hypothesis, we analyzed whether PIWIL4 RNA targets overlapped with known sites of R-loop enrichment and nascent transcription in AML cells. For this, we performed DNA:RNA hybrid immunoprecipitation followed by sequencing (DRIP-seq) on THP-1 cells, overlayed it with our PAR-CLIP data, and published Global Run-On sequencing of THP-1 cells (supplemental Figure 7A).67 Strikingly, 72% of PIWIL4 target (PAR-CLIP) promoters, 74% exons, and 67% introns overlapped with broad regions of R-loop enrichment (DRIP-seq, ±1.5 kb) and with nascent transcription (global run-on sequencing, ±1.5 kb) at promoters (46%) (Figure 6A; supplemental Figure 7B). In particular, centered peaks of PAR-CLIP showed a significant overlap with R-loops at promoters and exons and with nascent transcription at promoters but not at exons (Figure 6B). A total of 951 PIWIL4 targets showed strong overlap with R-loop enrichment and nascent transcription (Figure 6C). Collectively, overlapping promoter-, exon-, and intron-associated loci consisted of sites harboring several AML-specific enhancers and genes coding for factors critical for AML growth and LSC survival. These included CD93, TRIB1, BCL2, HBP1, FTO, ZEB1, and the HOXA locus that were also downregulated in our RNA-seq data set68-71 (Figure 6D-E; supplemental Figure 7C-D; supplemental Table 5). In all, 359 PIWIL4 RNA targets that were known sites of R-loop enrichment or nascent transcription were downregulated in our RNA-seq data set and once again belonged to hypoxia and TNF-α via NF-κB signaling pathways, suggesting that PIWIL4 depletion could halt transcription of a subset of AML-associated target genes, possibly because of R-loop accumulation (Figure 6E; supplemental Table 5).
PIWIL4 binds the RNA and prevents R-loop accumulation at AML- and LSC-associated genes. (A) Overlap of PAR-CLIP peaks with DRIP-seq peaks (±1.5 kb). (B) Heatmap of centered PAR-CLIP peaks at promoters and exons and its overlap with corresponding DRIP-seq and Global Run-On Sequencing (GRO-seq) peaks.67 (C) Heatmap of PAR-CLIP peaks at promoters sorted by peak distance to transcription start site and its overlap with DRIP-seq and GRO-seq peaks. (D) PAR-CLIP data overlayed with DRIP-seq, published GRO-seq, and H3K27Ac chromatin immunoprecipitation (ChIP)-seq data35 of THP-1 cells at select AML-associated PWIIL4 targets. (E) MSigDB pathway analysis of downregulated PIWIL4 RNA targets that overlapped with sites of R-loop enrichment or nascent transcription (P < .05, FDR < 0.05). (F) DRIP-qPCR of PIWIL4 targets, CD93 and TRIB1 promoter in THP-1 cells transduced with scrambled vs shRNAs against PIWIL4 vs shRNA coexpressed with PIWIL4 overexpression construct. (G) mRNA expression of selected PIWIL4 targets in THP-1 cells transduced with scrambled vs shRNAs against PIWIL4 vs shRNA coexpressed with PIWIL4 overexpression construct.

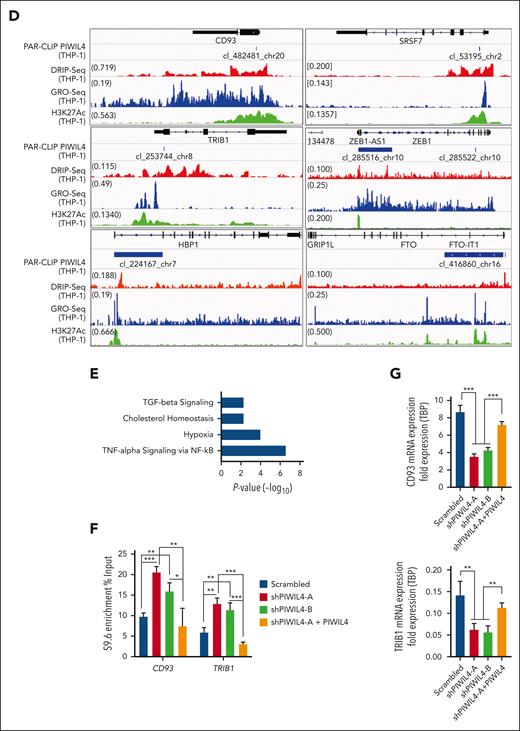
To further test this hypothesis, we tested whether PIWIL4 depletion induced R-loop accumulation at 2 such AML- and LSC-associated target genes, CD93 and TRIB1, that were downregulated in our RNA-seq data set, and at a known R-loop–enriched locus RPL13A, which was not identified as a PIWIL4 target in our PAR-CLIP data set. DRIP-qPCR revealed that PIWIL4 depletion indeed induced R-loop accumulation at the promoters of CD93 and TRIB1, but did not induce a significant increase in R-loop accumulation in RPL13A (Figure 6F; supplemental Figure 7E). Rescue of PIWIL4 expression via overexpression prompted the rescue of the R-loop accumulation phenotype at CD93 and TRIB1 promoters. Concurrently, the KD and rescue of PIWIL4 expression induced loss and rescue of CD93 and TRIB1 gene expression, respectively, demonstrating that PIWIL4 was required to regulate R-loop homeostasis at genes required for AML growth and thereby maintain their expression (Figure 6G). Finally, the KD of PIWIL4 in patients with primary AML induced downregulation of CD93, TRIB1, and BCL2, but KD in healthy CD34+ stem progenitors did not alter the expression of these genes, validating our findings in the AML cell line model and healthy hematopoiesis, respectively (supplemental Figure 7F-G). Collectively, these studies demonstrate that AML cells owe their dependency on PIWIL4 to its ability to resolve R-loops, which not only prevents DNA damage but also maintains the transcription of a subset of loci required for AML cell survival.
PIWIL4 depletion and ATR inhibition act synergistically to target AML cells
Previous studies have shown that the ATR response triggered by R-loops can render cells dependent on the ATR pathway for survival.62,72,73 Because PIWIL4 depletion preferentially impeded LSCs but spared healthy human HSPCs, the inhibition of PIWIL represents a viable therapeutic target for AML stem cells. We tested the combination of ATR inhibition with PIWIL4 depletion to induce a compound effect on AML cells using the ATR kinase inhibitor AZ20 (ATRi). PIWIL4 depletion rendered AML cells more sensitive to ATR inhibition in liquid culture, with a reduction of the 50% inhibitory concentration of the ATRi by 12- and 3.6-fold in PIWIL4-depleted THP-1 and NB-4 cells, respectively (Figure 7A). ATRi and PIWIL4 depletion antagonized the colony-forming potential of AML cell lines in vitro and primary AML growth in vivo (Figure 7B-C). The combination also provided survival benefits to xenografts in vivo (Figure 7D). These proof-of-principle experiments indicate that PIWIL4 depletion and R-loop accumulation potentiate ATR sensitivity in AML cells and that the combination of PIWIL4 depletion and ATR inhibition is an effective strategy for antagonizing AML stem cell growth.

PIWIL4 depletion and ATR inhibition act synergistically to target AML cells. (A) Fifty percent inhibitory concentration (IC50) of ATRi in scrambled and PIWIL4-depleted AML cells at 72 hours after treatment. (B) Percentage difference in clonogenicity in CFU assay of scrambled and shPIWIL4-transduced AML cell lines treated with respective IC50 concentrations (scrambled/shPIWIL4-A/shPIWIL4-B: THP-1-712/50/64 nM; NB-4-240/61/71 nM) of ATRi. (C) Flow cytometry analysis of human engraftment in BM of NSG mice sacrificed 12 weeks after transplantation with cells from patients with AML transduced with shRNA or scrambled, sorted and treated with dimethyl sulfoxide (DMSO) or respective IC50 concentrations of ATRi (712 nM for scrambled and 50 nM for shPIWIL4-A) for 48 hours. (D) Kaplan Meier plot depicting survival of NSG mice that underwent transplantation with scrambled control or shPIWIL4-transduced THP-1 cells treated with DMSO or respective IC50 concentrations of ATRi (712 nM for scrambled and 50 nM for shPIWIL4-A).
PIWIL4 depletion and ATR inhibition act synergistically to target AML cells. (A) Fifty percent inhibitory concentration (IC50) of ATRi in scrambled and PIWIL4-depleted AML cells at 72 hours after treatment. (B) Percentage difference in clonogenicity in CFU assay of scrambled and shPIWIL4-transduced AML cell lines treated with respective IC50 concentrations (scrambled/shPIWIL4-A/shPIWIL4-B: THP-1-712/50/64 nM; NB-4-240/61/71 nM) of ATRi. (C) Flow cytometry analysis of human engraftment in BM of NSG mice sacrificed 12 weeks after transplantation with cells from patients with AML transduced with shRNA or scrambled, sorted and treated with dimethyl sulfoxide (DMSO) or respective IC50 concentrations of ATRi (712 nM for scrambled and 50 nM for shPIWIL4-A) for 48 hours. (D) Kaplan Meier plot depicting survival of NSG mice that underwent transplantation with scrambled control or shPIWIL4-transduced THP-1 cells treated with DMSO or respective IC50 concentrations of ATRi (712 nM for scrambled and 50 nM for shPIWIL4-A).
Discussion
The development of targeted therapies aimed at eradicating LSCs in AML but sparing their normal counterpart has remained a challenge, largely because of the only subtle differences between LSCs and their normal counterparts.74 Oncogene-driven high transcriptional activity and resultant R-loop accumulation impede proliferation and induce genome instability. This could render AML LSCs preferentially dependent on enzymes that erase R-loop accumulation and represent a potential differential vulnerability between AML LSCs and healthy HSCs. Indeed, oncogene-induced hypertranscription correlates with increased R-loop formation and transcription-replication conflicts, resulting in replication stress and genomic instability, and the forced overexpression of RNase H alleviates this phenotype. This indicates that cancer cells need to develop mechanisms to minimize oncogene-induced R-loop accumulation, and these mechanisms could represent a therapeutic vulnerability.75,76 We demonstrate that, in contrast to normal HSCs, AML LSCs and leukemic bulk preferentially depend on PIWIL4, a protein that resolves R-loops via its enzymatic activity. The loss of nuclear expression of PIWIL4 in AML cells induces R-loop accumulation, replication stress, DNA damage, and activation of the ATR pathway, which is alleviated by PIWIL4 re-expression. These data are supported by studies of prokaryotic PIWI,57 archaebacterial PIWI,58 and the human PIWI domain of AGO2,59 which collectively suggest that PIWIL proteins possess R-loop resolving capabilities.
PIWIL4 binds the RNA of genes associated with the CMP/GMP lineage expression signature, hypoxia and TNFα-via NF-κB signaling, and several LSC maintenance factors, that are known sites of R-loop enrichment, nascent transcription, and open chromatin in AML cells. As proof-of-principle, we demonstrate that PIWIL4 impairs R-loop accumulation and promotes transcription via its R-loop resolving activity at 2 genes required for LSC function and AML growth, CD93 and TRIB1. Cycling LSCs will likely have a greater need for R-loop resolution and PIWIL4 expression because of the potential for increased transcription-replication conflict in proliferative cells, as the most prevalent source of R-loop–mediated damage seems to arise during the S-phase of the cell cycle.77 It is intriguing that CD93, the cell-surface marker that regulates LSC self-renewal and defines cycling LSCs in the MLL-AF9 model, is a target of PIWIL4.70 However, it is unclear why cycling healthy human HSPCs do not require PIWIL4, which was also shown in mice.30 A potential cause could be higher reactive oxygen species in AML cells which could inhibit canonical RNase H function and prompt PIWIL4 to take over RNase H function.78 Another reason could be the posttranslational modification of PIWIL4. The arginine methylase, protein arginine N-methyltransferase 5 methylase, which is also required for AML growth, LSC function, and the maintenance of genomic integrity of AML cells, symmetrically dimethylates the arginine residues of Piwi proteins and renders them stable.79-81 It would be of interest to test whether PIWIL4 protein methylation levels are increased in AML vs healthy cells.
Our data provide a strong rationale for developing inhibitors against the PIWI domain of PIWIL proteins and should fuel such efforts. Such an approach is likely to be most attractive in combination with other antileukemic drugs, such as ATR inhibitors. R-loop accumulation–inducing drugs render leukemia cells sensitive to ATR inhibition.72 Therefore, combining PIWI inhibitors with existing ATR inhibitors, some of which are in clinical trials, would be a promising therapeutic approach in multiple cancers, including AML (ClinicalTrials.gov identifier: NCT03770429).82 The combination of the ATR inhibitor with gemcitabine was shown to be highly efficient preclinically in an AML model. However, all these approaches do not implement preferential targeting of AML LSCs, in contrast to what we have seen with PIWIL4 depletion.
Overall, our data shed a light on the crucial role of R-loop formation in AML and characterize PIWIL4 as a dependency factor for AML bulk and LSCs, required for the resolution of R-loops, maintenance of LSC-associated gene expression, and safeguarding the genomic integrity of AML cells through its enzymatic function. Studies in the near future could be directed toward the role of PIWIL4 in clonal competition and the transition from MDS to secondary AML. The fitness and dominance of preleukemic or leukemic clones could depend on their ability to efficiently regulate R-loops. Therefore, it is worth assessing whether PIWIL4 is one such factor that promotes clonal survival and dominance via its R-loop–resolving activity.
Acknowledgments
The authors thank all members of the animal facility of the University of Ulm, Germany for breeding and maintenance of the animals. They thank all members of the Institute of Experimental Cancer Research for technical support, such as patient sample preparation, cell sorting, irradiation of mice, or injection of cells into mice. They thank Dominique Desef and Christian Desiderato for purifying the catalytically inactivated RNASEH protein fused with GFP (ΔRNH-GFP), and Oliver Goldbeck and Christian Riedel for troubleshooting and support. They thank the Core Facility Cytometry and Core Facility Microscopy, University of Ulm. They thank Lisa Wiesmüller for participation in fruitful discussions and Konstanze Döhner and Hartmut Döhner for providing patient samples.
The work was supported by grants received by S.B. from the Baustein program 3.2, University of Ulm, Germany; C.B. and V.P.S.R. from the Deutsche Forschungsgemeinschaft (SPP1463); C.B. from SFB1074, project A04; V.P.S.R. from the Department of Science and Technology, Science and Engineering Board, and Engineering Research Board, India, SQUID-1972-VR-5379; and A. Karsan from the New Frontiers Program Project grant of the Terry Fox Research Institute and the Canadian Institutes of Health Research.
Authorship
Contribution: C.B., V.P.S.R., and S.B. designed the project and discussed and analyzed the data; S.B. performed most of the experiments, with support from A.J.P., J. Mueller, and J. Mark; A.J.P., J. Mueller, and N.M.V. assisted with immunofluorescence and confocal microscopy; S.I. provided functionality tested human LSCs; gene expression data of functionally validated LSCs was analyzed by V.P.S.R. using the Basepair pipeline; A.J.P., N.M.V., T.M., K.F., and J. Mueller participated in in vivo experiments by transplanting cells and IV immunoglobulin, irradiating and bleeding mice; the R-loop enzyme assays were performed and analyzed by T.Y. and L.Z. at MGH, Harvard, and independently by C.S. at DKFZ, Heidelberg; PAR-CLIP was performed by J.I.H. and data analysis was done by A. Kloetgen; liquid chromatography/mass spectrometry and its analysis was performed by R.R. and S.W.; A. Karsan provided data of AML, secondary AML, and patients with MDS and logistical support; E.D. prepared DRIP-seq libraries and A.T. provided logistical support; S.B., A.U.S., A. Kumari, and L.W. analyzed RNA-seq, PAR-CLIP, Global Run-On sequencing, and DRIP-seq data using Basepair; E.K. and S.H. performed the in vivo test for PIWIL4 enzyme activity on R-loops; L.Q.-M. and I.G.-M. performed histopathology; and S.B., C.B., and V.P.S.R. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Christian Buske, Institute of Experimental Cancer Research, University Hospital of Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany; e-mail: christian.buske@uni-ulm.de; and Vijay P. S. Rawat, Special Centre for Molecular Medicine, Jawaharlal Nehru University, New Delhi, India; e-mail: vijaypsrawat@mail.jnu.ac.in.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal