

Key Points
Platelet necrosis regulated by cyclophilin D mediates platelet-neutrophil interactions in ischemic stroke.
Targeting cyclophilin D–mediated platelet necrosis improves stroke outcomes.

Abstract
Dysregulated platelet functions contribute to the development and progression of ischemic stroke. Utilizing mice with a platelet-specific deletion of cyclophilin D (CypD), a mediator of necrosis, we found that platelet necrosis regulates tissue damage and outcomes during ischemic stroke in vivo. Mice with loss of CypD in platelets (CypDplt−/−mice) exhibited significantly enhanced cerebral blood flow, improved neurological and motor functions, and reduced ischemic stroke infarct volume after cerebral ischemia-reperfusion injury. These effects were attributable, at least in part, to platelet-neutrophil interactions. Twenty-four hours after stroke, significantly more circulating platelet-neutrophil aggregates (PNAs) were found in CypDplt+/+ mice. Underscoring the role of platelet necrosis in PNA formation, we observed a significant number of phosphatidylserine (PS)+ platelets in PNAs in CypDplt+/+ mice. In contrast, significantly fewer platelets in PNAs were PS+ in CypDplt−/− counterparts. Accordingly, mice with CypD-deficient platelets had fewer neutrophils and PNAs recruited to their brain following stroke relative to wild-type counterparts. Neutrophil depletion in wild-type mice conferred protection from ischemic stroke to a similar degree as observed in mice with CypD-deficient platelets. Neutrophil depletion in CypDplt−/− mice did not further reduce infarct size. Transmission electron microscopy of ex vivo–formed PNAs revealed a propensity of necrotic platelets to interact with neutrophils. These results suggest that necrotic platelets interact with neutrophils to exacerbate brain injury during ischemic stroke. Because inhibiting platelet necrosis does not compromise hemostasis, targeting platelet CypD may be a potential therapeutic strategy to limit brain damage following ischemic stroke.
Introduction
Antiplatelet agents, including aspirin, are used to prevent ischemic stroke in patients. Nevertheless, these agents are incompletely effective.1 Emerging evidence highlights that platelets contribute to ischemic stroke through mechanisms and pathways that may not be targeted with classic antiplatelet agents.2,3 Platelets mediate heterotypic leukocyte interactions and recruitment, which contributes to the pathophysiology of stroke.4 In particular, platelet-neutrophil interactions promote brain injury following ischemic stroke. Preclinical5,6 and clinical studies7,8 have found that intravascular neutrophils accumulate in the brain after stroke and that neutrophil interactions with platelets drive injurious inflammatory signaling.5,9 Yet, the mechanism by which platelets interact with neutrophils and subsequently mediate thromboinflammation in ischemic stroke remains elusive.
Recently, a novel thrombotic mechanism between necrotic platelets and neutrophils was reported in models of gut ischemia-reperfusion injury.10 A key step in platelet necrosis is the formation of the mitochondrial permeability transition pore (MPTP) in the inner membrane of mitochondria. The MPTP allows the influx of cytosolic molecules into the mitochondria, disrupting the mitochondrial membrane and function. MPTP formation is regulated by cyclophilin D (CypD), and CypD in platelets regulates procoagulant platelet formation and platelet necrosis.11,12 However, the role of platelet CypD in thrombosis appears to be setting dependent,10-13 and it has never been established whether platelet CypD controls brain injury, neutrophil trafficking, and outcomes during ischemic stroke.
Methods
Mice
All animal experiments complied with the regulatory standards of the University of Utah. The generation of platelet-specific CypD mice has been described.10 Briefly, conditional deletion of CypD from platelets in mice (CypDplt−/− mice) was achieved by crossing C57BL/6J mice harboring a floxed allele of CypD (CypDplt+/+) with platelet factor 4-Cre–transgenic mice in which Cre recombinase is expressed under the control of the platelet factor 4 promoter. All experiments were performed using age- and sex-matched adult littermates.
Ischemic stroke model
Transient middle cerebral artery occlusion (tMCAO) was performed as described previously.14 Anesthesia was induced by inhalation of 5% isoflurane and maintained by inhalation of 2% isoflurane. After a midline incision in the neck, the proximal common carotid artery and the external carotid artery were ligated, and a standardized silicon rubber–coated 6.0 nylon monofilament (Doccol Corp., Redlands, CA) was inserted and advanced via the right internal carotid artery to occlude the origin of the right middle cerebral artery (MCA). The intraluminal suture was left in situ for 60 minutes. Next, animals were reanesthetized, and the occluding monofilament was withdrawn to allow reperfusion. In a subset of mice, the filament was left in place for 24 hours to induce permanent middle cerebral artery occlusion (pMCAO).
Neurological tests
Twenty-four hours after induction of MCAO, the modified Bederson test and the grip strength test were used to assess global neurological and motor functions in mice, respectively.14
Cerebral lesion quantification
To measure cerebral infarct volumes, mice were euthanized 24 hours after induction of MCAO. Brains were quickly isolated and cut into 2-mm-thick coronal sections using a mouse brain slice matrix. The slices were stained with 2% 2,3,5-triphenyl tetrazolium chloride (Sigma-Aldrich, St. Louis, MO) to distinguish healthy tissue from infarcted tissue. Stained slices were photographed, and infarct areas (white) were quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
Monitoring of cerebral blood flow
Cerebral blood flow (CBF) in the MCA territory was determined by laser Doppler flow measurements (moorVMS-LDF1; Moor Instruments; Devon, UK) via a fiber optic probe placed in the vascular territory of the right MCA.
Immunofluorescence
Twenty-four hours after stroke, mice were euthanized, and brains were dissected. Brains were cut into 2-mm-thick coronal sections, immediately placed in O.C.T. compound, and frozen in liquid nitrogen for sectioning. Next, 12-µm-thick sections were stained for the presence of neutrophils (rat anti-mouse Ly6G; eBioscience, San Diego, CA; or goat anti-mouse MPO R&D Systems, Minneapolis, MN) and platelets (rat anti-mouse GPIb–DyLight 649 conjugated; Emfret, Würzburg, Germany). Images were acquired using a high-resolution confocal reflection microscope (Olympus IX81, FV3000; Olympus, Melville, NY).
Flow cytometric analysis of cerebral leukocyte recruitment
Twenty-four hours after stroke, CypDplt+/+ and CypDplt−/− mice were euthanized, and brains were dissected. The hemispheres were mechanically disrupted using a scalpel and incubated for 30 minutes at 37°C with a digestion buffer (2.5 mg/mL collagenase D [Invitrogen, Carlsbad, CA], 5 U/mL DNase I [ThermoFisher, Waltham, MA] in RPMI 1640 + 10% inactivated fetal calf serum [Invitrogen]) and pressed through a cell strainer. Next, cells were separated from myelin and debris by Percoll (GE Healthcare, Chicago, IL) gradient centrifugation and incubated with CD45 APC-Cy7 (eBioscience), CD11b PE-Cy7 (eBioscience), Ly6G BV510 (eBioscience), Ly6C FITC (eBioscience), CD41 APC (MWREG30; ThermoFisher) and CD16/CD32 (Fc Block; eBioscience) for 30 minutes at room temperature in phosphate-buffered saline + 5% fetal bovine serum. After 30 minutes, the cells were washed and subsequently fixed in 200 μL of 0.5% paraformaldehyde and analyzed on a BD CytoFLEX (Beckman-Coulter, Pasadena, CA) located in the University of Utah Flow Cytometry Core.
Platelet-leukocyte aggregates
Whole blood was collected from CypDplt+/+ and CypDplt−/− mice 24 hours after tMCAO. Whole blood was diluted 1:10 into M199 (Lonza), and supplemented with 100 U/mL heparin to prevent exogenous thrombin generation. Diluted whole blood was stained with rat anti-mouse CD41 (APC, MWREG30; ThermoFisher) to detect platelets and rat anti-mouse Ly6G (BioLegend, San Diego, CA) to detect neutrophils or Ly6C (eBioscience) to detect monocytes. To detect phosphatidylserine (PS)+ platelets, FITC-labeled lactadherin (Haematologic Technologies, Essex Junction, VT) was added. In some experiments, only platelets were labeled, and P-selectin (Wug.E9-PE) expression and GPIb-α (Xia.G5-FITC; both from Emfret) were examined. Samples were stained for 15 minutes at 37°C and then fixed with BD FACS lysis buffer. Samples were analyzed on a BD CytoFLEX located in the University of Utah Flow Cytometry Core.
Neutrophil depletion
Neutrophils were depleted in CypDplt+/+ and CypDplt−/− mice by an anti-Ly6G antibody (10 mg/kg body weight; rat anti-mouse Ly6G, clone 1A8; Bio X Cell, West Lebanon, NH) given by intraperitoneal injection 24 hours before induction of cerebral ischemia.15 Control mice received an intraperitoneal injection of a rat IgG2a isotype control (10 mg/kg body weight; anti-trinitrophenol; Bio X Cell).
Platelet and neutrophil assays
Platelets were isolated from whole blood after cardiac puncture, as previously described,16-18 with specified modifications. Whole blood was drawn into sodium citrate (1:9) and diluted 1:1 with PIPES-saline-glucose buffer. Platelets were washed twice in PIPES-saline-glucose buffer before being resuspended in M199. Neutrophils were collected from bone marrow from femurs and tibias. Neutrophils were then isolated using a negative selection neutrophil isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and resuspended in M199. For transmission electron microscopy (TEM), platelets and neutrophils were mixed in a 100:1 ratio and stimulated with protease-activated receptor 4 (PAR4) peptide (150 μM, final) and collagen-related peptide (CRP; 150 ng/mL, final) for 15 minutes at 37°C. Samples were processed for TEM as previously described.19 For platelet-mixing experiments, isolated platelets from CypDplt+/+ and CypDplt−/− mice were stained with DyLight 488 GPIb-α or DyLight 649 GPIb-α (Emfret), respectively, and then washed twice with PIPES-saline-glucose buffer. Stained platelets were mixed with neutrophils in a 100:1 ratio and stimulated with PAR4 peptide (150 μM, final) and CRP (150 ng/mL, final) for 15 minutes. Neutrophils were stained with Ly6G BV510, and samples were run on a BD CytoFLEX. Platelet AKT activation was performed as previously described.20
Statistical analyses
Statistical analyses were performed with GraphPad Prism 8.1.1 (GraphPad Software, La Jolla, CA). Prior to statistical analysis, a D’Agostino-Pearson normality test was used to check data distribution. An unpaired Student t test or 1-way analysis of variance with Dunnett’s post hoc test or a Mann-Whitney test, as appropriate, was used for statistical comparison when applicable (infarct size, platelet-leukocyte aggregates, PS+ platelets in platelet-neutrophil aggregates (PNAs), P-selectin expression, GPIb-α expression, neutrophil counts, and neutrophil recruitment). In the case of nonparametric data (Bederson test and grip strength test score), a Kruskal-Wallis test with a post hoc Dunn correction was performed. Changes in laser Doppler blood flow were analyzed and compared by repeated-measures analysis of variance. All data are represented as mean ± standard deviation. A 2-tailed P < .05 was considered statistically significant.
Results
Loss of platelet CypD protects mice from cerebral ischemia-reperfusion injury
To investigate the role of platelet necrosis in cerebral ischemia-reperfusion injury, we subjected CypDplt−/− and CypDplt+/+ mice to tMCAO for 1 hour, followed by 23 hours of reperfusion. Brains were isolated 24 hours after stroke onset to determine ischemic stroke brain damage, as measured by infarct size. Infarct volumes were significantly reduced in the absence of platelet CypD (Figure 1A-B). Although CypDplt+/+ mice had an average infarct volume of 78.6 ± 27.7 mm3, infarct volumes in CypDplt−/− mice were 39 ± 15.7 mm3 (P < .0001). In agreement with reduced infarct size, CypDplt−/− mice exhibited significant improvements in neurological behavior (Bederson test score; Figure 1C) and motor function (grip strength test score; Figure 1D). In accordance with Stroke Therapy Academic Industry Roundtable recommendations,21 both female and male mice were studied. The protective effect observed in CypDplt−/− mice was observed similarly in both sexes (Figure 1E-F).
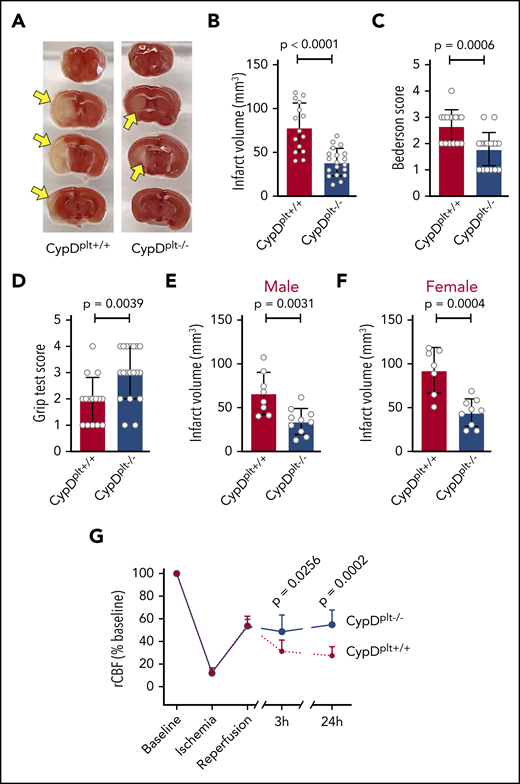
Deletion of platelet CypD reduces infarct size and improves neurologic and motor functions and blood flow following cerebral ischemia-reperfusion injury. Male and female platelet-specific CypD-deficient (CypDplt−/−) mice or littermate controls (CypDplt+/+) were subjected to transient (eg, 1 hour) cerebral ischemia, followed by 23 hours of reperfusion via tMCAO. (A) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white; arrows) indicates infarcted areas. (B) Brain infarct volumes were quantified by planimetric analysis. (C) Neurological outcome was assessed using the Bederson test; a lower score indicates better functioning. (D) Motor function was examined using the grip strength test; a higher score indicates better functioning. Brain infarct volumes in male (E) and female (F) mice 24 hours after tMCAO. (G) CBF was monitored longitudinally in the right MCA territory (the same side as occlusion) before ischemia (baseline), during ischemia, at the start of reperfusion, and at 3 and 24 hours after stroke onset.
Deletion of platelet CypD reduces infarct size and improves neurologic and motor functions and blood flow following cerebral ischemia-reperfusion injury. Male and female platelet-specific CypD-deficient (CypDplt−/−) mice or littermate controls (CypDplt+/+) were subjected to transient (eg, 1 hour) cerebral ischemia, followed by 23 hours of reperfusion via tMCAO. (A) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white; arrows) indicates infarcted areas. (B) Brain infarct volumes were quantified by planimetric analysis. (C) Neurological outcome was assessed using the Bederson test; a lower score indicates better functioning. (D) Motor function was examined using the grip strength test; a higher score indicates better functioning. Brain infarct volumes in male (E) and female (F) mice 24 hours after tMCAO. (G) CBF was monitored longitudinally in the right MCA territory (the same side as occlusion) before ischemia (baseline), during ischemia, at the start of reperfusion, and at 3 and 24 hours after stroke onset.
To determine whether improved stroke outcomes in CypDplt−/− mice were associated with improved local CBF during reperfusion, CBF was measured at time points during and after stroke induction (Figure 1G). During ischemia, as well as immediately after reperfusion, CBF was nearly identical in CypDplt+/+ and CypDplt−/− mice. Nevertheless, 3 hours after stroke onset (2 hours into the reperfusion phase), CBF was significantly greater in CypDplt−/− mice compared with CypDplt+/+ mice (48.6% ± 14.7% vs 31.3% ± 9.9%, P = .026). This difference in CBF persisted and was even more pronounced at 24 hours (54.8% ± 12.9% vs 27.4% ± 7.8%, P = .0002).
Platelet CypD is not critical in permanent cerebral ischemia
Because differences in CBF were observed during cerebral reperfusion, we further investigated whether platelet necrosis was contributing to brain injury during the ischemia or reperfusion phase. To accomplish this, we used a pMCAO model, in which the occluding filament is left in situ, resulting in permanent ischemia without any reperfusion. With permanent occlusion, no differences were observed in infarct volume between CypDplt−/− and CypDplt+/+ mice (Figure 2A-B). In addition, no differences were observed in neurological behavior or motor function (Figure 2C-D). These results suggest that platelet CypD mediates reperfusion injury following transient cerebral ischemia.

During permanent ischemic stroke, platelet CypD does not regulate infarct size or neurological and motor outcomes. Male and female CypDplt−/− mice or littermate controls (CypDplt+/+) were subjected to permanent (eg, 24 hours) cerebral ischemia (eg, pMCAO). (A) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white; arrows) indicates infarcted area. (B) Brain infarct volumes were quantified by planimetric analysis. (C) Neurological outcome was assessed using the Bederson test. (D) Motor function was examined using the grip strength test.
During permanent ischemic stroke, platelet CypD does not regulate infarct size or neurological and motor outcomes. Male and female CypDplt−/− mice or littermate controls (CypDplt+/+) were subjected to permanent (eg, 24 hours) cerebral ischemia (eg, pMCAO). (A) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white; arrows) indicates infarcted area. (B) Brain infarct volumes were quantified by planimetric analysis. (C) Neurological outcome was assessed using the Bederson test. (D) Motor function was examined using the grip strength test.
Cerebral ischemia-reperfusion injury induces the formation of circulating necrotic PNAs
Previous studies have demonstrated that platelet-leukocyte aggregates containing either monocytes or neutrophils play critical roles in the pathophysiology of ischemic stroke and reperfusion injury.5,10,22-24 To study the contribution of platelet CypD to platelet-leukocyte interactions, we first examined whether circulating platelet-monocyte aggregates (PMAs) were altered between CypDplt+/+ and CypDplt−/− mice after cerebral ischemia and reperfusion. In the transient MCAO model 24 hours after stroke, we observed no difference in circulating PMAs between CypDplt+/+ and CypDplt−/− mice, suggesting that CypD does not regulate PMA formation after ischemic stroke (Figure 3A). In contrast, circulating PNAs were nearly twofold higher in CypDplt+/+ mice compared with CypDplt−/− mice (27.3% ± 3.2% vs 16.9% ± 2.8%, P = .006; Figure 3B). Reperfusion of ischemic tissue initiates oxidative damage and cell death via the generation of reactive oxygen species (ROS).25,26 Oxidative stress induces CypD-mediated necrosis27,28 ; therefore, ROS generated by surrounding cells after reperfusion can induce platelet necrosis.11 Necrotic platelets express PS on their surface, become procoagulant, and have been shown to be more prone to interact with neutrophils.11,13,29,30 Underscoring the role of platelet necrosis in the formation of PNAs, we observed that 33% ± 4.0% of platelets in PNAs expressed PS on their surface in CypDplt+/+ mice 24 hours after ischemic stroke. In contrast, significantly fewer platelets (17.8% ± 5.1%) in PNAs were PS+ in the CypDplt−/− counterparts (Figure 3C).
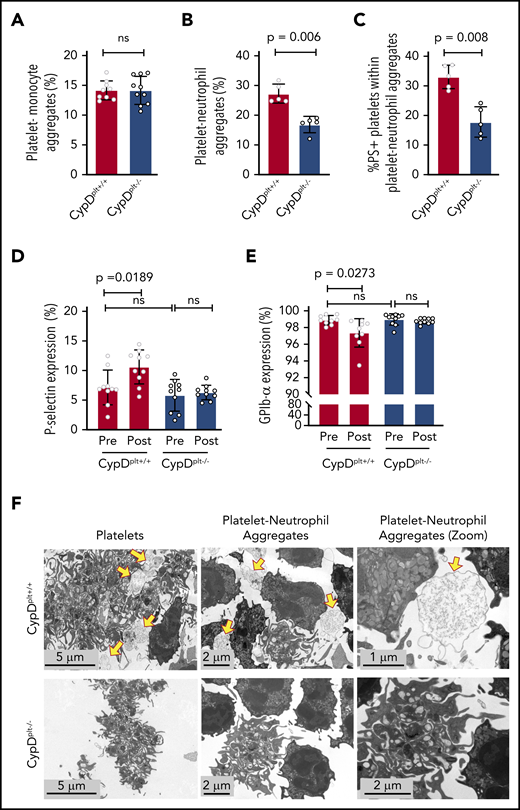
Cerebral ischemia-reperfusion injury induces the formation of circulating necrotic PNAs. Male and female CypDplt−/− mice or littermate controls (CypDplt+/+) were subjected to 1 hour of cerebral ischemia, followed by 23 hours of reperfusion via tMCAO. Twenty-four hours after tMCAO, PMAs (A), PNAs (B), and PS+ platelets within PNAs (C) were quantified by flow cytometry. P-selectin (D) and GPIb-α (E) expression on platelets was analyzed by flow cytometry immediately before and 24 hours after ischemic stroke. (F) Platelets and neutrophils were isolated from CypDplt+/+ and CypDplt−/− mice and activated with dual agonists against PAR4 and GPVI to induce platelet necrosis. PNAs were then imaged by TEM (panels are representative of n = 3 mice per group). Yellow arrows indicate necrotic platelets. ns, not significant.
Cerebral ischemia-reperfusion injury induces the formation of circulating necrotic PNAs. Male and female CypDplt−/− mice or littermate controls (CypDplt+/+) were subjected to 1 hour of cerebral ischemia, followed by 23 hours of reperfusion via tMCAO. Twenty-four hours after tMCAO, PMAs (A), PNAs (B), and PS+ platelets within PNAs (C) were quantified by flow cytometry. P-selectin (D) and GPIb-α (E) expression on platelets was analyzed by flow cytometry immediately before and 24 hours after ischemic stroke. (F) Platelets and neutrophils were isolated from CypDplt+/+ and CypDplt−/− mice and activated with dual agonists against PAR4 and GPVI to induce platelet necrosis. PNAs were then imaged by TEM (panels are representative of n = 3 mice per group). Yellow arrows indicate necrotic platelets. ns, not significant.
We next measured whether cerebral ischemia-reperfusion altered platelet P-selectin expression and GPIb-α levels because platelet necrosis can induce changes to these platelet markers.31,32 CypDplt+/+ and CypDplt−/− mice expressed similar levels of basal P-selectin, as well as GPIb-α (Figure 3D-E). Interestingly, after cerebral ischemia-reperfusion, platelets from CypDplt+/+ mice expressed significantly more platelet P-selectin (Figure 3D), whereas little change in P-selectin was observed in CypDplt−/− mice. Furthermore, we measured a small, but significant, decrease in GPIb-α levels on platelets from CypDplt+/+ mice after cerebral reperfusion. No change in GPIb-α expression was observed on platelets from CypDplt−/− mice (Figure 3E).
To investigate whether necrotic platelets were more likely to interact with neutrophils, platelets and neutrophils were isolated from CypDplt+/+ and CypDplt−/− mice and activated with dual agonists against PAR4 and glycoprotein VI (GPVI) to induce platelet necrosis.11 Coincubated cells were then imaged by TEM. Platelets from CypDplt+/+ and CypDplt−/− mice appeared activated, as evidenced by pseudopodia formation and the absence of granules (Figure 3F). However, large vacuolated platelets (which are consistent with platelet necrosis29,32 ) were primarily observed in CypDplt+/+ mice. TEM imaging demonstrated the formation of PNAs in CypDplt+/+ and CypDplt−/− mice. A majority of the platelets from CypDplt+/+ mice surrounding, and appearing to aggregate with, neutrophils appeared necrotic, whereas platelets from CypDplt−/− mice aggregating with neutrophils generally retained their intracellular structures (Figure 3F; supplemental Figure 1, available on the Blood Web site).
We next asked whether CypDplt+/+ platelets interacted preferentially with neutrophils compared with platelets from CypDplt−/− mice in the context of platelet necrosis. Platelets from CypDplt+/+ and CypDplt−/− mice were labeled with different fluorophores and combined at 1:1 ratios in the presence of neutrophils. Platelet necrosis was induced with dual-agonist stimulation (PAR4 peptide and CRP), and platelet-neutrophil interactions were measured by flow cytometry. Platelets from CypDplt+/+ mice were significantly more likely to bind to neutrophils compared with CypDplt−/− platelets (supplemental Figure 2). Finally, although necrotic platelets appeared to bind preferentially to neutrophils, neutrophil binding to platelets from CypDplt+/+ and CypDplt−/− mice induced similar outside-in signaling in platelets (supplemental Figure 3). Taken together, these results suggest that cerebral ischemia-reperfusion induces CypD-mediated platelet necrosis, which regulates the formation of circulating PNAs.
CypD-mediated platelet necrosis regulates neutrophil accumulation in the brain after cerebral ischemia-reperfusion
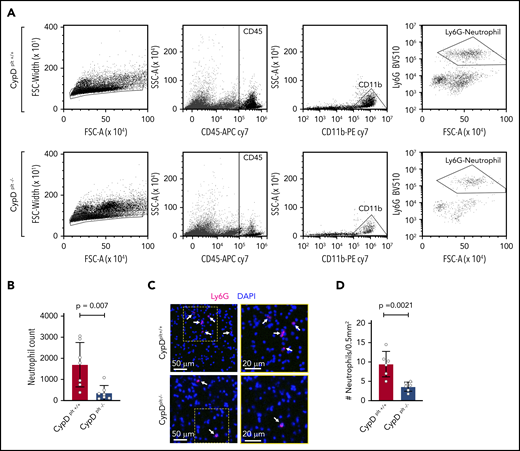
Neutrophil recruitment to the brain is critical to the pathophysiology of cerebral ischemia-reperfusion injury.7,33,34 Because CypD-mediated platelet necrosis altered the formation of circulating PNAs, we examined whether platelet necrosis affected neutrophil recruitment and PNA formation in the brain after ischemia. The ipsilateral side of the brain from CypDplt+/+ and CypDplt−/− mice was isolated and lysed into a single-cell suspension after ischemia and reperfusion and stained for neutrophil-specific markers, including CD45high, CD11b+, and Ly6G+. Flow cytometry analysis revealed an ∼70% reduction in neutrophil accumulation in the ischemic brains of CypDplt−/− mice compared with CypDplt+/+ mice (1699 ± 397 vs 368 ± 131, P = .007; Figure 4A-B; supplemental Figure 4). Next, we histologically examined the brain sections to visually observe and quantify neutrophil recruitment. Neutrophil recruitment to areas of ischemic brain following stroke was significantly reduced in CypDplt−/− mice compared with CypDplt+/+ animals, confirming our observations by flow cytometry (Figure 4C-D). The numbers of circulating platelets (969 000 ± 124 000 per microliter vs 1 041 000 ± 124 000 per microliter) and neutrophils (1680 ± 830 per microliter vs 1670 ± 1030 per microliter) were similar in CypDplt+/+ and CypDplt−/− mice after ischemic stroke, indicating that reduced numbers of platelets or neutrophils in CypDplt−/− mice do not account for these differences. Of note, the number of Ly6Chigh monocytes recruited to the brain was also reduced in CypDplt−/− mice compared with CypDplt+/+ mice (supplemental Figure 5).
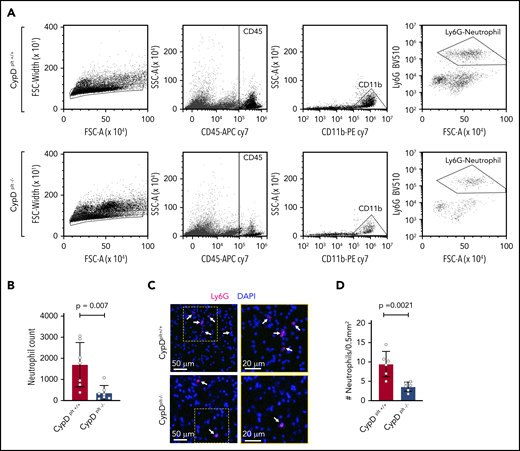
Platelet CypD mediates the recruitment of neutrophils to the brain following cerebral ischemia-reperfusion injury. Male and female CypDplt−/− mice or littermate controls (CypDplt+/+) were subjected to 1 hour of cerebral ischemia followed by 23 hours of reperfusion (tMCAO). (A) Representative dot plots displaying gating strategy for the flow cytometric quantification of neutrophils recruited to the brain. Neutrophils were identified as single cells that were CD45high, CD11b+, and Ly6G+. (B) Quantification of the number of neutrophils in the ischemic hemisphere analyzed by flow cytometry 24 hours after stroke. (C) Representative images of neutrophil (Ly6G, magenta) staining in ischemic brain tissues 24 hours after stroke. Neutrophils are denoted by white arrows. Blue staining shows nuclei (DAPI). Boxes (in left panels) are shown enlarged (right panels). (D) Quantification of the number of neutrophils in ischemic brain tissues analyzed by immunohistochemistry 24 hours after stroke. Four sections throughout the brain were analyzed in each mouse.
Platelet CypD mediates the recruitment of neutrophils to the brain following cerebral ischemia-reperfusion injury. Male and female CypDplt−/− mice or littermate controls (CypDplt+/+) were subjected to 1 hour of cerebral ischemia followed by 23 hours of reperfusion (tMCAO). (A) Representative dot plots displaying gating strategy for the flow cytometric quantification of neutrophils recruited to the brain. Neutrophils were identified as single cells that were CD45high, CD11b+, and Ly6G+. (B) Quantification of the number of neutrophils in the ischemic hemisphere analyzed by flow cytometry 24 hours after stroke. (C) Representative images of neutrophil (Ly6G, magenta) staining in ischemic brain tissues 24 hours after stroke. Neutrophils are denoted by white arrows. Blue staining shows nuclei (DAPI). Boxes (in left panels) are shown enlarged (right panels). (D) Quantification of the number of neutrophils in ischemic brain tissues analyzed by immunohistochemistry 24 hours after stroke. Four sections throughout the brain were analyzed in each mouse.
We next examined whether platelet necrosis altered the formation of PNAs in the brain, similar to our findings in the circulation. Histological examination of the brain sections following stroke revealed that PNAs were readily observed in CypDplt+/+ mice but were generally absent in CypDplt−/− counterparts (Figure 5A). We then quantified the number of PNAs using flow cytometry by staining neutrophils in the brain for CD41, a platelet-specific marker. Brain PNAs were significantly higher in CypDplt+/+ mice compared with CypDplt−/− mice (38.3% ± 3.7% vs 24.9% ± 3.3%, P = .02; Figure 5B-C). In contrast, PMAs in the brain were similar in CypDplt+/+ mice and CypDplt−/− mice (supplemental Figure 5). These results demonstrate that platelet necrosis regulates the recruitment and accumulation of PNAs in the ischemic stroke brain.

Platelet CypD mediates PNA accumulation in brain tissue following cerebral ischemia-reperfusion injury. Male and female CypDplt−/− mice or littermate controls (CypDplt+/+) were subjected to 1 hour of cerebral ischemia followed by 23 hours of reperfusion (tMCAO). (A) Representative images of platelet-neutrophil complexes, as costained by GPIb (platelet, green) and myeloperoxidase (neutrophils, red), in ischemic brain tissue 24 hours after stroke. Blue staining shows nuclei (DAPI). The boxed areas in each panel are shown enlarged (insets). (B) Representative dot plots of flow cytometric analysis of PNAs in brain tissue. (C) Quantification of the number of PNAs in the ischemic hemisphere analyzed by flow cytometry 24 hours after stroke.
Platelet CypD mediates PNA accumulation in brain tissue following cerebral ischemia-reperfusion injury. Male and female CypDplt−/− mice or littermate controls (CypDplt+/+) were subjected to 1 hour of cerebral ischemia followed by 23 hours of reperfusion (tMCAO). (A) Representative images of platelet-neutrophil complexes, as costained by GPIb (platelet, green) and myeloperoxidase (neutrophils, red), in ischemic brain tissue 24 hours after stroke. Blue staining shows nuclei (DAPI). The boxed areas in each panel are shown enlarged (insets). (B) Representative dot plots of flow cytometric analysis of PNAs in brain tissue. (C) Quantification of the number of PNAs in the ischemic hemisphere analyzed by flow cytometry 24 hours after stroke.
Platelet CypD–mediated recruitment of neutrophils controls CBF and reperfusion injury
To confirm whether the protective effect observed in our CypDplt−/− mice was due to reduced platelet-neutrophil interactions, we depleted circulating neutrophils 24 hours before the induction of stroke. Depletion of neutrophils significantly reduced infarct size and neurological damage following ischemic stroke in CypDplt+/+ mice (Figure 6A-E). Neutrophil depletion in CypDplt+/+ mice also resulted in significantly higher CBF 3 and 24 hours after cerebral ischemia (Figure 6F), similar to CBF observed in CypDplt−/− mice (Figure 1G). We next examined neutrophil depletion in the setting of platelet CypD deficiency. Neutrophil counts at baseline and following neutrophil depletion were similar in CypDplt−/− and CypDplt+/+ mice (Figure 6G; data not shown). Neutrophil depletion in CypDplt+/+ mice provided significant protection against brain infarct volume compared with isotype control–treated animals. However, neutrophil depletion conferred no additional protective effect in CypDplt−/− animals. These results suggest that the interplay between necrotic platelets and neutrophils regulates cerebral ischemia-reperfusion injury (Figure 6G-I; supplemental Figure 7).
![Platelet CypD–mediated neutrophil recruitment regulates CBF, infarct size, and neurological outcomes following cerebral ischemia-reperfusion. Wild-type C57BL/6J mice (A-F) or CypDplt−/− mice and littermate controls (CypDplt+/+) (G-I) were treated with a neutrophil-depleting antibody (anti-Ly6G) or a control antibody (immunoglobulin G [IgG]) 24 hours before the induction of ischemic stroke (eg, tMCAO). (A) The number of circulating neutrophils in IgG- or anti-Ly6G–treated mice, demonstrating that neutrophil depletion with anti-Ly6G was effective. (B) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white, arrows) indicates infarcted areas. (C) Brain infarct volumes were quantified by planimetric analysis. (D) Neurological outcome was assessed using the Bederson test. (E) Motor function was examined using the grip strength test. (F) CBF was monitored longitudinally in the right MCA territory (the same side as occlusion) before ischemia (baseline), during ischemia, at the start of reperfusion, and at 3 and 24 hours after stroke onset. (G) Circulating neutrophils in CypDplt−/− and CypDplt+/+ mice treated with IgG control or an anti-Ly6G antibody to deplete neutrophils. (H) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white, arrows) indicates infarcted areas. (I) Brain infarct volumes were quantified by planimetric analysis. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/6/10.1182_blood.2019002124/2/m_bloodbld2019002124f6.png?Expires=1765071632&Signature=jTdrSv1PUMJ8TPyzP8wIQECbfuvATr~RaQLJVioxrASj3tZG0ViXLhfeCbJHc0-O~fwUwRcHuNZ4Gg6lpWuaij590jlB5sbTZ1gOaZKH9gbVo75IGQ0UpbOhzJAlvCim-eEWZkQVDjuDZ8mq6Z~WUmMMc1DK-xJqnQaPjYQdVXmdPsd2gxvsVz0uiOnFBoXoRMYhZwrKGSsI5ii4k4S1i8oSBXxoW-mSPYyYAX~tifn89uq65OvIPjrUOhKEYVgzUQxYPr~lZfhEVFEg4NDECdk0hvE6XDzj7bEtMIRUQxYpk~mUalicpVrCE87T967cqjdcS5EGIZveVJjeSTPGVQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Platelet CypD–mediated neutrophil recruitment regulates CBF, infarct size, and neurological outcomes following cerebral ischemia-reperfusion. Wild-type C57BL/6J mice (A-F) or CypDplt−/− mice and littermate controls (CypDplt+/+) (G-I) were treated with a neutrophil-depleting antibody (anti-Ly6G) or a control antibody (immunoglobulin G [IgG]) 24 hours before the induction of ischemic stroke (eg, tMCAO). (A) The number of circulating neutrophils in IgG- or anti-Ly6G–treated mice, demonstrating that neutrophil depletion with anti-Ly6G was effective. (B) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white, arrows) indicates infarcted areas. (C) Brain infarct volumes were quantified by planimetric analysis. (D) Neurological outcome was assessed using the Bederson test. (E) Motor function was examined using the grip strength test. (F) CBF was monitored longitudinally in the right MCA territory (the same side as occlusion) before ischemia (baseline), during ischemia, at the start of reperfusion, and at 3 and 24 hours after stroke onset. (G) Circulating neutrophils in CypDplt−/− and CypDplt+/+ mice treated with IgG control or an anti-Ly6G antibody to deplete neutrophils. (H) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white, arrows) indicates infarcted areas. (I) Brain infarct volumes were quantified by planimetric analysis. ns, not significant.
Platelet CypD–mediated neutrophil recruitment regulates CBF, infarct size, and neurological outcomes following cerebral ischemia-reperfusion. Wild-type C57BL/6J mice (A-F) or CypDplt−/− mice and littermate controls (CypDplt+/+) (G-I) were treated with a neutrophil-depleting antibody (anti-Ly6G) or a control antibody (immunoglobulin G [IgG]) 24 hours before the induction of ischemic stroke (eg, tMCAO). (A) The number of circulating neutrophils in IgG- or anti-Ly6G–treated mice, demonstrating that neutrophil depletion with anti-Ly6G was effective. (B) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white, arrows) indicates infarcted areas. (C) Brain infarct volumes were quantified by planimetric analysis. (D) Neurological outcome was assessed using the Bederson test. (E) Motor function was examined using the grip strength test. (F) CBF was monitored longitudinally in the right MCA territory (the same side as occlusion) before ischemia (baseline), during ischemia, at the start of reperfusion, and at 3 and 24 hours after stroke onset. (G) Circulating neutrophils in CypDplt−/− and CypDplt+/+ mice treated with IgG control or an anti-Ly6G antibody to deplete neutrophils. (H) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white, arrows) indicates infarcted areas. (I) Brain infarct volumes were quantified by planimetric analysis. ns, not significant.
Discussion
CypD-mediated formation of the mitochondrial permeability transition pore initiates necrotic cell death through the activation of scramblases and calpains, resulting in exposure of PS on the cell surface.28 In platelets, CypD-mediated necrosis contributes to the formation of a subpopulation of highly activated procoagulant platelets that express high levels of PS on their surface.11,35,36 In this study, we report, for the first time, a critical role for CypD-mediated platelet necrosis in mediating cerebral ischemia-reperfusion injury. Our results demonstrate that cerebral ischemia-reperfusion induces platelet necrosis, which, in turn, modulates injurious neutrophil recruitment and PNA formation in the brain, impairing CBF. The impairments in CBF are associated with greater infarct size and more severe neurological deficits. Thus, our results identify a novel biological target to potentially reduce reperfusion injury in ischemic stroke patients.
The formation of necrotic procoagulant platelets occurs when platelets are strongly stimulated, resulting in increased PS exposure that enhances thrombin generation and strengthens the platelet plug.11,35 Clinical studies have correlated the formation of necrotic procoagulant platelets with adverse outcomes following stroke, as well as with an increased risk for recurrent stroke.37-40 Our study now establishes a detrimental role for CypD-mediated procoagulant platelet formation in a model of cerebral ischemia-reperfusion injury. This is in agreement with earlier reports of a critical role for thrombin, GPVI, and ROS in mediating cerebral ischemia-reperfusion injury.2,25,41 Dual-agonist stimulation by thrombin and collagen, or by thrombin and ROS, is an established inducer of platelet necrosis.11,12,29 Our results now imply that a combination of thrombin, GPVI signaling, and ROS contributes to CypD platelet–mediated necrosis during ischemic stroke, resulting in procoagulant platelet formation and reperfusion injury. Interestingly, platelet necrosis appears to regulate brain damage only during reperfusion injury, because CypD-deficient animals were not protected during permanent ischemia. These results suggest that the rapid exposure of platelets to ROS during cerebral reperfusion is critical for inducing CypD-mediated platelet necrosis and inducing necrosis-dependent brain injury.
A key finding from our study is that CypD-mediated platelet necrosis is critically involved in regulating neutrophil infiltration during cerebral reperfusion. Twenty-four hours after stroke, we observed a significant reduction in the number of neutrophils in the brain in CypD-deficient mice. Interestingly, ∼40% of the neutrophils in the brain after stroke in CypDplt+/+ mice were associated with platelets. In addition to the substantial reduction in neutrophils in the brains of CypDplt−/− mice after ischemia-reperfusion injury, neutrophils in the brain had significantly fewer platelets associated with them. Similar observations were made within the circulation after ischemic stroke: significantly more PNAs were identified in whole blood from CypDplt+/+ mice compared with CypDplt−/− mice. A significant number of platelets within the circulating PNAs were PS+, whereas this number was reduced by ∼50% in CypD-deficient mice. This implies a critical role for CypD-dependent platelet necrosis in the formation of PNAs trafficking to the brain, mediating cerebral ischemia-reperfusion injury.
Neutrophils play an essential function by providing a rapid innate immune response against invading pathogens; however, neutrophils can also damage tissue as a result of unwanted granule release and generation of ROS.34 This is of particular relevance in situations of sterile inflammation, such as ischemic stroke. Ischemic stroke triggers an immune response that promotes fast immune cell migration and infiltration to the brain.7,33,42 Neutrophils are among the first immune cells recruited to the brain and have a remarkable destructive potential, either through direct neurotoxic effects from the release of proteolytic enzymes43 or through indirect effects resulting from intravascular neutrophil accumulation and capillary blood flow obstruction resulting in the no-reflow phenomenon.15,44,45 Increased neutrophil counts are frequently reported in stroke patients, and several studies have reported a link between increased neutrophil counts and poor stroke outcomes.33,46-48 Intriguingly, a recent study found that high neutrophil counts at admission could predict poor outcomes in stroke patients who underwent successful thrombectomy.48 Our experimental data corroborate these clinical findings, because neutrophil depletion in our mouse models provided significant protection from reperfusion injury, implying a causal role for neutrophils in aggravating ischemic stroke brain damage after successful reperfusion. Furthermore, our data support previous publications demonstrating that neutrophil depletion limits ischemic stroke brain injury.5,15,45,49
An additional novel finding in our study is the observation that neutrophil depletion improved CBF during reperfusion after transient ischemia. Several studies have observed a significant number of intravascular neutrophils in murine,6,7 baboon,44 and human stroke brains,7,8 indicating that neutrophils may mediate CBF. Based on our findings, and these published studies, we postulate that neutrophils may contribute to the no-reflow phenomenon during reperfusion by interacting with necrotic platelets, forming platelet-neutrophil microthrombi that partial occlude the cerebral microvasculature. This is in agreement with earlier studies that showed that necrotic platelets had a propensity to interact with neutrophils.10,30,50 In particular, Yuan and colleagues10 elegantly visualized the injurious interaction between necrotic platelets and neutrophils during gut ischemia-reperfusion injury. Their recent study provides evidence that the formation of platelet-neutrophil macroaggregates can result in widespread pulmonary thrombosis. Similarly, we found that platelet CypD significantly enhanced detrimental PNA formation following stroke. Our observation that neutrophil depletion conferred protection to a similar extent as platelet-specific CypD deficiency, as well as that neutrophil depletion did not further reduce ischemic stroke brain injury in platelet CypD-deficient animals, implies a causative role for necrotic platelets to interact with neutrophils and exacerbate ischemic stroke brain damage.
The interaction between necrotic platelets and neutrophils is likely multifactorial. Platelet-neutrophil interactions are mediated by platelet P-selectin and GPIb-α, which bind neutrophil P-selectin glycoprotein-1 and MAC-1 (CD11b/CD18), respectively.51,52 These receptors are critical in ischemic stroke, because deletion of P-selectin,53 GPIb-α,2 or MAC-154 offers protection in murine tMCAO models. To elucidate the role of these receptors, we measured the expression of surface P-selectin and GPIb-α in platelet CypD–deficient and CypD-sufficient mice and observed similar baseline expression of both receptors. However, a modest, but significant, increase in platelet P-selectin was observed after cerebral ischemia-reperfusion in CypDplt+/+ mice. In contrast, platelet P-selectin did not increase in CypDplt−/− mice. This coincided with a modest, but significant, decrease in surface GPIb-α expression on platelets from CypDplt+/+ mice, suggesting greater GPIb-α shedding following stroke. Both observations are consistent with increased platelet necrosis after ischemic stroke, because P-selectin expression and GPIb-α shedding are markers of platelet necrosis.13,31,55 The increase in platelet PS expression in PNAs is also associated with increased platelet necrosis. Moreover, our data indicate that necrotic platelets are more prone to interact with neutrophils. Indeed, in vitro experiments using flow cytometry and TEM revealed an apparent preference of necrotic platelets to interact with neutrophils. The observation of increased platelet PS expression mediating increased PNAs in our model is supported by previous reports demonstrating that PNA formation during gut ischemia-reperfusion is dependent on CypD and platelet PS exposure.10 Yuan et al10 observed that PS+ platelet membrane fragments were key in promoting neutrophil aggregation and vascular occlusion. We hypothesized, based on their report and our findings, that CypD-mediated platelet necrosis triggers PS+ PNAs, resulting in vascular occlusion, decreased CBF, and increased infarct volume. This mechanism is supported by a recent report demonstrating that administration of annexin A1, which blocks PS, reduced PNAs and infarct size after cerebral ischemia-reperfusion.56 Taken together, these results indicate that platelet necrosis and PS regulate platelet-neutrophil interactions; however, future studies are necessary to fully elucidate the role of platelet necrosis–dependent PS exposure in regulating PNA formation.
The role for platelet-monocyte interactions in ischemic stroke is less clear. Previous studies have demonstrated that PMAs are increased in ischemic stroke patients compared with healthy controls early on, whereas PNAs remain more durably elevated in stroke patients throughout a 10-day period.24 Experimental studies, however, have suggested that monocytes are not critical in the early phases of ischemia-reperfusion injury, because depletion of all monocyte subtypes did not have any effect on infarct size or neurological scores after ischemic stroke.57 In our model, there was no significant difference in circulating or brain PMAs after stroke between CypDplt+/+ and CypDplt−/− mice, suggesting that platelet necrosis does not mediate these interactions during our conditions of ischemic stroke. This is further supported by the observation that platelets associated with PMAs expressed very little PS after stroke (data not shown). Future studies are necessary to determine whether platelet-monocyte interactions are causal or consequential in ischemic stroke.
Global CypD deficiency is protective in cerebral ischemia-reperfusion injury.27 This protective effect was hypothesized to be the result of reduced necrosis in tissue surrounding the infarct. In contrast, our results provide new evidence that platelet CypD regulates neutrophil recruitment and cerebral infarct size during ischemia-reperfusion injury. Because CypDplt−/− mice do not display any hemostatic defects,10,12 targeting platelet necrosis has potential in ischemic stroke therapy, in particular when reperfusion is achieved. Cyclosporine is a therapeutic agent that is used in clinical settings and is known to inhibit CypD-dependent necrosis. Several studies have previously reported neuroprotective effects of cyclosporine in ischemic stroke,58 resulting in a clinical trial in which cyclosporine was given in combination with thrombolytic therapy.59 Although cyclosporine did not significantly improve overall stroke outcomes, upon subgroup analysis, cyclosporine therapy was beneficial in patients with a proximal occlusion where reperfusion was achieved. These results are consistent with our findings that targeting platelet necrosis improved stroke outcomes attributable to reperfusion injury. Because reperfusion is the primary goal of stroke treatment, identifying therapies that may help to prevent reperfusion injury is imperative to improve stroke outcome. Because tissue plasminogen activator is the only primary therapeutic agent used in patients with acute ischemic stroke, our findings suggest that cyclosporine (or other therapies targeting platelet necrosis) may be beneficial and worth further study. In light of this, aspirin pretreatment was recently shown to potentiate the effects of cyclosporine in reducing necrotic platelet formation.13 Furthermore, aspirin also reduces platelet-leukocyte interactions in stroke patients.60 Future studies are needed to investigate the therapeutic potential of combined CypD inhibition and aspirin treatment in the setting of stroke.
In conclusion, our data demonstrate that cerebral ischemia-reperfusion induces CypD-mediated platelet necrosis, which, in turn, regulates injurious neutrophil recruitment and PNA formation in the brain, impairing CBF and increasing cerebral infarction. These findings identify platelet CypD as a potential therapeutic target to reduce reperfusion injury in ischemic stroke patients.
For original data, please contact Robert A. Campbell (rcampbell@u2m2.utah.edu).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Diana Lim for assistance with creating the figures and Neal Tolley, and Toni Blair for editorial assistance.
This work was supported by grants from the National Institutes of Health National Heart, Lung, and Blood Institute (HL112311, HL126547 [M.T.R.]), the National Institutes of Health National Institute on Aging (AG059877, AG048022 [M.T.R.], K01AG059892 [R.A.C.]), and the National Institutes of Health National Institute of Neurological Disorders and Stroke (U10NS086606 [R.A.C.]); Merit Review Award I01 CX001696 from the US Department of Veterans Affairs Clinical Sciences R&D Service; program grants from the Australian National Health and Medical Research Council (1016647, 1113577 [B.T.K.]); and the University of Utah Flow Cytometry Facility, in addition to the National Institutes of Health National Cancer Institute, through grant 5P30CA042014-24. F.D. is a postdoctoral fellow of the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (12U7818N).
The sponsor had no role in the design or preparation of paper.
The contents do not represent the views of the US Department of Veterans Affairs or the US government.
Authorship
Contribution: F.D., B.K.M., Y.K., I.P., A.S.E., B.T.K., M.T.R., and R.A.C. designed and performed experiments; F.D., M.T.R., and R.A.C. analyzed results, created the figures, and wrote the manuscript; and all authors reviewed and critically edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Robert A. Campbell, University of Utah Health Sciences Center, Eccles Institute of Human Genetics, 15 North 2030 East, Building 533, Suite 4225, Salt Lake City, UT 84112; e-mail: rcampbell@u2m2.utah.edu; and Matthew T. Rondina, University of Utah Health Sciences Center, Eccles Institute of Human Genetics, 15 North 2030 East, Building 533, Suite 4225, Salt Lake City, UT 84112; e-mail: matthew.rondina@hsc.utah.edu






![Platelet CypD–mediated neutrophil recruitment regulates CBF, infarct size, and neurological outcomes following cerebral ischemia-reperfusion. Wild-type C57BL/6J mice (A-F) or CypDplt−/− mice and littermate controls (CypDplt+/+) (G-I) were treated with a neutrophil-depleting antibody (anti-Ly6G) or a control antibody (immunoglobulin G [IgG]) 24 hours before the induction of ischemic stroke (eg, tMCAO). (A) The number of circulating neutrophils in IgG- or anti-Ly6G–treated mice, demonstrating that neutrophil depletion with anti-Ly6G was effective. (B) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white, arrows) indicates infarcted areas. (C) Brain infarct volumes were quantified by planimetric analysis. (D) Neurological outcome was assessed using the Bederson test. (E) Motor function was examined using the grip strength test. (F) CBF was monitored longitudinally in the right MCA territory (the same side as occlusion) before ischemia (baseline), during ischemia, at the start of reperfusion, and at 3 and 24 hours after stroke onset. (G) Circulating neutrophils in CypDplt−/− and CypDplt+/+ mice treated with IgG control or an anti-Ly6G antibody to deplete neutrophils. (H) Representative brain sections stained with 2,3,5-triphenyl tetrazolium chloride. Healthy tissue stains red, whereas an absence of staining (white, arrows) indicates infarcted areas. (I) Brain infarct volumes were quantified by planimetric analysis. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/6/10.1182_blood.2019002124/2/m_bloodbld2019002124f6.png?Expires=1765511364&Signature=LaIQ7Ur4T0nIIzxzj6HwLTsRaUvv4OuQNtwpSc7qtUTy8Aq77bekY5uU4E3SADfuyk41v2pauVyLwcJiQhzQsGC4HnIAaUlqGSWTzmTk~0tD6a0OST3Yo6ajlRgHqF2PCpz5xBesZmlfH2bWgWtL~DhBjkxmqSS90KmPNSycgHTVPa0-aQTERiINaUGhZRO6om6fCxZghWqn2B-aekInJ6LAQ3d2vZvGvANs4K3w4bmLb9n1dcPEHbuTwDJWJSi0poLnUcEFMR2U6TUQP1jVhivzqJl3lCyYqFVd1XoB2pECheFd~~EY4seIpAqFbPrdTSdmXwoJdHL1BdvSTDBWuw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
