

Key Points
Hepatocyte-derived tPA contributes to basal circulating tPA activity and affects injury-induced fibrinolysis.
Hepatocyte tPA is induced by ATF6 and subjected to negative regulation by the corepressor DACH1.

Abstract
Tissue-type plasminogen activator (tPA) is a major mediator of fibrinolysis and, thereby, prevents excessive coagulation without compromising hemostasis. Studies on tPA regulation have focused on its acute local release by vascular cells in response to injury or other stimuli. However, very little is known about sources, regulation, and fibrinolytic function of noninjury-induced systemic plasma tPA. We explore the role and regulation of hepatocyte-derived tPA as a source of basal plasma tPA activity and as a contributor to fibrinolysis after vascular injury. We show that hepatocyte tPA is downregulated by a pathway in which the corepressor DACH1 represses ATF6, which is an inducer of the tPA gene Plat. Hepatocyte-DACH1–knockout mice show increases in liver Plat, circulating tPA, fibrinolytic activity, bleeding time, and time to thrombosis, which are reversed by silencing hepatocyte Plat. Conversely, hepatocyte-ATF6–knockout mice show decreases in these parameters. The inverse correlation between DACH1 and ATF6/PLAT is conserved in human liver. These findings reveal a regulated pathway in hepatocytes that contributes to basal circulating levels of tPA and to fibrinolysis after vascular injury.
Introduction
Tissue-type plasminogen activator (tPA) is a secreted serine protease that initiates the dissolution of a fibrin clot in a process called fibrinolysis.1,2 When a fibrin clot forms on the wall of an injured vessel, tPA binds to the fibrin and converts plasminogen to plasmin, which proteolytically degrades the fibrin clot.3,4 tPA is released locally by vascular endothelial cells in response to injury, preventing excessive fibrin deposition and thrombosis.5,6 However, freshly isolated blood from healthy subjects undergoes spontaneous fibrinolysis,7,8 suggesting the presence of basal tPA (ie, tPA activity present before injury occurs). In this context, a major gap in fibrinolysis research is centered on the roles, regulation, and sources of basal plasma tPA. A key question in this area, and one that raises the possibility that a nonendothelial source of tPA may be important, is how medium-sized and large vessels respond to injury with respect to fibrinolysis. Endothelial cells of these vessels express less tPA than small vessels,9-11 and the tPA that is secreted by these cells gets rapidly diluted owing to the rapid flow and a much lower surface-to-volume ratio of large vessels. Thus, a nonendothelial source of tPA may be important in limiting clot extension and thrombosis after injury to medium-sized and large arteries.
The fibrinolytic activity of plasma is determined, in large part, by the relative concentrations of plasma tPA and its inhibitor, plasminogen activator inhibitor-1 (PAI-1).1,2 Longitudinal studies have revealed an association between lower plasminogen activator and fibrinolytic activity in plasma and future recurrent myocardial infarction.12 Moreover, basal plasma fibrinolytic activity was shown to have a diurnal variation, with a nadir in the morning, which is the time of highest risk for coronary artery disease.13 More recently, studies have shown that low plasma tPA activity per se predicts cardiovascular disease in humans.14,15 Collectively, these studies suggest that basal plasma fibrinolytic activity, determined in part by tPA concentration, is a functionally important mechanism to prevent pathological fibrin clot formation and thrombosis.
Although hepatocytes have been shown to express tPA protein and messenger RNA (mRNA),16,17 the fibrinolytic function of hepatocyte tPA, either in the liver or systemically, and its mode of regulation remain unknown. In this study, we show that hepatocytes are a significant source of plasma tPA under basal conditions and that hepatocyte-derived tPA complements locally released vessel wall–derived tPA to limit clotting after arterial injury. Moreover, we show that hepatocyte-derived tPA is negatively regulated by a corepressor called DACH1. DACH1 functions by repressing the transcription factor ATF6, which we show is an inducer of the tPA gene Plat. These findings add new insight into the decades-old mystery of the roles, regulation, and sources of basal tPA.
Methods
Mice
Dach1fl/fl mice were generated as previously described18 and crossed onto the C57BL/6J background. Male and female Dach1fl/fl mice were injected IV with adeno-associated virus 8 (AAV8) viruses containing hepatocyte-specific TBG-Cre recombinase (AAV8-TBG-Cre) at 3 months of age to deplete DACH1 in hepatocytes19 (HC-DACH1–knockout [KO] mice). Control mice were Dach1fl/fl mice injected with AAV8 viruses containing the control vector virus (AAV8-TBG-LacZ). Atf6fl/fl mice20 on the C57BL/6J background were purchased from The Jackson Laboratory. Male and female Atf6fl/fl mice were injected IV at 3 months of age with AAV8-TBG-Cre to deplete ATF6 in hepatocytes (HC-ATF6–KO mice). Control animals were Atf6fl/fl mice injected with AAV8-TBG-LacZ virus. Male and female wild-type (WT) C57BL/6J mice were purchased from The Jackson Laboratory and maintained for 1 week in the Columbia University Medical Center (CUMC) animal facility before IV injection of AAV8-H1–short hairpin Plat (shPlat) to silence hepatocyte Plat. Female WT C57BL/6J mice and holo-tPA–KO mice21 were purchased from The Jackson Laboratory and maintained for 1 week in the CUMC animal facility before IV injection of AAV8-TBG-Plat to express Plat in hepatocytes. AAV8 vectors were delivered at a titer of 1 × 1011 genome copies per mouse, and experiments were commenced 3 to 6 weeks later. For all experiments, mice were maintained on a 12-hour light–dark cycle with free access to a normal chow diet and water. Male and female mice of the same age and similar weight were randomly assigned to experimental and control groups. All mouse experiments were performed with the approval of the Institutional Animal Care and Use Committee of CUMC.
Viral constructs
Adenovirus expressing LacZ (adeno-LacZ), adenovirus expressing ΔDS-DACH1, adenovirus expressing short hairpin RNA targeting human ATF6 (adeno-shATF6), and adenovirus expressing a nuclear-active form of ATF6 (adeno–ATF6-N) were described previously19,22,23 and amplified by Viraquest (North Liberty, IA). AAV8-TBG-Cre and AAV8-TBG-LacZ were purchased from the Penn Vector Core. The AAV8–H1–short hairpin RNA (shRNA) construct targeting murine Plat was made by annealing complementary oligonucleotides and then ligating them into the pAAV-RSV-GFPH1 vector, as described previously.24 The resultant constructs were amplified by Salk Institute Gene Transfer Targeting and Therapeutics Core. AAV8-TBG-Plat was purchased from Vector Biolabs.
Mouse thrombosis assays
Carotid artery thrombosis was the result of FeCl3-induced injury or Rose bengal/laser-induced photochemical injury, as described previously.25,26 Briefly, mice were anesthetized with isoflurane and placed on a thermo-controlled blanket (37°C), followed by surgical exposure of the carotid artery. For the FeCl3 procedure, a filter paper soaked in 10% FeCl3 was applied to the artery for 3 minutes, followed by rinsing with normal saline. Blood flow was monitored with an ultrasound flow probe (Transonics) and recorded using LabChart software (ADInstruments). The time to total occlusion was defined as the time interval between application of FeCl3 and stable occlusion of the artery, with no blood flow for 3 minutes.25 For the Rose bengal/laser procedure, 0.15 mL of Rose bengal in 0.9% saline was injected IV through the tail vein to achieve a dose of 50 mg/kg. A green laser light (Melles Griot) at a wavelength of 540 nm was applied 6 cm from the carotid artery, and blood flow was monitored continuously from the onset of photochemical injury.26 The time to total occlusion was defined as the time interval between application of the laser and stable occlusion of the artery, with no blood flow for 10 minutes. The mice were euthanized immediately after the procedure.
Mouse tail bleeding assay
Mice were anesthetized with isoflurane and positioned horizontally on a platform that allowed the tail to descend ∼2 cm from the top of the platform. A segment of tail on the distal tail tip was transected with a no. 11 surgical scalpel to induce wounds ∼ 2 mm in diameter. Bleeding was monitored by gently dabbing the tail tip on Whatman paper at 10-second intervals until the cessation of bleeding.27 The time to stable cessation of bleeding was defined as the time interval between the tail incision and cessation of bleeding, with no evidence of rebleeding for 60 seconds. Bleeding exceeding 15 minutes was stopped by applying pressure.
Plasma collection and analyses
Blood from cardiac puncture in 10% volume of sodium citrate (3.8%, weight-to-volume ratio) was centrifuged for 15 minutes at 2300g, and plasma was carefully collected from the supernatant fraction. Plasma samples were divided into aliquots, snap-frozen, and stored at −80°C until analyses. Total antigen levels of tPA, PAI-1, plasminogen, fibrinogen/fibrin, fibrin degradation products (FDPs), and thrombin–antithrombin complexes in plasma were measured by enzyme-linked immunosorbent assay (ELISA) kits, according to manufacturer’s instructions (catalog numbers are listed in supplemental Methods, available on the Blood Web site). Plasma PAI-1–free tPA was measured by an assay in which the ELISA plate was coated with PAI-1 antigen to enable detection of only the functional PAI-1–free form of tPA. tPA enzymatic activity in plasma or tissue lysates was assayed by chromatographically measuring the release of p-nitroaniline chromophore from a plasmin-specific synthetic substrate. Results were recorded and analyzed using a VersaMax Microplate Reader and SoftMax Pro software (Molecular Devices).
tPA release
For the basal value, plasma (∼15 µL) was collected via tail bleed, with care taken not to subject the tail to pressure. One week later, plasma was collected 20 minutes after FeCl3-induced carotid injury. All blood was immediately suspended in 10% volume of sodium citrate (3.8%, weight-to-volume ratio), followed by a 15-minute centrifugation at 2300g at room temperature, and plasma was carefully collected from the supernatant fraction. Plasma samples were divided into aliquots, snap-frozen, and stored at −80°C until analyses. Basal and post-FeCl3 plasma tPA antigen levels were measured on the same ELISA plate. Results were recorded using a VersaMax Microplate Reader and analyzed by SoftMax Pro software (Molecular Devices). The tPA release data were calculated by subtracting the post-FeCl3 value from the basal value for each patient.28
Clauss fibrinogen assay
Comparison of clottable fibrinogen in plasma between control and hepatocyte (HC)-DACH1–KO mice was carried out using a Clauss fibrinogen assay kit (Helena Laboratories; catalog number 5376). This kit, which is representative of commercially available kits to assay clottable fibrinogen in mouse plasma, uses bovine thrombin to cleave fibrinogen and human fibrinogen for the standard curve. Briefly, 0.2 mL of plasma diluted 1:10 in Owren’s Veronal Buffer provided in the kit was incubated for 2 minutes at 37°C. Thrombin reagent (0.1 mL) was then added, and clotting time was measured. Fibrinogen levels were determined from a standard curve prepared from known dilutions of fibrinogen. The data are presented as clottable fibrinogen relative to the control mouse cohort.
Euglobulin clot lysis assay
The euglobulin fraction from 50 µL of citrated mouse plasma was resuspended in 900 µL of 0.017% acetic acid, placed on ice for 20 minutes, and then centrifuged for 20 minute at 2000g at 4°C. After careful removal of the supernatant fraction, each pellet (euglobulin fraction) was resuspended in 55 µL of sodium borate/NaCl (pH 9.0) and transferred into a single well on a flat-bottom 96-well microtiter assay plate. A total of 50 µL of 25 mM CaCl2 was added to each well, and the plates were recorded at 405 nm every 10 minutes, with 3-second shakes before each reading, at room temperature for 16 hours. Clot lysis time was calculated as the time to achieve 50% of clot lysis (half-lysis time).29
Human and mouse primary hepatocyte experiments
Human primary hepatocytes were obtained from the Liver Tissue Cell Distribution System at the University of Pittsburgh. Primary mouse hepatocytes were isolated from 10-week-old WT C57BL/6J mice, as described previously.19,30 All cells were cultured in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum and then transduced with various adenoviral constructs, as described in the figure legends. The cells were harvested after 18 hours in RIPA buffer (Thermo Fisher; catalog number 89900) with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher; catalog number #78444) for immunoblotting or in RNA lysis buffer (QIAGEN; catalog number 79216) for mRNA quantification. Culture media were collected, snap-frozen in liquid nitrogen, and stored at −80°C until processing.
Human liver specimens
Deidentified human liver specimens were acquired from the Liver Tissue Cell Distribution System at the University of Minnesota. The specimens were collected postmortem on the date of liver transplantation and preserved as frozen samples. The diagnostic information is included in supplemental Table 1. Phenotypic and pathological characterization was conducted by medical physicians and pathologists associated with the Liver Tissue Cell Distribution System. The protocol was approved by the Institutional Review Board at CUMC.
Statistical analysis
All results are presented as mean ± standard error of the mean (SEM). The D’Agostino-Pearson omnibus test was used to test for normality. Two-tailed P values were calculated using the Student t test for normally distributed data and the Mann-Whitney U rank-sum test for nonnormally distributed data. One-way analysis of variance, followed by the Tukey test, was used to evaluate differences among groups when ≥3 groups were analyzed. Linear regression was used for determining the correlation of variables. Statistical analyses were conducted using Prism (GraphPad) and R software.
Results
Mice lacking hepatocyte DACH1 have increased fibrinolysis
DACH1 is a helix-turn-helix transcriptional corepressor that determines cell fate and retrains aberrant cell growth,31 and we showed recently that it regulates an insulin receptor signaling pathway in hepatocytes by repressing the gene encoding the transcription factor ATF6.19 To obtain a more global understanding of genes regulated by DACH1 in hepatocytes, we conducted a liver RNA sequencing study in Dach1fl/fl mice injected with hepatocyte-specific AAV8-TBG-Cre (HC-DACH1–KO; Cre) vs control AAV8-TBG-LacZ (LacZ)19 (Figure 1A). AAV8-TBG-Cre deletes floxed genes specifically in hepatocytes vs other types of liver cells or other organs.30,32 Among the more striking finding was that Plat was markedly higher in HC-DACH1–KO livers (Figure 1B), whereas the expression of other major coagulation-related factors was unchanged (supplemental Figure 1A-B). To assess the functional significance of this finding, we examined total tPA antigen and enzymatic activity in the plasma and observed significant increases in both end points in the KO mice (Figure 1C-D). Consistent with previous reports that plasma plasminogen decreases after fibrinolytic therapy,33,34 we found lower plasminogen concentration in the plasma of HC-DACH1–KO mice vs control mice (supplemental Figure 1C). In contrast, the plasma concentration of PAI-1, the major inhibitor of tPA activity, was similar in control and HC-DACH1–KO mice (supplemental Figure 1D). Further, PAI-1–free tPA, the form of tPA that is functionally active, was significantly increased in the KO mice (Figure 1E). Increased plasma tPA can result from decreased receptor-mediated tPA clearance by the liver by LRP1 and the mannose receptor,35 but there were no differences in the expression of the mRNAs encoding these receptors (Lrp1 and Mrc1) between control and HC-DACH1–KO mouse livers (supplemental Figure 1E). As additional evidence for increased fibrinolytic activity in the plasma of HC-DACH1–KO mice, KO plasma also showed decreases in fibrinogen-fibrin antigen (Figure 1F), clottable fibrinogen levels (Figure 1G), and euglobulin clot lysis time (Figure 1H), which is the time it takes to dissolve a clot formed in vitro in the euglobulin fraction of plasma in the absence of plasmin inhibitors.29 There was also an increase in FDPs in KO plasma (Figure 1I). This increase in FDPs likely was not due to an increase in thrombin-mediated fibrin formation, because the concentration of plasma thrombin–antithrombin complexes, a marker of activated thrombin, was similar between the KO and control mice (supplemental Figure 2A). These combined data demonstrate that mice lacking DACH1 in their hepatocytes have increased fibrinolysis.6,35,36
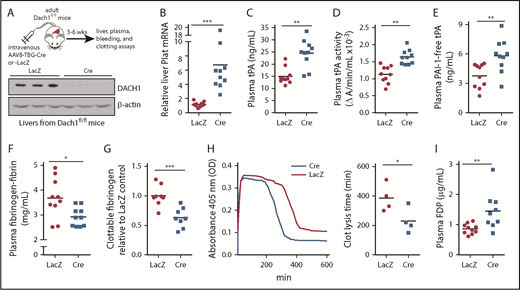
DACH1 deletion in hepatocytes increases liver tPA, plasma tPA and systemic fibrinolytic activity in mice. (A) Experimental scheme for depleting hepatocyte DACH1 in adult mice and immunoblot of liver DACH1 in these mice, with β-actin as loading control. Dach1fl/fl mice were injected IV with AAV8-TBG-Cre (Cre) to create HC-DACH1–KO mice, with AAV8-TBG-LacZ–injected Dach1fl/fl mice (LacZ) serving as controls. (B) Plat mRNA in liver normalized to Rplp0 and expressed as relative to the value in LacZ mice. (C) Plasma tPA concentration by ELISA. (D) Plasma tPA activity by enzymatic assay. (E) Plasma PAI-1–free tPA concentration by ELISA. (F) Plasma fibrinogen-fibrin antigen concentration by ELISA. (G) Plasma clottable fibrinogen concentration by Clauss fibrinogen assay. The data are expressed relative to the value in plasma of LacZ mice. The absolute values using human fibrinogen for the standard curve were 2.27 ± 0.14 (LacZ) and 1.43 ± 0.14 (Cre) mg/mL. (H) Concentration of fibrin degradation products in plasma by ELISA. (I) Fibrinolytic activity measured by euglobulin clot lysis assay from plasma. Horizontal lines in dot-density plots indicate mean values. n = 8 to 10 mice per group (A-H); 4 mice per group (I). *P < .05, **P < .01, ***P < .001, Mann-Whitney U test (B), Student t test (C-F,I).
DACH1 deletion in hepatocytes increases liver tPA, plasma tPA and systemic fibrinolytic activity in mice. (A) Experimental scheme for depleting hepatocyte DACH1 in adult mice and immunoblot of liver DACH1 in these mice, with β-actin as loading control. Dach1fl/fl mice were injected IV with AAV8-TBG-Cre (Cre) to create HC-DACH1–KO mice, with AAV8-TBG-LacZ–injected Dach1fl/fl mice (LacZ) serving as controls. (B) Plat mRNA in liver normalized to Rplp0 and expressed as relative to the value in LacZ mice. (C) Plasma tPA concentration by ELISA. (D) Plasma tPA activity by enzymatic assay. (E) Plasma PAI-1–free tPA concentration by ELISA. (F) Plasma fibrinogen-fibrin antigen concentration by ELISA. (G) Plasma clottable fibrinogen concentration by Clauss fibrinogen assay. The data are expressed relative to the value in plasma of LacZ mice. The absolute values using human fibrinogen for the standard curve were 2.27 ± 0.14 (LacZ) and 1.43 ± 0.14 (Cre) mg/mL. (H) Concentration of fibrin degradation products in plasma by ELISA. (I) Fibrinolytic activity measured by euglobulin clot lysis assay from plasma. Horizontal lines in dot-density plots indicate mean values. n = 8 to 10 mice per group (A-H); 4 mice per group (I). *P < .05, **P < .01, ***P < .001, Mann-Whitney U test (B), Student t test (C-F,I).
These findings and those presented in the next section (“Silencing hepatocyte tPA decreases fibrinolysis in HC-DACH1–KO and WT mice”) in WT mice suggest that hepatocytes are an important source of functionally active plasma tPA, which is under negative regulation by DACH1, in the basal state. To determine whether this pathway also impacts the fibrinolytic response to vascular injury, we assayed tail bleeding time and coagulation and thrombosis after vascular injury in HC-DACH1–KO mice and control mice. HC-DACH1–KO mice had significantly longer tail bleeding times and showed a rebleeding pattern compared with control mice (Figure 2A). Moreover, occlusive carotid artery thrombosis induced by FeCl3 or photochemical injury was prolonged in the KO mice, and there was a pattern of transient occlusions, followed by rapid increases in blood flow, which was suggestive of transient recanalization of the clotted vessel (Figure 2B-C). Platelet count, adenosine 5′-diphosphate–stimulated platelet aggregation, and platelet surface levels of activated αIIbβ3 (JON/A) and P-selectin were similar in HC-DACH1–KO and control mice (supplemental Figure 2B-D).
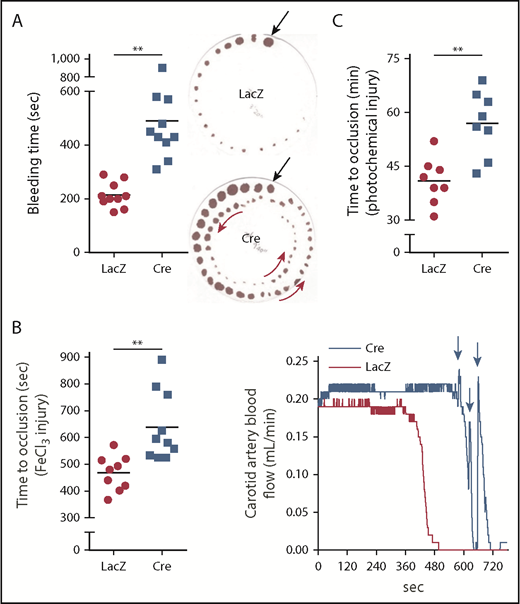
DACH1 deletion in hepatocytes increases bleeding time and time to thrombotic carotid occlusion in mice. (A) Tail bleeding time and representative images of bleeding patterns on filter paper. Black arrows indicate beginning bleeding time course, and red arrows depict episodes of rebleeding. Time to occlusive carotid arterial thrombosis induced by 10% FeCl3 injury (B) or Rose bengal/laser photochemical injury (C). (B) Representative blood flow pattern. The red arrows depict rapid increases in blood flow after transient occlusions, suggestive of transient recanalization of the clotted vessel. **P < .01, 2-tailed Student t test (n = 8-10 mice per group).
DACH1 deletion in hepatocytes increases bleeding time and time to thrombotic carotid occlusion in mice. (A) Tail bleeding time and representative images of bleeding patterns on filter paper. Black arrows indicate beginning bleeding time course, and red arrows depict episodes of rebleeding. Time to occlusive carotid arterial thrombosis induced by 10% FeCl3 injury (B) or Rose bengal/laser photochemical injury (C). (B) Representative blood flow pattern. The red arrows depict rapid increases in blood flow after transient occlusions, suggestive of transient recanalization of the clotted vessel. **P < .01, 2-tailed Student t test (n = 8-10 mice per group).
Silencing hepatocyte tPA decreases fibrinolysis in HC-DACH1–KO and WT mice
To directly test the role of hepatocyte tPA in the HC-DACH1–KO phenotype, we compared mice with normal HC-DACH1 (LacZ), HC-DACH1–KO mice (Cre), and HC-DACH1–KO mice treated with shPlat in an AAV8-H1 vector (Cre-shPlat). AAV8-H1-shRNA vectors silence genes specifically in hepatocytes.24,31,37,38 As before, HC-DACH1–KO mice had increases in liver Plat mRNA, plasma tPA activity and protein, bleeding time, and time to FeCl3-induced occlusive carotid arterial thrombosis, but treatment of these KO mice with shPlat normalized all of these parameters (Figure 3). As an indicator of the hepatocyte specificity of HC-DACH1–KO mice and AAV8-H1-shPlat mice, we showed that carotid intimal and medial-adventitial Plat levels were similar in the 3 groups of mice (supplemental Figure 3A). Note that the intima is enriched in endothelial cells, the cell type thought to be the major source of tPA,5,6,39 and the media-adventitia is enriched in smooth muscle cells (supplemental Figure 3B), which is another source of tPA.40 Thus, the increase in circulating tPA protein and activity in mice with deleted hepatocyte DACH1 can be directly and causally linked with the increase in liver Plat in these mice.
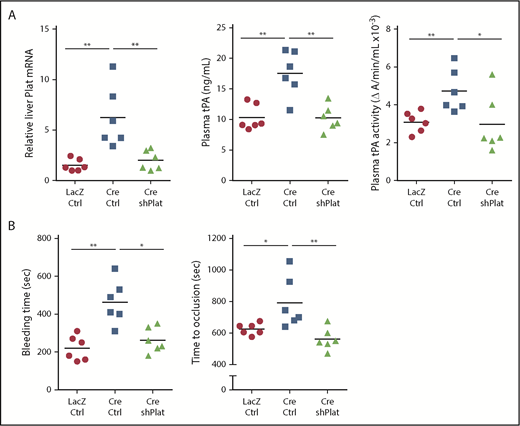
Silencing of hepatocyte Plat mRNA in HC-DACH1–KO mice decreases liver tPA, plasma tPA, and systemic fibrinolytic activity. Liver Plat mRNA and plasma tPA protein concentration and activity (A) and tail bleeding time and time to occlusive carotid arterial thrombosis induced by photochemical injury (B) in Dach1fl/fl mice administrated LacZ or Cre as in Figure 1, together with control AAV8-H1 virus (Ctrl) or AAV8-H1-shPlat. Horizontal lines indicate mean values. *P < .05, **P < .01, 1-way analysis of variance, followed by the Tukey test (n = 6 mice per each group).
Silencing of hepatocyte Plat mRNA in HC-DACH1–KO mice decreases liver tPA, plasma tPA, and systemic fibrinolytic activity. Liver Plat mRNA and plasma tPA protein concentration and activity (A) and tail bleeding time and time to occlusive carotid arterial thrombosis induced by photochemical injury (B) in Dach1fl/fl mice administrated LacZ or Cre as in Figure 1, together with control AAV8-H1 virus (Ctrl) or AAV8-H1-shPlat. Horizontal lines indicate mean values. *P < .05, **P < .01, 1-way analysis of variance, followed by the Tukey test (n = 6 mice per each group).
Although previous studies have shown evidence of tPA expression by hepatocytes, to our knowledge, none has shown that hepatocyte tPA plays a role in systemic fibrinolysis. Therefore, we conducted a series of additional tests to further explore the relative level of tPA expression by hepatocytes and its role in WT mice. Using flow cytometry, immunoblot, and quantitative polymerase chain reaction, we found that hepatocytes express high levels of tPA relative to liver nonparenchymal cells, which are enriched in endothelial cells, macrophages, and hepatic stellate cells; consistent with the hepatocyte specificity of AAV8-H1-shRNA (above), only hepatocyte tPA was decreased by AAV8-H1-shPlat (supplemental Figure 4A-D). In terms of comparisons with well-known sources of tPA, the levels of Plat mRNA, immunoreactive tPA protein, and tPA activity in liver were comparable to those in arterial tissue, and arterial tPA was not affected by AAV8-H1-shPlat (Figure 4A-C). We also assessed the role of hepatocyte tPA in mice with normal DACH1. As shown in Figure 4B-C, we were able to lower liver tPA in WT mice by ∼40% using AAV8-H1-shPlat. This level of silencing led to an ∼50% lowering of plasma tPA protein and an ∼30% lowering of plasma tPA activity (Figure 4D-E). Despite this only partial silencing efficiency, there was a 43% decrease in bleeding time (P = .0003) and a 25% decrease in the time to occlusive carotid arterial thrombosis induced by photochemical injury (P = .001) (Figure 4F-G). Additionally, AAV8-H1-shPlat did not significantly alter thrombin-antithrombin complexes (supplemental Figure 4E), indicating that the reduced time to occlusion and bleeding time were likely not due to an increase in fibrin formation.
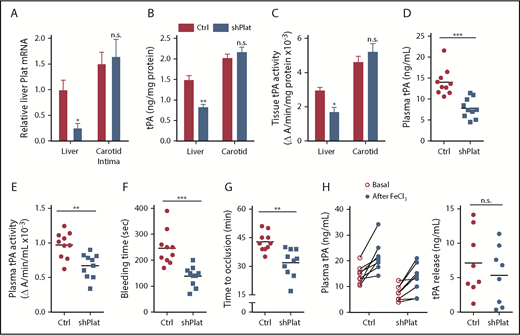
Silencing of hepatocyte Plat mRNA in WT mice decreases liver tPA, plasma tPA, and systemic fibrinolytic activity.Plat mRNA (A), tPA concentration by ELISA (B), and tPA activity by enzymatic assay (C) in the liver and carotid arterial lysates of WT mice injected with control AAV8-H1 virus or AAV8-H1-shPlat. Results are shown as mean ± SEM (n = 4 mice per each group). Plasma tPA protein concentration (D), plasma tPA activity (E), tail bleeding time (F), and time to occlusive carotid arterial thrombosis (G) induced by photochemical injury in WT mice injected with control AAV8-H1 virus or AAV8-H1-shPlat (n = 8-10 mice per each group). (H) Plasma tPA concentration 1 week before (basal) and 20 minutes after FeCl3-induced carotid artery thrombosis; tPA release was calculated by subtracting the basal value from the after-FeCl3 value for each mouse. Horizontal lines in dot-density plots indicate mean values. *P < .05, **P < .01, ***P < .001, 2-tailed Student t test (A-C,E-H), Mann-Whitney U test (D). n.s., not significant (P ≥ .05).
Silencing of hepatocyte Plat mRNA in WT mice decreases liver tPA, plasma tPA, and systemic fibrinolytic activity.Plat mRNA (A), tPA concentration by ELISA (B), and tPA activity by enzymatic assay (C) in the liver and carotid arterial lysates of WT mice injected with control AAV8-H1 virus or AAV8-H1-shPlat. Results are shown as mean ± SEM (n = 4 mice per each group). Plasma tPA protein concentration (D), plasma tPA activity (E), tail bleeding time (F), and time to occlusive carotid arterial thrombosis (G) induced by photochemical injury in WT mice injected with control AAV8-H1 virus or AAV8-H1-shPlat (n = 8-10 mice per each group). (H) Plasma tPA concentration 1 week before (basal) and 20 minutes after FeCl3-induced carotid artery thrombosis; tPA release was calculated by subtracting the basal value from the after-FeCl3 value for each mouse. Horizontal lines in dot-density plots indicate mean values. *P < .05, **P < .01, ***P < .001, 2-tailed Student t test (A-C,E-H), Mann-Whitney U test (D). n.s., not significant (P ≥ .05).
Endothelial granule–stored tPA can be rapidly released in response to endothelial injury–induced thrombosis,5 and we predicted that this process would not be affected by silencing hepatocyte tPA. To test this prediction, we compared tPA before and after FeCl3-induced carotid injury and found that the relative increase after FeCl3 was similar between control and hepatocyte tPA–silenced mice (Figure 4H). These data suggest that the release of tPA from the endothelium after injury is independent of hepatocyte tPA and, in view of the previous data above, that hepatocyte tPA works in concert with tPA released from the endothelium to affect postinjury fibrinolysis.
As a final test of the role of hepatocyte tPA, we treated holo-tPA–KO mice21 with AAV8-TBG-Plat (tPA-KO + HC-Plat) to restore only hepatocyte tPA; WT mice + HC-LacZ and tPA-KO + HC-LacZ were used as the controls (supplemental Figure 5). The hepatocyte tPA–restored KO mice showed a substantial increase in plasma tPA protein and a significant decrease in clot lysis time (supplemental Figure 6). These combined data further demonstrate the importance of hepatocytes as a source of systemic functionally active tPA.
DACH1 lowers hepatocyte tPA in primary human hepatocytes and mouse liver by repressing ATF6, which is a transcriptional inducer of the tPA gene Plat
To understand how DACH1 regulates Plat expression in hepatocytes, with a focus on human relevance, we compared primary human hepatocytes transduced with a dominant-negative mutant of DACH1 (ΔDS-DACH1)41 with control LacZ-transduced hepatocytes. We first validated this model by showing that ΔDS-DACH1 increased the cellular mRNA and protein of tPA and ATF6, a transcription factor that is normally repressed by DACH119 (Figure 5A). ΔDS-DACH1 also increased tPA protein and activity in the media of the hepatocytes (Figure 5B). We next considered the hypothesis that DACH1 directly represses PLAT, but there were no conserved DACH1 binding consensus sequences42 within 15 kb upstream or downstream of the PLAT genomic region. However, we did find a highly conserved ATF6 binding motif in exon 9 of PLAT (Figure 5C), suggesting the hypothesis that DACH1 decreases tPA by first repressing ATF6, which would then lead to decreased PLAT transcription. Chromatin immunoprecipitation assays in mouse liver revealed enrichment of ATF6 at the aforementioned consensus site but not at a nonconsensus site in the Plat gene (Figure 5D). In further support of this hypothesis, silencing of ATF6 in primary human hepatocytes using adeno-shATF6 decreased tPA protein and PLAT mRNA (Figure 5E; supplemental Figure 7A). Conversely, transduction of human hepatocytes with ATF6-N43 increased tPA protein and PLAT mRNA (Figure 5F; supplemental Figure 7B). Using adeno-shATF6 and adeno–ATF6-N, similar results were obtained in mouse primary hepatocytes (supplemental Figure 7C-D).
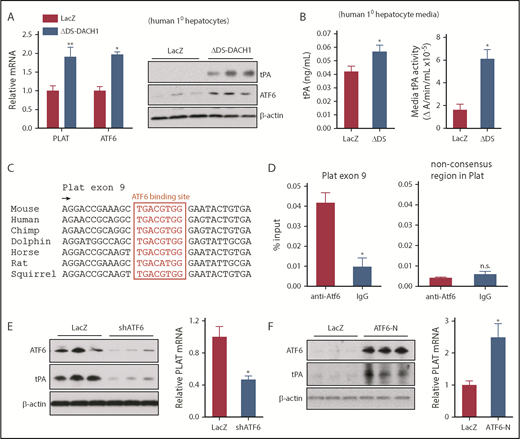
DACH1 decreases tPA expression in hepatocytes by repressing the Plat inducer ATF6. (A) Human primary hepatocytes were transduced with adeno-LacZ or adenovirus expressing dominant-negative ΔDS-DACH1 and then assayed for PLAT and ATF6 mRNA and for tPA and ATF6 by immunoblot. (B) tPA concentration by ELISA and tPA activity by enzymatic assay in the culture medium of the cells in (A). In (A-B), results are shown as mean ± SEM (n = 3 sets of cells per each group). (C) A conserved ATF6 binding consensus sequence in exon 9 of the PLAT gene. (D) Mouse liver nuclear extracts were subjected to a chromatin immunoprecipitation assay using anti-ATF6 or control immunoglobulin G. The exon 9 region containing the ATF6 binding sequence and a nonconsensus sequence in the Plat gene as control were amplified by quantitative polymerase chain reaction and normalized to the values obtained from input DNA. Results are shown as mean ± SEM (n = 3 mice per group). (E) Immunoblots of ATF6 and tPA and quantification of relative PLAT mRNA in primary human hepatocytes transduced with adeno-LacZ or adeno-shATF6. (F) Immunoblots of ATF6 and tPA and PLAT mRNA in primary human hepatocytes treated with control adeno-LacZ or adeno–ATF6-N. In (E-F), results are shown as mean ± SEM (n = 3 sets of cells per each group). Shorter film exposure time was used for developing immunoblots in panel D vs panel E to obtain signals within linear range. *P < .05, **P < .01, 2-tailed Student t test. n.s., not significant (P ≥ .05).
DACH1 decreases tPA expression in hepatocytes by repressing the Plat inducer ATF6. (A) Human primary hepatocytes were transduced with adeno-LacZ or adenovirus expressing dominant-negative ΔDS-DACH1 and then assayed for PLAT and ATF6 mRNA and for tPA and ATF6 by immunoblot. (B) tPA concentration by ELISA and tPA activity by enzymatic assay in the culture medium of the cells in (A). In (A-B), results are shown as mean ± SEM (n = 3 sets of cells per each group). (C) A conserved ATF6 binding consensus sequence in exon 9 of the PLAT gene. (D) Mouse liver nuclear extracts were subjected to a chromatin immunoprecipitation assay using anti-ATF6 or control immunoglobulin G. The exon 9 region containing the ATF6 binding sequence and a nonconsensus sequence in the Plat gene as control were amplified by quantitative polymerase chain reaction and normalized to the values obtained from input DNA. Results are shown as mean ± SEM (n = 3 mice per group). (E) Immunoblots of ATF6 and tPA and quantification of relative PLAT mRNA in primary human hepatocytes transduced with adeno-LacZ or adeno-shATF6. (F) Immunoblots of ATF6 and tPA and PLAT mRNA in primary human hepatocytes treated with control adeno-LacZ or adeno–ATF6-N. In (E-F), results are shown as mean ± SEM (n = 3 sets of cells per each group). Shorter film exposure time was used for developing immunoblots in panel D vs panel E to obtain signals within linear range. *P < .05, **P < .01, 2-tailed Student t test. n.s., not significant (P ≥ .05).
To determine the role of ATF6 in vivo, we studied Atf6fl/fl mice treated with AAV8-TBG-Cre or control AAV8-TBG-LacZ, which we validated by showing successful knockdown of ATF6 in the livers of the Cre mice (supplemental Figure 8). In support of the hypothesis, the Cre mice showed decreases in liver Plat mRNA, plasma tPA protein and activity, bleeding time, and time to occlusive carotid arterial thrombosis induced by photochemical injury (Figure 6). These combined data support the existence of a DACH1 → ↓ATF6 → ↓tPA pathway in hepatocytes that has a significant effect on systemic tPA activity.
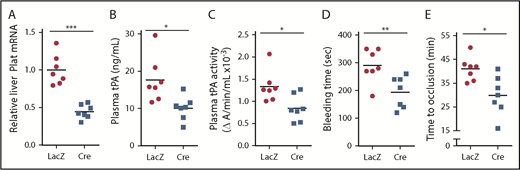
ATF6 deletion in hepatocytes decreases systemic fibrinolytic activity, bleeding time, and time to thrombotic carotid occlusion. Liver Plat mRNA (A), plasma tPA protein concentration (B), plasma tPA activity (C), tail bleeding time (D), and time to occlusive carotid arterial thrombosis (E) induced by photochemical injury in Atf6fl/fl mice treated with AAV8-TBG-LacZ or AAV8-TBG-Cre. Horizontal lines in dot-density plots indicate mean values. *P < .05, **P < .01, ***P < .01, 2-tailed Student t test (n = 7 mice per each group).
ATF6 deletion in hepatocytes decreases systemic fibrinolytic activity, bleeding time, and time to thrombotic carotid occlusion. Liver Plat mRNA (A), plasma tPA protein concentration (B), plasma tPA activity (C), tail bleeding time (D), and time to occlusive carotid arterial thrombosis (E) induced by photochemical injury in Atf6fl/fl mice treated with AAV8-TBG-LacZ or AAV8-TBG-Cre. Horizontal lines in dot-density plots indicate mean values. *P < .05, **P < .01, ***P < .01, 2-tailed Student t test (n = 7 mice per each group).
High DACH1 in human liver is associated with low levels of ATF6 and PLAT
We were able to obtain 25 random human liver specimens from the National Institutes of Health–sponsored Liver Tissue Cell Distribution System (supplemental Table 1) and observed a spectrum of DACH1 protein levels (Figure 7A). Without regard to the clinical characteristics of the donors or possible reasons for DACH1 variation, we saw this as an opportunity to test unbiased correlations of DACH1 with ATF6 and PLAT mRNA in human liver. We found that all livers with relatively high DACH1 expression (densitometric ratio > 4) had relatively low levels of ATF6 and PLAT mRNA (relative mRNA <4 and <15, respectively) (Figure 7B). There was also a significant inverse relationship between a DACH1 level > 4 and ATF6 and PLAT mRNA levels >4 and >15, respectively. However, some livers had relatively low ATF6 and PLAT at low DACH1, suggesting that when gene repression by DACH1 is low, ≥1 other pathway may be able to suppress ATF6 and PLAT. Finally, in relationship to the new finding in this study that ATF6 is an inducer of PLAT, we found a very tight correlation between ATF6 and PLAT in human liver (Figure 7C). These human liver data complement the causation data with primary human hepatocytes, which together provide evidence that key components of the new pathway revealed in this study are present in hepatocytes from human liver.
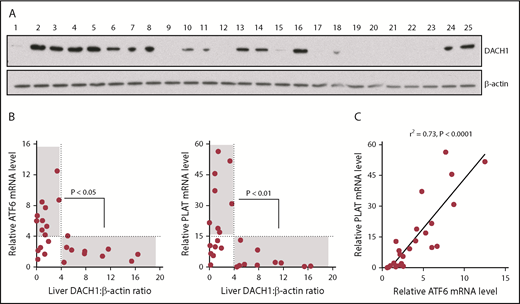
Relationships among DACH1, ATF6, and PLAT in human liver. (A) Immunoblot of DACH1, with β-actin as a loading control, in lysates of liver specimens from 25 human subjects (supplemental Table 1). (B) ATF6 or PLAT mRNA was normalized to RPLP0. Liver DACH1:β-actin was quantified by densitometric analysis of the immunoblots in (A). Data were analyzed by the Fisher exact 2-tail tests for DACH1 <4 or >4 vs ATF6 <4 or >4 and vs PLAT <15 or >15. Statistically significant inverse associations were found for the gray areas in each graph, with the P values indicated. (C) Correlation of liver ATF6 and PLAT mRNA. The data were analyzed by linear regression, with the r2 and P values indicated.
Relationships among DACH1, ATF6, and PLAT in human liver. (A) Immunoblot of DACH1, with β-actin as a loading control, in lysates of liver specimens from 25 human subjects (supplemental Table 1). (B) ATF6 or PLAT mRNA was normalized to RPLP0. Liver DACH1:β-actin was quantified by densitometric analysis of the immunoblots in (A). Data were analyzed by the Fisher exact 2-tail tests for DACH1 <4 or >4 vs ATF6 <4 or >4 and vs PLAT <15 or >15. Statistically significant inverse associations were found for the gray areas in each graph, with the P values indicated. (C) Correlation of liver ATF6 and PLAT mRNA. The data were analyzed by linear regression, with the r2 and P values indicated.
Discussion
The distinction between acutely released and “constitutive” tPA was made years ago,5 but the sources, regulation, and functional significance of constitutive tPA have remained largely unknown. The new findings in this study suggest a scenario in which hepatocyte-derived tPA, regulated by a fascinating corepressor/transcription factor pathway, is a key source of functionally active plasma tPA under basal conditions and, most importantly, influences fibrinolysis when a subsequent injury does occur. We find it particularly interesting that hepatocytes are a functionally important source of systemic tPA-mediated fibrinolytic activity, which is a scenario that was not previously appreciated. Previous work has shown that the liver produces most factors relevant to blood coagulation, but the role of the liver in fibrinolysis represents an area of uncertainty, including the roles and regulation of liver-derived PAI-1.44 Plasma PAI-1 was not affected by deleting DACH1 in hepatocytes, but other processes that affect liver PAI-1 will influence the net contribution of the liver to fibrinolysis.
The findings in this study raise the possibility that preexisting tPA may “prime” the fibrinolytic system so that when an injury does occur, the response can be immediate and complement the local release of vessel wall tPA. The evolutionary advantage of such an integrated program of fibrinolysis would be to prevent pathologic clot extension beyond the immediate site of injury, particularly in larger vessels in which locally released endothelial-derived tPA may be diluted.5,45 In support of this concept, there are a number of articles in the literature suggesting, in humans, the importance and regulation of plasma tPA that does not simply arise from acute vessel injury. As one example, plasma tPA protein has been shown to be elevated in the setting of thrombotic risk, which has been interpreted as a compensatory response.46,47 In another aspect of physiology, plasma tPA is affected by circadian rhythm: the time of lowest tPA correlates with the peak time of portal vein congestion in subjects with cirrhosis.48 Although the cellular source of the plasma tPA in these examples is not known, new findings from this study raise the interesting possibility that it may originate from hepatocytes. Finally, it should be mentioned that tPA and tPA-generated plasmin also have a number of nonfibrinolytic functions (eg, in wound repair, angiogenesis, tissue remodeling, neurotransmission, synaptic plasticity, and regulation of inflammation)45,49-51 ; thus, the role of hepatocyte-derived tPA in these processes represents a topic for future study.
In conclusion, our findings add new insights into the regulation of tPA and its role in fibrinolysis. We imagine that these insights and ones that will follow from future studies in this area will suggest new therapeutic strategies for diseases of hemostatic imbalance and perhaps diseases that are influenced by other functions of tPA.
Presented in abstract form at the 60th annual meeting of the American Society of Hematology, San Diego, CA, 1 December 2018.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Barry S. Coller (Rockefeller University) for assistance with the platelet aggregation study; José A. López (University of Washington) for helpful discussions; Keiko Nakamura (University of Tokyo) for technical assistance; Amanda Doran (CUMC) for editing of the manuscript; Marion Namenwirth and Abigail Knoble (University of Minnesota) and Nicole Martik (University of Pittsburgh) for arranging and providing human liver samples and primary hepatocytes, respectively; Ana Rivas (Columbia University Institute of Comparative Medicine Diagnostic Laboratory) for assistance with the platelet count assay; and Marc Montminy (Salk Institute for Biological Studies) for adeno-shATF6 and adeno–ATF6-N.
Z.Z. was funded by a Berrie Scholar Postdoctoral Fellowship Award from the Russell Berrie Foundation and through a National Institutes of Health, National Heart, Lung, and Blood Institute (NIH-NHLBI) postdoctoral fellow institutional training grant (T32HL007343). L.N. was supported by NIH-NHLBI grant HL121131. A.Y. was supported by an NIH-NHLBI postdoctoral fellow institutional training grant (T32HL007343). X.W. was supported by a Liver Scholar Award from the American Liver Foundation. B.C. was supported by a postdoctoral fellowship from the American Heart Association and by NIH, National Institute of Diabetes and Digestive and Kidney Diseases (NIH-NIDDK) grant K99DK115778. L.O. was supported by NIH-NIDDK grant R01DK106045 and a Pilot and Feasibility grant from the Columbia University Diabetes Research Center (P30DK063608). R.G.P. was supported by NIH, National Cancer Institute (NCI) grant R01CA132115, the Breast Cancer Research Foundation, and a generous grant from the Dr. Ralph and Marian C. Falk Medical Research Trust. M.K.J. was supported by NIH-NHLBI grants R35HL135789 and R01HL123098. I.T. was supported by NIH-NHLBI grant HL087123. Human liver samples and primary hepatocytes were obtained from the Liver Tissue Cell Distribution System (University of Minnesota and University of Pittsburgh), which was funded by NIH contract HHSN276201200017C. The flow cytometry analyses used the resources of the Columbia University Cancer Center Flow Core Facility funded in part by NIH-NCI P30CA013696 and by the NIH Office of the Director under Shared Instrumentation Grant S10RR027050.
Authorship
Contribution: Z.Z. and I.T. designed the research; Z.Z., L.N., W.W., A.Y., X.W., B.C., and S.L. conducted the research; L.O. and R.G.P. provided critical reagents and advice related to DACH1; Z.Z., L.N., W.W., B.C., R.R., M.K.J., and I.T. analyzed the data; Z.Z., A.Y., X.W., B.C., and I.T. wrote the manuscript; and all authors read and commented on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ze Zheng, Department of Medicine, Columbia University Medical Center, 630 West 168th St, New York, NY 10032; e-mail: zz2391@columbia.edu; or Ira Tabas, Department of Medicine, Columbia University Medical Center, 630 West 168th St, New York, NY 10032; e-mail: iat1@columbia.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal