

Key Points
Opsonization of HIT ULICs by complement facilitates FcγRIIA-dependent cellular effector function.
Inhibition of the proximal steps in complement activation downregulates the prothrombotic effects of HIT ULICs.

Abstract
Heparin-induced thrombocytopenia (HIT) is a prothrombotic disorder mediated by ultra-large immune complexes (ULICs) containing immunoglobulin G (IgG) antibodies to a multivalent antigen composed of platelet factor 4 and heparin. The limitations of current antithrombotic therapy in HIT supports the need to identify additional pathways that may be targets for therapy. Activation of FcγRIIA by HIT ULICs initiates diverse procoagulant cellular effector functions. HIT ULICs are also known to activate complement, but the contribution of this pathway to the pathogenesis of HIT has not been studied in detail. We observed that HIT ULICs physically interact with C1q in buffer and plasma, activate complement via the classical pathway, promote codeposition of IgG and C3 complement fragments (C3c) on neutrophil and monocyte cell surfaces. Complement activation by ULICs, in turn, facilitates FcγR-independent monocyte tissue factor expression, enhances IgG binding to the cell surface FcγRs, and promotes platelet adhesion to injured endothelium. Inhibition of the proximal, but not terminal, steps in the complement pathway abrogates monocyte tissue factor expression by HIT ULICs. Together, these studies suggest a major role for complement activation in regulating Fc-dependent effector functions of HIT ULICs, identify potential non-anticoagulant targets for therapy, and provide insights into the broader roles of complement in immune complex–mediated thrombotic disorders.
Introduction
Heparin-induced thrombocytopenia (HIT) is a potentially life-threatening thrombotic disorder caused by ultralarge immune complexes (ULICs) composed of antibodies (Abs) bound to multimeric antigens containing platelet factor 4 (PF4) and heparin. HIT ULICs bind and cross-link cellular FcγRIIA on multiple effector cells, activating pathways that initiate prothrombotic processes.1-8 HIT ULICs activate platelets in an FcγRIIA-dependent manner to generate procoagulant microparticles,2-4 release polyphosphates,9,10 and form “coated” platelets4 to facilitate assembly of clotting factors that promote thrombus formation. FcγRIIA-dependent monocyte activation by HIT ULICs upregulates expression of tissue factor (TF) and generates thrombin,4-6 whereas engagement of neutrophil FcγRs by HIT ULICs stimulates degranulation7,8 and release of neutrophil extracellular traps11-13 that further stabilize and propagate thromboses.
Many ICs also activate the complement system. Complement activation by ICs can promote their solubilization14 and clearance from the circulation,15 and it recruits inflammatory cells by generating C3a and C5a.15-17 Opsonization of complement components also invests ICs with the capacity to interact with cells through complement receptors that cooperate with cellular FcγRs to promote effector functions such as phagocytosis, cell activation, and antigen presentation.18-22
It has been reported that HIT sera and isolated immunoglobulin G (IgG) fractions activate the classical pathway of complement and promote heparin-dependent complement deposition on platelets23 and endothelial cells.24 Increased levels of C3 as well as IgG were detected on platelets from patients with HIT compared with platelets from heparinized subjects without thrombocytopenia or from healthy control subjects.23
However, the contribution of complement to the pathogenesis of thrombocytopenia and thrombosis in patients with HIT has not been studied in detail. Such studies are now warranted given the data indicating suboptimal clinical outcomes of patients with HIT treated with direct thrombin inhibitors or alternative forms of anticoagulation. Indeed, recent retrospective25-27 and epidemiologic28 studies show that direct thrombin inhibitors, fondaparinux, or direct oral anticoagulants have not fully abrogated the trajectory of thrombotic complications (23% to 36%) or death (26% to 53%) in severely affected patients, and they may compound morbidity by increasing the risk of bleeding.25,27 This clinical impasse in dosing of antithrombotic agents highlights the need to explore alternative non-anticoagulant therapeutic options based on a deeper understanding of pathogenesis of the clinical syndrome. Complement inhibition provides one potential strategy, given the intersection of complement and coagulation in thromboinflammatory disorders29,30 and the effectiveness of anti-complement therapy in reducing thrombotic complications associated with paroxysmal nocturnal hemoglobinuria,31 atypical hemolytic uremic syndrome,32 and the catastrophic antiphospholipid Ab syndrome.33,34 We therefore undertook these studies to examine the contribution of complement to FcγRIIA-mediated cellular activation and procoagulant effects of HIT Abs, as well as to identify optimal potential targets in the complement system for therapeutic inhibition in HIT.
Methods
Reagents and Abs
Information regarding commercially available reagents and Abs are provided in the supplemental Methods (available on the Blood Web site). Murine monoclonal Ab KKO [IgG2bκ recognizing human PF4/heparin>>PF4 and RTO (IgG2bκ recognizing PF4>>PF4/heparin) were developed, purified, and isolated in the laboratory according to published methods.35 Complement inhibitors used in these studies include: BBK32, a classical pathway inhibitor to C1r [herein referred to as C1r(i)] derived from Borrelia burgdorferi, kindly provided by Dr. Brandon Garcia, East Carolina University36; anti-C1q Fab (IgG1 Fab; ANXM1-Fab, referred to as αC1q-Ab), a monoclonal Fab that targets the classical pathway component C1q, generously provided by Annexon Biosciences (San Francisco, CA); Cp40, a small molecule inhibitor that binds C3/C3b/iC3b/C3c37; and eculizumab, an anti-C5 monoclonal Ab (Alexion Pharmaceuticals, Boston, MA; referred to as αC5-Ab).
Complement assays in plasma and whole blood
Complement activation by KKO or HIT ULICs was performed in plasma or whole blood (WB) from healthy donors as follows. Unless otherwise specified, plasma (100 µL) or WB (200 µL) was incubated with buffer (phosphate-buffered saline), PF4 (25 μg/mL) ± heparin (1 U/mL), and Abs (KKO 50 µg/mL or isotype [ISO] control 50 µg/mL, HIT or control human IgG [500-750 µg/mL]) for 30 to 45 minutes at 37°C followed by the addition of 10 mM EDTA. In some experiments, plasma or blood was preincubated with and without inhibitors [C1r(i), 25 µg/mL; αC1q-Ab, 50 µg/mL; αC5-Ab, 50 μg/mL] or Cp40 (20 µM) for 15 minutes at room temperature before adding a source of antigens and detection Abs. EDTA-treated plasma was used directly or separated from WB by centrifugation in complement immunoassays. To detect C3 activation initiated by ULICs, microtiter plates were coated with KKO (2 µg/mL) and incubated with plasma at a 1:50 dilution followed by detection with anti-C3c Ab (detecting the fixed C3b/iC3b fragments), as previously described.38 To detect sC5b-9, microtiter plates were coated with an anti–C5b-9 Ab and incubated with plasma at 1:50 dilution followed by detection with biotinylated anti–sC5b-9 Ab and enzyme-labeled streptavidin.
Monocyte procoagulant/TF assays
FXa activity expressed on the surface of isolated monocytes was measured as previously described.39 Briefly, HIT complexes were formed on the cell surface by adding PF4 (10 µg/mL) and KKO or ISO control Ab (40 µg/mL). C1q was then added at C1q:KKO molar ratios of 1:8 and 1:800 in serum-free media for 5 hours at 37°C under 5% carbon dioxide, and aliquots from cell suspensions were analyzed for FXa activity by using a chromogenic assay. To measure TF expression by monocytes, WB was incubated with control or blocking agents: anti-FcγRIIA Ab (IV.3, 10 µg/mL), Cp40 (20 µM), αC5-Ab (50 µg/mL), αC1q-Ab (300 µg/mL), or controls for 15 to 30 minutes at room temperature. This was followed by incubation with antigen and Ab for 4 hours at 37°C. Expression of TF was assayed by flow cytometry40 using a BD FACS Canto Flow cytometer (BD Biosciences, Franklin Lakes, NJ). Analyses were performed by using FCS express software (Version 5 Flow Research Edition; De Novo Software, Pasadena, CA).
Flow cytometry assays
Binding of complement fragments or human/murine IgG to cells was measured after incubation of WB with KKO or HIT ULICs for 45 minutes at 37°C (as described earlier). After red blood cell (RBC) lysis, blocking, and washing, the remaining white blood cells were incubated with fluorescently conjugated anti-mouse IgG or anti-human IgG Abs at 4°C for 30 minutes. The cells were then washed and incubated with Abs to cell-specific markers (anti-CD19 [B cells], anti-CD3 [T cells], anti-CD66b [neutrophils], anti-CD14 [monocytes]) and anti-C3c (detecting the C3b/iC3b fragments) at 4°C for 30 minutes. The cells were washed, fixed with 1% paraformaldehyde, and flow analysis performed as described earlier for TF.
Statistical analysis
Data are expressed as means ± standard deviation (SD). Significance was calculated by using 1-way analysis of variance with Tukey’s multiple comparison test or 2-way analysis of variance. Statistical analyses were performed by using GraphPad Prism version 7.0 (Graph Pad Software, La Jolla, CA).
Results
Physical interactions of complement component C1q with HIT ULICs
The addition of HIT plasma and heparin to healthy donor platelets activates complement via the classical pathway as evidenced by consumption of plasma C1 and a requirement for C4.23 To examine how the initiation of complement activation affects the intrinsic structure of HIT immune complexes, we used dynamic light scattering (supplemental Methods) to study the interaction of C1q with ULICs formed between PF4/heparin and KKO35 in buffer using purified components (PF4/heparin ± KKO ± C1q). In preliminary studies, C1q exhibited minimal binding to PF4/heparin (data not shown). Addition of KKO to PF4/heparin complexes approximately doubled the size of complexes formed in solution (PF4/heparin 274 [169-347] nm vs PF4/heparin + KKO 605 [210-838] nm; median [range] size at 24 hours) (Figure 1A). However, addition of C1q to ULICs at molar ratios ranging from 1:16 to 1:4 relative to KKO caused a further threefold to fourfold increase in the average size of the complexes. At a molar ratio of 1:4, the median size of the ULICs increased to 2032 nm (range, 669-2228 nm) by 1 hour and remained stable in size for at least 24 hours.
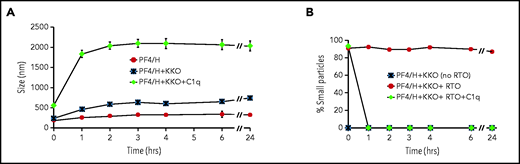
Physical interactions of HIT ULICs and C1q. (A) C1q increases the size of HIT ULICs. PF4 (10 µg/mL) and heparin (H; 0.1 U/mL) were incubated alone or with KKO (50 µg/mL) to generate ULICs. C1q was added to KKO ULICs at molar ratio of 1:4, and size of the complexes was measured by dynamic light scattering. The mean ± standard error of the mean of 3 independent experiments is shown. P < .001 for PF4/heparin + KKO + C1q vs all other conditions starting at 1 hour (2-way analysis of variance). (B) C1q restores formation of ULICs. PF4 (10 µg/mL), KKO (30 µg/mL), RTO (350 µg/mL), and heparin (0.2 U/mL) were preincubated, and buffer alone or buffer containing C1q (1:16 ratio with KKO) was added 1 hour later. The percentage of small particles (<20 nm) is shown at the indicated times. The mean ± standard error of the mean of 3 experiments is shown.
Physical interactions of HIT ULICs and C1q. (A) C1q increases the size of HIT ULICs. PF4 (10 µg/mL) and heparin (H; 0.1 U/mL) were incubated alone or with KKO (50 µg/mL) to generate ULICs. C1q was added to KKO ULICs at molar ratio of 1:4, and size of the complexes was measured by dynamic light scattering. The mean ± standard error of the mean of 3 independent experiments is shown. P < .001 for PF4/heparin + KKO + C1q vs all other conditions starting at 1 hour (2-way analysis of variance). (B) C1q restores formation of ULICs. PF4 (10 µg/mL), KKO (30 µg/mL), RTO (350 µg/mL), and heparin (0.2 U/mL) were preincubated, and buffer alone or buffer containing C1q (1:16 ratio with KKO) was added 1 hour later. The percentage of small particles (<20 nm) is shown at the indicated times. The mean ± standard error of the mean of 3 experiments is shown.
We next examined the effect of complement fixation on ULIC stability using a murine monoclonal Ab RTO that recognizes PF4 monomers and inhibits their tetramerization and subsequent multimerization.41 Formation of ULICs by PF4, heparin, and KKO was markedly inhibited by RTO (350 µg/mL), with ∼80% of the particles remaining between 10 and 15 nm in size both immediately and at 24 hours. In contrast, ULIC formation was resistant to RTO when C1q was present (1:16 molar ratio to KKO) (Figure 1B). Indeed, the ULICs continued to increase in size from ∼1100 nm at 1 hour to >2000 nm on average by 24 hours (data not shown). Second, addition of C1q to PF4 and heparin restored formation of ULICs at concentrations of RTO (350-400 µg/mL) sufficient to prevent their formation in the absence of complement (data not shown). Third, preformed ULICs containing C1q remained intact even after addition of RTO at concentrations >500 µg/mL that were sufficient to cause their complete disruption in the absence of complement (data not shown). Thus, C1q not only increases the size of ULICs but improves their stability as well.
In other studies, we found that C1q colocalizes with KKO within ULICs in plasma according to confocal microscopy (supplemental Figure 1). C1q was not visualized if the plasma was preincubated with inhibitory αC1q-Ab (data not shown). Together, these studies show that C1q incorporates on and/or within ULICs in plasma.
Complement activation by KKO and HIT ULICs in plasma and WB
We then questioned whether fixation of C1q to HIT ULICs initiates complement activation in plasma. To measure ULIC-associated or soluble complement fragments, we incubated KKO ULICs in plasma (Figure 2A) or WB (Figure 2B) and measured generation of C3 fragments using a previously described antigen capture assay,38 or soluble C5b-9 (sC5b-9) by immunoassay to detect activation of the terminal steps in the pathway. ULICs formed with KKO, but not an ISO control, generated C3 fragments and sC5b-9 (supplemental Figure 2A) in plasma. Similar, albeit less robust, generation of C3c and sC5b-9 (supplemental Figure 2B) by KKO ULICs was also seen in WB. To examine PF4/heparin-dependent activation of complement by patient or control IgG, plasma or WB was incubated with healthy donor IgG (“CON IgG”) and anti-PF4/heparin IgG from patients with (“HIT IgG”; n = 6) or without (“αPF4/H IgG”, n = 3) HIT, either in buffer or with PF4/heparin. Several HIT IgGs, but none of the IgGs from control subjects or patients with asymptomatic anti-PF4/heparin Abs, activated complement in plasma and WB (Figure 2C-D). These findings indicate that complement activation by KKO/HIT ULICs occurs in plasma and proceeds to the terminal steps in the complement pathway.
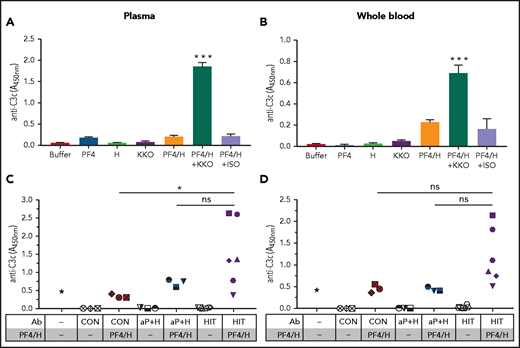
Complement activation by KKO and HIT ULICs occurs in plasma and WB. Plasma (A,C) or WB (B,D) were incubated with KKO or ISO (A,B) or anti-PF4/heparin IgG from patients with (“HIT IgG”; n = 6) or without (“αPF4/H IgG”, n = 3) HIT or control IgG either in buffer or with PF4/heparin (H) as shown on the x-axis. Complement degradation product C3c was measured. Results shown are representative of 3 independent experiments. ***P < .0001, with all other conditions (A,B) and *P < .05 (C,D) (1-way analysis of variance with Tukey’s multiple comparison test). ns, not significant.
Complement activation by KKO and HIT ULICs occurs in plasma and WB. Plasma (A,C) or WB (B,D) were incubated with KKO or ISO (A,B) or anti-PF4/heparin IgG from patients with (“HIT IgG”; n = 6) or without (“αPF4/H IgG”, n = 3) HIT or control IgG either in buffer or with PF4/heparin (H) as shown on the x-axis. Complement degradation product C3c was measured. Results shown are representative of 3 independent experiments. ***P < .0001, with all other conditions (A,B) and *P < .05 (C,D) (1-way analysis of variance with Tukey’s multiple comparison test). ns, not significant.
Susceptibility of HIT ULICs to complement inhibitors
To identify potential complement-directed therapeutic targets in HIT, WB was preincubated with complement inhibitors targeting proximal steps [αC1q-Ab, C1r(i), C1-INH, or Cp40] or terminal steps in complement activation (αC5-Ab) before the addition of KKO or HIT ULICs. Plasma or WB was also incubated with the monoclonal Ab IV.3, an FcRγIIA-blocking Ab, to examine the contributions of FcγRIIA to complement activation. Incubation was followed by detection of C3c (Figure 3A) and sC5b-9 (Figure 3B) by immunoassays. Inhibition of the classical pathway [αC1q-Ab, C1r(i), C1-INH, and Cp40] in WB markedly attenuated the generation of C3c-containing fragments (∼100% reduction) and sC5b-9 (∼100% reduction) by KKO ULICs, whereas IV.3 had much lower effects on C3c (∼26% reduction) and C5b-9 (∼14% reduction) generation. As expected, incubation of WB with αC5-Ab reduced sC5b-9 production by ∼99%, while having a more modest effect on C3 generation (∼33% reduction). Comparable results were noted for C3c/sC5b-9 when KKO ULICs were added to plasma (supplemental Figure 3) or when complement-activating HIT IgGs (HIT-1 and HIT-2) were added to WB (supplemental Figure 4). These findings indicate that: (1) classical pathway inhibitors [C1r(i), αC1q-Ab, and C1-INH] and the C3 inhibitor Cp40 have comparable effects on complement inhibition by HIT ULICs; and (2) inhibitors of the proximal, but not the terminal, steps in complement activation reduce both C3 and C5 generation by HIT ULICs. Given the equivalence of classical pathway inhibitors in these assays, αC1q-Ab was used in subsequent experiments.
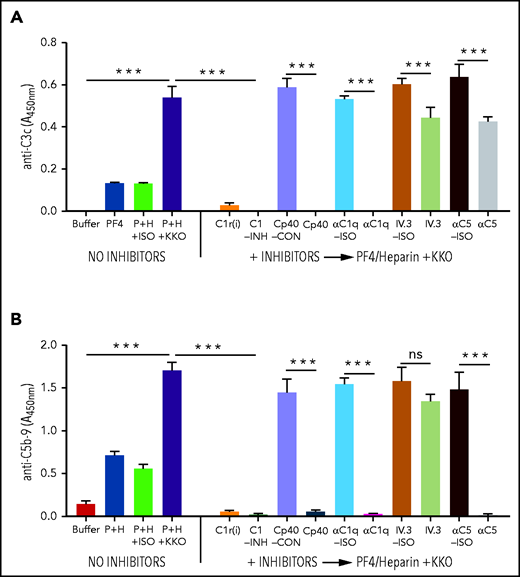
Effect of inhibitors targeting the proximal steps in the complement pathway in the presence of KKO ULICs. WB was preincubated with the complement inhibitors or respective controls (ISO=isotype or CON=control) shown on the x-axis before adding KKO ULIC, and complement activation was measured by generation of C3c (A) or C5b-9 (B). Results shown are representative of 3 independent experiments. ***P < .0001 (1-way analysis of variance with Tukey’s multiple comparison test). ns, not significant.
Effect of inhibitors targeting the proximal steps in the complement pathway in the presence of KKO ULICs. WB was preincubated with the complement inhibitors or respective controls (ISO=isotype or CON=control) shown on the x-axis before adding KKO ULIC, and complement activation was measured by generation of C3c (A) or C5b-9 (B). Results shown are representative of 3 independent experiments. ***P < .0001 (1-way analysis of variance with Tukey’s multiple comparison test). ns, not significant.
Complement activation by HIT ULICs increases deposition of C3 and IgG on cell surfaces
Complement-coated ICs can facilitate both antigen deposition on cell surfaces and subsequent clearance from the circulation via cellular FcγRs.19,42,43 To determine if complement activation enhances binding of HIT ULICs to cell surfaces, WB was incubated with various agonists (buffer, PF4 ± heparin ± KKO/HIT ULICs, or control Abs) followed by staining with labeled α-human C3c, α-mouse/human IgG, or cell-specific markers; binding was measured by flow cytometry.
Incubation of WB with KKO ULICs, but not ISO ULICs or antigens alone, led to increased IgG binding and marked deposition of C3c on neutrophils, monocytes, and B cells (Figure 4), with minimal binding of C3c/IgG to T cells and RBCs (data not shown). As noted in previous studies,23 KKO ULICs also increased C3 deposition on platelets, which was attenuated by classical pathway inhibitors and Cp40 but not by anti-C5 or IV.3 (data not shown). Similar cell surface binding to neutrophils, monocytes, and B cells was seen with complement-activating HIT ULICs; representative HIT ULICs are shown in the insets of Figure 4. Indeed, there was a strong correlation between deposition of C3 and IgG, but not IgM (data not shown), on various cells incubated with PF4/heparin and KKO or HIT ULICs but not ISO ULICs (supplemental Figure 5).
![KKO/HIT ULICS activate complement and lead to deposition of complement on neutrophils, monocytes, and B cells. Main figures show binding (mean fluorescence intensity [MFI]) of α-C3c (top) or α-mouse IgG (bottom) to defined cell populations after incubating WB with conditions indicated on the x-axis and analyzed by flow cytometry. Cell populations were gated by labeled Abs to CD66 (neutrophils), CD14 (monocytes), and CD19 (B cells). Insets for each graph show respective cell population when WB was incubated with buffer (B) or HIT IgG (HIT) alone or with PF4/heparin (P+H+HIT) or ISO/control with PF4/heparin (P+H+ISO). The graphic is representative of ≥3 independent experiments using KKO and complement-activating HIT IgG. **P < .005, ***P < .0001 (compared with all other conditions using 1-way analysis of variance with Tukey’s multiple comparison test).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/21/10.1182_blood.2020009487/5/m_bloodbld2020009487f4.png?Expires=1768343951&Signature=wcNcgnvba8sHtWRDPI7Tm95gytN5TnyvcZ2zSoG3wP2gbQpQ7hWBJElesRxTWSqK8ywvPQ1khEJvAmEZh0ilHvPgpihkyUBGNfNOKrplgrfY-ibfztnIs5O1dFHqhXaT-7Nj3S21KB5Br2dNV7UZJKLVXyis1Nck~hKbhHAML9E~EhTFqsMfKJAa-qdQYg0d5ncVuYbfarJrmcbUwLy6KSoOU8N4Rc8pxtFUHYN1D6Jbux34Atv4qtIGcrbfh1Xsbvk~071IIkPlI49JOPuq3fEZV15UGNzGvH15~xnIzApfz63h-GZfrLUvRtDTcPICq5wqCLpdp3eGAtXCwqsyEg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
KKO/HIT ULICS activate complement and lead to deposition of complement on neutrophils, monocytes, and B cells. Main figures show binding (mean fluorescence intensity [MFI]) of α-C3c (top) or α-mouse IgG (bottom) to defined cell populations after incubating WB with conditions indicated on the x-axis and analyzed by flow cytometry. Cell populations were gated by labeled Abs to CD66 (neutrophils), CD14 (monocytes), and CD19 (B cells). Insets for each graph show respective cell population when WB was incubated with buffer (B) or HIT IgG (HIT) alone or with PF4/heparin (P+H+HIT) or ISO/control with PF4/heparin (P+H+ISO). The graphic is representative of ≥3 independent experiments using KKO and complement-activating HIT IgG. **P < .005, ***P < .0001 (compared with all other conditions using 1-way analysis of variance with Tukey’s multiple comparison test).
KKO/HIT ULICS activate complement and lead to deposition of complement on neutrophils, monocytes, and B cells. Main figures show binding (mean fluorescence intensity [MFI]) of α-C3c (top) or α-mouse IgG (bottom) to defined cell populations after incubating WB with conditions indicated on the x-axis and analyzed by flow cytometry. Cell populations were gated by labeled Abs to CD66 (neutrophils), CD14 (monocytes), and CD19 (B cells). Insets for each graph show respective cell population when WB was incubated with buffer (B) or HIT IgG (HIT) alone or with PF4/heparin (P+H+HIT) or ISO/control with PF4/heparin (P+H+ISO). The graphic is representative of ≥3 independent experiments using KKO and complement-activating HIT IgG. **P < .005, ***P < .0001 (compared with all other conditions using 1-way analysis of variance with Tukey’s multiple comparison test).
HIT ULICs promote complement-dependent monocyte procoagulant and TF activity
Activation of monocytes contributes to thrombosis in HIT. Binding of HIT ULICs upregulates monocyte procoagulant activity, monocyte TF expression, and release of TF-positive microparticles.6,40,44 To examine the contribution of complement, isolated monocytes were incubated in serum-free media in the presence of KKO/ISO ULICs with or without exogenous C1q (C1q:KKO molar ratios of 1:8 and 1:800) for 5 hours. Procoagulant activity was assessed by measuring TF- and FVIIa-dependent cell-associated FXa activity.39 Monocytes incubated with C1q alone generated little to no procoagulant activity, and cells incubated with KKO/ISO ULICs in the absence of C1q generated minimal FXa activity. In contrast, addition of C1q (C1q:KKO molar ratios of 1:8) caused a striking increase in FXa generation (Figure 5A).
![HIT ULICs promote monocyte procoagulant activity and TF expression in a complement-dependent manner. (A) C1q binding to HIT ULICs enhances monocyte procoagulant activity. KKO/ISO ULICs and human C1q were added to monocytes in serum-free media for 5 hours at 37°C under 5% carbon dioxide. C1q was added at C1q:KKO molar ratios of 1:8 [C1q (hi)] and 1:800 [C1q (lo)]. Aliquots from the cell suspensions were analyzed for FXa activity by using a chromogenic assay. Results of 2 independent experiments, each done in quadruplicate (mean ± standard error of the mean), are shown as a fold increase in FXa generation relative to cells incubated with media alone. ***P < .0001 (compared with all other conditions using the 1-way analysis of variance with Tukey’s multiple comparison test). (B) HIT ULICs induce monocyte TF expression upstream of C5: WB blood was preincubated without or with complement inhibitors/controls (α-C1q ISO Ab/α-C1q Ab 300 μg/mL, CP40-CON/Cp40 20 μM, IV.3-ISO/IV.3 10 μg/mL, α-C5-ISO ab/α-C5 Ab 50 μg/mL) before adding PF4/heparin/KKO ULICs as shown on the x-axis. After 4 hours of incubation, RBCs were lysed, leukocytes were stained with α-CD14 and α-TF Ab, and the percentage of TF-expressing monocytes was measured. The graphic shows the results of 1 of 3 independent experiments tested in duplicate (mean ± SD). ***P < .0001 (1-way analysis of variance with the Tukey’s multiple comparison test). ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/21/10.1182_blood.2020009487/5/m_bloodbld2020009487f5.png?Expires=1768343951&Signature=uc5igcZBru4ig3DxI2G0mML3G3P7542fCu01-C21CiXh6Os-3n7kEUmb3XrhuyHrEd7~xLUAC~5CyLHoBpSm4RlZeNJqoleFVAkAZq-R7FLoNKp7-oJkR3SMImjzPG5m2zeaE3lo7TIIJynqzljOKQmTJGqSTfBDDo8BvrSoOesaTmNuefPmRs2c-kQB9P0DtnuEWsmJJ9ZMTd4ckqI~Rc7NyJZvZjRDhPakV0GPK2qng1LlyEuMcEZfGHAv2Q70R0jH1byjTJvYlKSZ0Ewwv3lFPkP0t09B2TVDwfGis8vCZar0n~NbYW4W4Xx8ztuB0b1T1ecAYH74EoBMYNMa-A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HIT ULICs promote monocyte procoagulant activity and TF expression in a complement-dependent manner. (A) C1q binding to HIT ULICs enhances monocyte procoagulant activity. KKO/ISO ULICs and human C1q were added to monocytes in serum-free media for 5 hours at 37°C under 5% carbon dioxide. C1q was added at C1q:KKO molar ratios of 1:8 [C1q (hi)] and 1:800 [C1q (lo)]. Aliquots from the cell suspensions were analyzed for FXa activity by using a chromogenic assay. Results of 2 independent experiments, each done in quadruplicate (mean ± standard error of the mean), are shown as a fold increase in FXa generation relative to cells incubated with media alone. ***P < .0001 (compared with all other conditions using the 1-way analysis of variance with Tukey’s multiple comparison test). (B) HIT ULICs induce monocyte TF expression upstream of C5: WB blood was preincubated without or with complement inhibitors/controls (α-C1q ISO Ab/α-C1q Ab 300 μg/mL, CP40-CON/Cp40 20 μM, IV.3-ISO/IV.3 10 μg/mL, α-C5-ISO ab/α-C5 Ab 50 μg/mL) before adding PF4/heparin/KKO ULICs as shown on the x-axis. After 4 hours of incubation, RBCs were lysed, leukocytes were stained with α-CD14 and α-TF Ab, and the percentage of TF-expressing monocytes was measured. The graphic shows the results of 1 of 3 independent experiments tested in duplicate (mean ± SD). ***P < .0001 (1-way analysis of variance with the Tukey’s multiple comparison test). ns, not significant.
HIT ULICs promote monocyte procoagulant activity and TF expression in a complement-dependent manner. (A) C1q binding to HIT ULICs enhances monocyte procoagulant activity. KKO/ISO ULICs and human C1q were added to monocytes in serum-free media for 5 hours at 37°C under 5% carbon dioxide. C1q was added at C1q:KKO molar ratios of 1:8 [C1q (hi)] and 1:800 [C1q (lo)]. Aliquots from the cell suspensions were analyzed for FXa activity by using a chromogenic assay. Results of 2 independent experiments, each done in quadruplicate (mean ± standard error of the mean), are shown as a fold increase in FXa generation relative to cells incubated with media alone. ***P < .0001 (compared with all other conditions using the 1-way analysis of variance with Tukey’s multiple comparison test). (B) HIT ULICs induce monocyte TF expression upstream of C5: WB blood was preincubated without or with complement inhibitors/controls (α-C1q ISO Ab/α-C1q Ab 300 μg/mL, CP40-CON/Cp40 20 μM, IV.3-ISO/IV.3 10 μg/mL, α-C5-ISO ab/α-C5 Ab 50 μg/mL) before adding PF4/heparin/KKO ULICs as shown on the x-axis. After 4 hours of incubation, RBCs were lysed, leukocytes were stained with α-CD14 and α-TF Ab, and the percentage of TF-expressing monocytes was measured. The graphic shows the results of 1 of 3 independent experiments tested in duplicate (mean ± SD). ***P < .0001 (1-way analysis of variance with the Tukey’s multiple comparison test). ns, not significant.
We next assessed the role of complement and FcγR-dependent and -independent pathways on monocyte procoagulant activity. WB was preincubated with inhibitors of the proximal and terminal complement pathways, or their respective controls, followed by incubation with buffer, PF4/heparin (H), or KKO/ISO ULICs for 4 hours, and expression of TF on CD14+ cells was measured by flow cytometry and FXa procoagulant activity determined as described earlier. Addition of KKO ULICs to WB lead to a significantly greater percentage of TF-positive cells (29.7 ± 0.8%; mean ± standard deviation) compared with buffer (3.7 ± 1.5%), PF4/H (6.2 ± 0.3%), or ISO-ULICs (6.8 ± 1.4%) (Figure 5B). Preincubation with IV.3 reduced monocyte TF expression ∼75% relative to its ISO control (IV.3 ISO; P < .0001), as expected.4,45 However, preincubation of WB with αC1q-Ab or Cp40 in the absence of FcγRIIA blockade also attenuated TF expression by ∼60% and 54%, respectively (P < .0001 relative to ISO), whereas anti-C5 was less effective (∼5% inhibition; P = not significant). TF protein expression by KKO was accompanied by functional procoagulant activity, as measured by FXa generation (supplemental Figure 6). Limited patient sample availability permitted evaluation of procoagulant activity to only one HIT IgG (HIT-1). Together, these findings show that complement augments FcγRIIA-dependent monocyte TF activity through components upstream of C5.
Complement facilitates binding of ULICs to FcRs which enhances effector function
Activated complement components generated by ICs incorporate onto and/or within antigen/Ab lattices.46 The findings of codeposition of fragments of C3 and ULIC-IgG on various cell surfaces (Figure 4; supplemental Figure 5) and FcγRIIA-independent regulation of TF expression by complement (Figure 5) led us to hypothesize that opsonization of complement by ULICs contributes to IgG binding and subsequent FcγR-mediated cellular effector function. To examine this possibility, we incubated WB with varying amounts of KKO ULICs (50-200 µg/mL) in the presence or absence of αC1q-Ab or ISO control and examined ULIC-IgG binding and concomitant monocyte TF expression. WB incubated in the absence of inhibitors displayed strong IgG binding to monocyte cell surface (Figure 6A) and monocyte TF expression (Figure 6B). αC1q-Ab, but not its ISO control, markedly reduced IgG binding and accompanying TF expression. Increasing KKO IgG (twofold to fourfold excess) did not overcome the effects of complement inhibition by αC1q-Ab. Inhibition of the classical pathway also markedly attenuated binding of HIT ULICs to multiple cell surfaces (supplemental Figure 7). These studies establish that activation of complement promotes binding of ULICs to cell surface FcγRIIA and likely regulates Fc-dependent effector functions.
![Opsonization by complement facilitates ULIC binding and thereby FcR effector functions. (A) Complement inhibition by α-C1qAb reduces deposition of KKO ULICs. WB was preincubated with buffer, α-C1q (300 µg/mL), or ISO control (300 µg/mL) before adding ULICs containing KKO or ISO IgG (50-200 µg/mL). Surface-bound mouse (m)IgG was measured by flow cytometry. α-C1q-Ab, but not its ISO control, prevented deposition of mIgG on the cell surface. Effects of complement inhibition could not be overcome with higher doses of ULIC-IgG. (B) Complement inhibition inhibits TF expression: Similar incubation conditions from panel A were used, followed by detection of monocyte TF by flow cytometry. Inhibition of complement reduced TF expression. The graphic shows results representative of 3 independent experiments tested in duplicate (mean ± SD). P < .0001 (1-way analysis of variance with the Tukey’s multiple comparison test for KKO+PF4/heparin [H] at each concentration vs each concentration of ISO controls and similarly for conditions using α-C1q at each concentration vs each concentration of ISO controls for α-C1q). *Buffer. **PF4/KKO (200 µg/mL). MFI, mean fluorescence intensity.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/21/10.1182_blood.2020009487/5/m_bloodbld2020009487f6.png?Expires=1768343951&Signature=UKG38W2hsHdnfgmxaygRPQBJ~Yn0QU0px5wKpOi8UF8ygLvE-1p2xoZCqutd1xSdKHfc20V3R9A8Px-4Ewm5ExZ76jTVw7XcSwLHcccCVlsViEu-9aeEmLkrlaLgcGIHl-gH8uKA7-PT5MIGEuu0AX85zCylduF~M6K37OVumMmOw40GfBjLSDTrNg-ELgMtGkaW05Uk1psxj3dZ0~To62Y4w4s6u8OoirXYKi~XACESqd6nj-5bPwzY9DZ2OnKqDG9NfHxk-CK479danbZeJgZ7g2u2nLrNBEw-p3lpo8Rq6k6ef-Yq2IX6YIJnKzprpJwxDVV5BN4h~IhFTc9Uhg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Opsonization by complement facilitates ULIC binding and thereby FcR effector functions. (A) Complement inhibition by α-C1qAb reduces deposition of KKO ULICs. WB was preincubated with buffer, α-C1q (300 µg/mL), or ISO control (300 µg/mL) before adding ULICs containing KKO or ISO IgG (50-200 µg/mL). Surface-bound mouse (m)IgG was measured by flow cytometry. α-C1q-Ab, but not its ISO control, prevented deposition of mIgG on the cell surface. Effects of complement inhibition could not be overcome with higher doses of ULIC-IgG. (B) Complement inhibition inhibits TF expression: Similar incubation conditions from panel A were used, followed by detection of monocyte TF by flow cytometry. Inhibition of complement reduced TF expression. The graphic shows results representative of 3 independent experiments tested in duplicate (mean ± SD). P < .0001 (1-way analysis of variance with the Tukey’s multiple comparison test for KKO+PF4/heparin [H] at each concentration vs each concentration of ISO controls and similarly for conditions using α-C1q at each concentration vs each concentration of ISO controls for α-C1q). *Buffer. **PF4/KKO (200 µg/mL). MFI, mean fluorescence intensity.
Opsonization by complement facilitates ULIC binding and thereby FcR effector functions. (A) Complement inhibition by α-C1qAb reduces deposition of KKO ULICs. WB was preincubated with buffer, α-C1q (300 µg/mL), or ISO control (300 µg/mL) before adding ULICs containing KKO or ISO IgG (50-200 µg/mL). Surface-bound mouse (m)IgG was measured by flow cytometry. α-C1q-Ab, but not its ISO control, prevented deposition of mIgG on the cell surface. Effects of complement inhibition could not be overcome with higher doses of ULIC-IgG. (B) Complement inhibition inhibits TF expression: Similar incubation conditions from panel A were used, followed by detection of monocyte TF by flow cytometry. Inhibition of complement reduced TF expression. The graphic shows results representative of 3 independent experiments tested in duplicate (mean ± SD). P < .0001 (1-way analysis of variance with the Tukey’s multiple comparison test for KKO+PF4/heparin [H] at each concentration vs each concentration of ISO controls and similarly for conditions using α-C1q at each concentration vs each concentration of ISO controls for α-C1q). *Buffer. **PF4/KKO (200 µg/mL). MFI, mean fluorescence intensity.
Complement inhibitors prevent platelet adhesion to injured endothelium
HIT Abs bind to ultralarge von Willebrand factor multimers at sites of endothelial injury in vitro and in vivo to facilitate brisk platelet adhesion and accumulation.47,48 To examine the contribution of complement to ULIC-mediated platelet deposition on injured endothelial cells, we performed microfluidic studies using WB containing KKO ULICs in the presence of increasing amounts of C1r(i) (0-100 μg/mL). Addition of KKO ULICs to WB resulted in marked platelet deposition on injured human umbilical vein endothelial cells (supplemental Figure 8), which was abrogated by pretreatment of WB with increasing doses of C1r(i) (Figure 7). These findings indicate that maximal platelet adhesion to von Willebrand factor and/or endothelial cells in the presence of ULICs seems, in large part, complement dependent.
![Inhibiting the proximal steps in the complement pathway impairs platelet adhesion to human umbilical vein endothelial cells induced by KKO. Microfluidic channels coated with human umbilical vein endothelial cells were perfused with recalcified citrated WB stimulated with monoclonal Ab KKO (50 µg/mL) in the presence of PF4 (25 µg/mL). Some samples were preincubated with C1r inhibitor [C1r(i); 5 µg/mL, 25 µg/mL, or 100 µg/mL] 15 minutes before the addition of KKO (50 µg/mL) and PF4 (25 µg/mL). Adhesion of calcein-labeled platelets was measured as total fluorescent intensity at 5-minute intervals, and the effect of C1r(i) is expressed as percentage of platelet adhesion in the presence of inhibitor compared with platelet adhesion without inhibitor (100%). *P < .05, **P < .005, ***P < .0005 (2-way analysis of variance with Dunnett’s correction); n = 4.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/21/10.1182_blood.2020009487/5/m_bloodbld2020009487f7.png?Expires=1768343951&Signature=KC8qmUXsB8hkArvk5wGk4yuZGi~n~LTxbJlugQ7MORKo3lQxs9dWE4F~-uFAC4J5nnqHF2EOnOdF4-hfncP0m8a~~PS9oHTKYlsjvz~xxJIQhbccrufs~EI1zpItUdiF2PYwvmI1hID8xCk0-JFMCZLhv~vrxSSAVRi59GWWh9vRJALrIzbF3ccqPKx5KJV2IhS08srPSPZzS51PhPysp-CawJR7fxe3qqUYPGCWo5ZuKcFqT-KFbVHWUP09OU7U2MnGxKIEj1sFI6aYBy98LEtTeeoMqiN95PQ~LRtCZJzag0FdeQm38vW~lj7Kj0Ia2YYQqifKx4l68lM9eseT3w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Inhibiting the proximal steps in the complement pathway impairs platelet adhesion to human umbilical vein endothelial cells induced by KKO. Microfluidic channels coated with human umbilical vein endothelial cells were perfused with recalcified citrated WB stimulated with monoclonal Ab KKO (50 µg/mL) in the presence of PF4 (25 µg/mL). Some samples were preincubated with C1r inhibitor [C1r(i); 5 µg/mL, 25 µg/mL, or 100 µg/mL] 15 minutes before the addition of KKO (50 µg/mL) and PF4 (25 µg/mL). Adhesion of calcein-labeled platelets was measured as total fluorescent intensity at 5-minute intervals, and the effect of C1r(i) is expressed as percentage of platelet adhesion in the presence of inhibitor compared with platelet adhesion without inhibitor (100%). *P < .05, **P < .005, ***P < .0005 (2-way analysis of variance with Dunnett’s correction); n = 4.
Inhibiting the proximal steps in the complement pathway impairs platelet adhesion to human umbilical vein endothelial cells induced by KKO. Microfluidic channels coated with human umbilical vein endothelial cells were perfused with recalcified citrated WB stimulated with monoclonal Ab KKO (50 µg/mL) in the presence of PF4 (25 µg/mL). Some samples were preincubated with C1r inhibitor [C1r(i); 5 µg/mL, 25 µg/mL, or 100 µg/mL] 15 minutes before the addition of KKO (50 µg/mL) and PF4 (25 µg/mL). Adhesion of calcein-labeled platelets was measured as total fluorescent intensity at 5-minute intervals, and the effect of C1r(i) is expressed as percentage of platelet adhesion in the presence of inhibitor compared with platelet adhesion without inhibitor (100%). *P < .05, **P < .005, ***P < .0005 (2-way analysis of variance with Dunnett’s correction); n = 4.
Discussion
The high complication rates associated with non-heparin anticoagulants in patients with severe HIT highlight the need for newer therapies that address the underlying disease pathobiology. In this study, we identify complement activation as an important and pliable mechanism by which HIT ULICs promote cellular activation and thrombosis. Using targeted complement inhibitors, we show that HIT ULICs activate complement via the classical pathway, which leads to deposition of activated complement components on multiple cell types, mediates binding of HIT ULICs to FcγRs, augments FcγR-dependent effector function relevant to HIT such as induction of TF on monocytes, and promotes platelet adhesion to injured endothelium. Inhibition of the classical pathway proximal to the activation of C3 attenuates downstream FcγR-mediated cellular effector functions, whereas blocking of C5 was less effective.
Although the ability of HIT Abs to activate complement was first recognized >40 years ago, interest in this pathway waned soon after descriptions of the platelet FcγRIIA as an essential mediator of disease.49,50 Subsequent studies showed that inhibition of FcγRIIA function with monoclonal Abs or downstream receptor signaling with Syk or tyrosine kinase inhibitors markedly attenuated platelet activation and thrombin generation by HIT ULICs.4 The requirement for FcγRIIA was further reinforced through findings in murine models. Wild-type mice, which do not express the genetic equivalent of human FcγRIIA, or single transgenic mice, expressing hPF4 or human FcγRIIA, did not develop thrombocytopenia or thrombosis when injected with a murine HIT-like monoclonal Ab, KKO. In contrast, double transgenic mice, expressing both hPF4 and human FcγRIIA on platelets and monocytes, developed profound thrombocytopenia and thrombosis when injected with KKO.1 However, these studies did not address nor exclude the involvement of mechanisms acting upstream or downstream of FcγR engagement, such as activation of complement.
The current study found that opsonization of complement augments engagement of cell surface FcγRIIAs by HIT ULICs. This finding is in line with previous studies which have shown that activation of complement enhances the efficiency of IC interactions with FcγRs in other settings. Superoxide generation, degranulation, and phagocytosis are markedly enhanced when neutrophils encounter RBCs coated with C3b and IgG, compared with RBCs coated with C3b or IgG alone.51 Moreover, the concentration of IgG-coated RBCs required to elicit these responses is lower when cells are opsonized with C3b. Other studies using C3b-IgG conjugates show that comparable phagocytic function of neutrophils cannot be induced by an equal number of unconjugated C3b or IgG molecules.52,53 In addition to size, antigen density, and IgG subclass, opsonization of complement is one of the most important factors that contributes to cell activation by ICs. For example, fixation of complement and interactions with complement receptors are a prerequisite for neutrophil activation by ICs of various subclasses (IgG1, IgG2, and IgG4) and epitope density.54 These findings are in keeping with our observation that αC1q-Ab markedly reduced binding of KKO IgG to monocyte surfaces, with attendant loss of procoagulant activity. Whether mechanisms relevant to soluble or circulating ULICs, as studied in this article, apply to cell-bound ICs formed in the absence of heparin, as occurs in autoimmune HIT,55 will require additional study.
The use of undiluted plasma and WB in our assays provided a physiological environment with which to assess complement and HIT ULIC interactions. These studies found that complement activation by HIT ULICs occurs in plasma and proceeds to terminal complement activation (Figure 2; supplemental Figure 2). The lower signals seen in WB relative to plasma are possibly due to dilutional effects of RBCs and/or additional cellular binding sites for complement fragments. Because there was no evidence of hemolysis (data not shown), it is likely that complement regulatory proteins limit formation or inactivate membrane attack complexes.
In our assays, only some HIT IgG mediated complement activation in plasma and WB. These differences were not explained by the distribution of IgG subclasses or IgG titers among patients (supplemental Table 1). Use of healthy donors rather than patients as a source of plasma or WB introduces additional biological variables that likely influence ULIC behavior. Differences in levels of complement regulatory proteins caused by common polymorphisms,56 levels of plasma proteins that influence PF4/heparin assembly (ie, fibronectin57), the FcγIIA-R131H polymorphism,58 and/or regulators of FcγR function such as TULA59 will need to be systematically evaluated in future studies.
Studies in WB also revealed significant deposition of ULICs on B cells relative to other FcγR-bearing cells such as monocytes and neutrophils (Figure 4). Although the effects of ULICs on monocytes and neutrophils expressing cellular FcγRIIA have been reported in HIT,4,8,11,13,60 the functional consequences of ULICs engaging B-cell FcR, which only express the inhibitory FcγRIIB, are less clear. B-cell FcγRIIB helps maintain peripheral tolerance,22 and mice lacking FcγRIIB develop autoimmunity.61 Simultaneous engagement of the inhibitory FcγRIIB and the B-cell receptor by ICs can override B-cell receptor activation signals caused by antigen and trigger B-cell apoptosis.62 This coinhibitory mechanism is postulated to mediate the protective effects of anti-D Abs in preventing rhesus D hemolytic disease of the newborn.63,64 Whether the self-limited nature of HIT Ab response65 or response to intravenous immunoglobulin66 can be explained in part by inhibitory effects of ULICs binding to FcγRIIB on antigen-specific B cells remains to be studied.
Our findings indicate that potential therapeutic agents targeting proximal steps of complement activation (the classical pathway or C3) may be better suited for future clinical investigations in HIT than those designed to block more terminal steps. Inhibitors of the classical pathway targeting C1q (anti-C1q), C1r (BBK32), and C1s (C1INH) as well as a C3 inhibitor (Cp40) reduced complement activation (Figure 3), diminished ULIC binding to cell surfaces (Figure 6; supplemental Figure 7), downregulated expression of TF by monocytes more than anti-C5 (Figures 5B, 6B), and attenuated platelet interactions with injured endothelium (Figure 7; supplemental Figure 8). The pathophysiological relevance of our findings requires further validation in murine models. However, the species specificity of all inhibitors used in these studies limits animal testing only to Cp40,37 which can be used in non-human primates. Therefore, future studies to examine the vivo relevance of our findings in the murine HIT model will likely require strategies that involve genetic editing and/or breeding the HIT double transgenic mice onto specific complement-deficient backgrounds.
Based on our findings, we propose the following model of complement’s involvement in HIT, as shown in the Visual Abstract. HIT ULICs activate complement in blood and on cell surfaces, leading to the incorporation of complement components. These engage cell surface complement receptors and facilitate binding and clustering of IgG ULICs on signal-transducing Fcγ receptors that promote procoagulant pathways on neutrophils and monocytes. When the proximal steps in complement activation are inhibited, ICs do not opsonize complement, which inhibits their binding to cell surface complement receptors and subsequent engagement of FcRs.
In summary, we describe a potentially significant role for complement in regulating FcγRIIA-mediated effector functions in HIT, including induction of monocyte procoagulant activity and platelet adhesion at sites of vascular injury, and we identify C1 and/or C3 as possible therapeutic targets. Our studies also provide insights into potential mechanisms by which complement may contribute to other immune-mediated thrombotic disorders, such as the recently described vaccine-induced thrombotic thrombocytopenia syndrome,67-70 systemic lupus erythematosus, and the antiphospholipid Ab syndrome. The significance of complement to the pathogenesis of these immunothrombotic disorders will require additional validation in preclinical models.
Acknowledgments
The authors thank Brandon Garcia, East Carolina University, for providing BBK32 and Annexon Biosciences for providing α-C1q and ISO control Abs.
Support was provided by the National Institutes of Health, National Heart, Lung, and Blood Institute, grants R01HL136512 (G.M.A.), R01HL151730 (L.R.), R01HL139448 (D.B.C.), R01HL142122 (D.B.C.), HL128895 (D.B.C.), R35 HL150698 (L.R.), R01HL139448 (L.R.), and K08-HL127183 (G.M.L.).
Authorship
Contribution: S.K., G.M.A., A.B., L.R., A.H.R., S.V.Y., S.Z., J.D.L., S.M., A.J., G.M.L., M.D., M.P., and D.B.C. conceived of and designed the study; G.M.A., S.K., A.B., A.J., S.M., L.R., D.B.C., M.P. J.D.L., A.H.R., and S.Z. provided study materials or patients; S.K., G.M.A., A.S., A.B., L.R., A.H.R., S.V.Y., S.Z., S.M., A.J., D.B.C., and M.P. collected and assembled the data; S.K., G.M.A., A.S., L.R., A.H.R., S.V.Y., S.Z., D.B.C., and M.P. analyzed and interpreted the data; G.M.A., D.B.C., S.K., M.P., L.R., A.H.R., S.V.Y., S.Z., S.M., and J.D.L. wrote the manuscript; and all authors gave final approval of the manuscript.
Conflict-of-interest disclosure: G.M.A. has an awarded patent for KKO (US application NO 60/143 536); G.M.A., M.P., and D.C. have pending intellectual property applications. J.D.L. is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors for therapeutic purposes and is the inventor of patents or patent applications that describe the use of complement inhibitors for therapeutic purposes, some of which are developed by Amyndas. J.D.L. is also the inventor of the Compstatin technology licensed to Apellis Pharmaceuticals [ie, 4(1MeW)7W/POT-4/APL-1 and PEGylated derivatives such as APL-2/pegcetacoplan and APL-9]. The remaining authors declare no competing financial interests.
Correspondence: Douglas B. Cines, Departments of Pathology and Laboratory Medicine and Medicine, Perelman School of Medicine, University of Pennsylvania, 513A Stellar-Chance Pavilion, 422 Curie Blvd, Philadelphia, PA 19104; e-mail: dcines@pennmedicine.upenn.edu.
Presented in abstract form at the 62nd annual meeting of the American Society of Hematology, 5-8 December 2020 (held virtually).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal