Abstract
Interrogation of hematopoietic tissue at the clonal level has a rich history spanning over 50 years, and has provided critical insights into both normal and malignant hematopoiesis. Characterization of chromosomes identified some of the first genetic links to cancer with the discovery of chromosomal translocations in association with many hematological neoplasms. The unique accessibility of hematopoietic tissue and the ability to clonally expand hematopoietic progenitors in vitro has provided fundamental insights into the cellular hierarchy of normal hematopoiesis, as well as the functional impact of driver mutations in disease. Transplantation assays in murine models have enabled cellular assessment of the functional consequences of somatic mutations in vivo. Most recently, next-generation sequencing–based assays have shown great promise in allowing multi-“omic” characterization of single cells. Here, we review how clonal approaches have advanced our understanding of disease development, focusing on the acquisition of somatic mutations, clonal selection, driver mutation cooperation, and tumor evolution.
Introduction
Every cell division throughout life, starting from the first division of the fertilized egg, requires the accurate replication of the entire genome, which in humans comprises 3 billion nucleotide base pairs. Mammalian DNA replication and repair systems, although highly complex and precise, are not infallible. Ongoing exposure to endogenous and extrinsic DNA-damaging insults inevitably results in the acquisition of somatic mutations in individual cells.1 Thus, the DNA composition of cells within a tissue can be compared with a fine mosaic, each distinct tile of which is akin to an individual cell that differs from its neighbor by virtue of its unique catalog of somatic mutations. The vast majority of such mutations occur in noncoding regions of the DNA and are believed to have a neutral effect on cellular fitness.2 However, mutations can lead to positive selection and clonal expansion if they land in genomic regions, for example, those with oncogenic potential, which result in cellular phenotypes that enhance fitness with respect to competing normal cells (termed “driver” mutations). Clonal expansion provides a reservoir for the acquisition of further driver mutations, and ultimately carcinogenesis. Thus, increasing somatic mutation poses a real threat to the organism, particularly when it concerns highly replicative tissues and long-lived species.
Complex adaptive systems have evolved to reduce the burden of somatic mutation. Within the hematopoietic system, which produces billions of cells each day, long-term self-renewal capacity is imparted only to hematopoietic stem cells (HSCs). The necessary exponential burst of proliferative capacity required to maintain tissue output is restricted to progenitor cells that are short-lived. However, despite protective DNA repair mechanisms and a reduced replicative burden, HSCs also accumulate mutations over their longer lifespan, which get immortalized in subsequent generations of daughter HSCs. The impact of such mutations within the HSC pool is challenging to characterize due to the rarity of the population, and the requirement for single-cell resolution. Indeed, much of our understanding of the consequences of somatic mutations on hematological disease development comes from studying hematopoietic cancers, where mutations in the tumor cell of origin are captured by virtue of the clonal expansion that subsequently occurs.
Recent decades have witnessed genetic characterization of tumors at an unprecedented scale, resulting in an almost complete compendium of somatic mutations that drive human cancers.3 However, 2 major challenges have become evident. First, cancer drivers are increasingly being identified in normal tissues, both solid organs4,5 and blood, from healthy individuals.6-9 This finding substantially blurs the distinction between normal physiology and the diseased state, and raises the possibility that mutation acquisition per se may not be the rate-limiting step for disease development. Second, intratumor heterogeneity and subclonal evolution within tumors provide a major route to therapeutic resistance.10 Genomic approaches applied to bulk tumor samples can estimate diversity within a tumor and, in some cases, the timing of genetic events.11 However, a detailed understanding of the factors driving mutation acquisition, clonal selection, and tumor evolution requires assessment of both normal, premalignant, and malignant hematopoietic cell populations at the level of individual cells.
In this review, we discuss a selection of clonal approaches and how they have provided valuable insights into both normal hematopoiesis and the development of hematological disorders. The review is biased toward the myeloid malignancies, to which clonal approaches have predominantly been applied to date. To avoid semantic confusion, we refer to a clone as comprising cells that descend from a common ancestor and share heritable genetic features. We use “lineage” to refer to a cell’s ancestral line, as opposed to the various differentiated hematopoietic cell types. “Mutation” is used in its broadest sense, and includes somatically acquired DNA single base-pair substitutions, insertions and deletions, copy-number changes, and other chromosomal rearrangements.
Clonal approaches
Interrogation of tumors at the clonal level was pioneered in blood and has a rich history spanning over 50 years (Figure 1). The identification of the Philadelphia chromosome as a clonal abnormality in cells from patients with chronic myeloid leukemia was seminal in implicating the first specific genetic mutation as a cause of cancer.12 Subsequent cytogenetic techniques, such as Giemsa banding of metaphase chromosomes and fluorescence in situ hybridization, identified a range of chromosomal lesions present in many leukemias and lymphomas.13

Timeline showing a selection of hematopoietic clonal approaches. Clonal approaches illustrated include chromosome characterization, hematopoietic colony assays, transplantation studies, and sequencing-based techniques. Major milestones in the development of these approaches are shown in the timeline.15,19,20,23,38,40,41,106-110 G&T-seq, genome and transcriptome sequencing; Trio-seq, single-cell triple omics sequencing.
Timeline showing a selection of hematopoietic clonal approaches. Clonal approaches illustrated include chromosome characterization, hematopoietic colony assays, transplantation studies, and sequencing-based techniques. Major milestones in the development of these approaches are shown in the timeline.15,19,20,23,38,40,41,106-110 G&T-seq, genome and transcriptome sequencing; Trio-seq, single-cell triple omics sequencing.
Studies of X-chromosome inactivation patterns have been another cornerstone of our understanding of the clonal origin of hematopoietic neoplasms. Expression studies of X-linked glucose-6-phosphate dehydrogenase (G6PD) in female patients genetically heterozygous for the G6PD locus were first used to study cancer in patients with leiomyomas. This work identified that tumor cells expressed only 1 G6PD allele, suggesting their unicellular, or clonal, origin.14 Similar findings were made in female patients with lymphoma15 and chronic myeloid leukemia,16 and in the latter, the presence of monoallelic expression of G6PD across the different differentiated hematopoietic cell types further suggested that the tumor arose from a multipotent stem cell.16 Such studies in polycythemia vera, a disease not previously recognized as being neoplastic, also established it as a clonal disorder arising in a multipotent progenitor or stem cell.17 Indeed, the recognized skewing of X-inactivation patterns often found in blood from elderly women paved the way for the subsequent discovery of age-related clonal hematopoiesis.6
Clonogenic assays involving the in vitro expansion of single myeloid progenitors were developed around the same time,18 and helped define the hematopoietic cellular hierarchy we are familiar with today. Diluted bone marrow or peripheral blood–derived mononuclear cells plated in semisolid media and cultured in the presence of colony-stimulating growth factors, result in the growth of distinct individual “colonies.” Each colony comprises a cluster of differentiated cells derived from a single progenitor cell. Using this approach, many myeloid diseases were dissected at the colony level in the 1970s. More recently, colonies of cells can also be grown in liquid culture within individual wells seeded with single cells of interest. The ability to isolate specific hematopoietic cell populations using flow cytometry, and the availability of an array of in vitro culture conditions, has since allowed the growth of colonies from specific starting hematopoietic populations and finer resolution of genotype-phenotype relationships. These colony-based approaches overcome many of the challenges of single-cell experiments by generating a larger amount of clonal material. However, they do restrict assessment to those diseases in which the abnormal cells are able to grow in vitro. For example, acute leukemia blasts have been much more challenging to expand in such conditions than progenitors from the myeloproliferative neoplasms.
The growth of clones in vivo rather than in vitro, using transplantation approaches in mice, offers the potential to assess the cellular consequences of mutations on disease development. Growth of splenocyte colonies in the 1960s using donor cells traceable in recipient mice identified for the first time that different myeloid cell types are derived from a common multipotent hematopoietic progenitor cell.19 Technical advances enabling single-cell transplantation assays are able to assess stem cell fitness and engraftment potential of individual cells harboring specific mutations, albeit in the environmental context of an irradiated recipient.20 Clones derived from human HSCs can also be characterized following xenotransplantation into mice21 using endogenous markers (eg, specific genetic mutations or rearrangements), or by introducing markers22 (eg, lentiviral vectors,23 genetic barcodes24 ). More recently, clustered regularly interspaced short palindromic repeats (CRISPR) scratchpads have been used for single-cell clonal tracing in zebrafish to dissect the embryonic relationships between adult cell types.25
Single-nucleotide polymorphism arrays and, subsequently, next-generation sequencing techniques have revolutionized our ability to interrogate and characterize the genomes of tumors. Sequencing can cover the whole exome, whole genome, target specific genetic regions of interest, and characterize chromosomal copy-number changes within blood samples. Such sequencing approaches have identified subclonal copy-number alterations26-28 and somatic mutations7,8,29,30 at increasing frequency with age in the blood of healthy individuals. Initially termed clonal mosaicism,26,27,31 this is now recognized as the presence of age-related clonal hematopoiesis that is distinct from clinically apparent hematological neoplasms.32 Although studies using increasing sensitivity and error-corrected sequencing have identified much higher rates of clonal hematopoiesis,33,34 the true incidence of mutant HSCs within the marrow and how this is influenced by age currently remain unknown.
Over the past few years, molecular characterization of single cells has provided a powerful approach to assessing clonal evolution and clonal diversity across a range of hematological disorders.35-37 Single-cell whole-exome sequencing can be performed but the data suffer from both false-negative and false-positive mutations due to the paucity of genomic material and requirement for DNA amplification.38,39 Given the recognition of molecular and cellular heterogeneity in tissues, combining information from both the genome and epigenome within the same cell is an area of intense technical development. Genomic and transcriptomic information can now be assessed in single cells (genome and transcriptome sequencing),40 and simultaneous measurement of genomic copy-number, DNA methylation, and RNA content in single cells (single-cell triple omics sequencing) is also feasible.41 Due to technical limitations, these methods have not yet been applied widely. However, such endeavors hold significant promise as it is likely they will ultimately allow us to understand the functional consequences of mutations in the context of epigenetic and cellular heterogeneity.
Clonal approaches have provided critical insights into both normal and malignant hematopoiesis. Here, we review how such approaches have contributed to our understanding of driver mutation acquisition, clonal expansion, the role of cooperating mutations, and the dynamics of tumor evolution.
Somatic mutations in HSCs
Somatic mutations in cells are acquired in several ways. Cell-extrinsic mutagens include chemicals (such as tobacco, aflatoxin, etc), ionizing radiation, and UV light. Additionally, DNA is damaged via cell-endogenous exposures to reactive oxygen species, inadequate function of DNA repair enzymes, abnormal activity of DNA-editing enzymes, and activity of viruses and retrotransposons.1 Interestingly, hematopoietic tumors and some pediatric brain tumors carry the smallest number of somatic mutations across all human cancers,42 suggesting that, compared with many other tissues, HSCs are relatively well protected from this mutagenic onslaught. One key study performed whole-exome sequencing on hematopoietic colonies grown from cord blood or from individuals of varying ages, and identified a linear increase in mutation burden with age.43 The number of somatic mutations found in acute myeloid leukemia (AML) in their study was close to that expected in a normal individual of the same age. These data provided 2 important insights: first, that most mutations detected in bulk AML samples represent those that have accumulated with age and were present prior to malignant transformation; and second, that background somatic mutation acquisition in HSCs can be viewed as a molecular clock, with the total number of mutations present reflecting the age of the individual. Whole-genome sequencing of colonies from normal bone marrow has identified that a large component of these background mutations involve cytosine to thymine transitions in a cytosine guanine dinucleotide context, which is the result of the time-dependent tendency toward spontaneous deamination of 5-methylcytosines over life.44 In addition, there is an excess of nucleotide transitions over transversions, typical of polymerase errors during DNA replication. Some myeloid tumors show evidence of additional mutational processes, for example, mutations driven by chemotherapeutic agents.10 Such DNA-damaging processes have been found to leave distinct “mutational signatures” in the DNA,1,45 and the mechanisms of DNA damage for some mutational processes have been further clarified using mutagenesis assays in cell lines and model organisms.46-48 Future studies of these signatures at a clonal level are likely to identify intrinsic and extrinsic mechanisms that contribute to mutation acquisition and reveal how they vary during development, with aging, by microenvironmental context, and across different hematopoietic cell types.
Driver mutation acquisition
Driver mutations are a tiny subset of somatic mutations that are found recurrently in tumors at specific genomic loci at a much higher frequency than would be expected by chance. In recent years, sequencing studies of tissues have also identified the presence of driver mutations in healthy skin, esophagus, colon, endometrium and blood.4,5,7,8,29,39,49,50 The overwhelming majority of driver mutations identified to date affect protein-coding regions of the genome, with just a few reported in noncoding regions. However, most sequencing studies of cancers have focused on tumor exomes, thus, leading to a discovery bias. From a smaller number of whole-genome studies, noncoding driver mutations have been discovered in cancers including hematological neoplasms. These include mutations in regulatory regions affecting the expression of NOTCH1 and PAX5 in chronic lymphocytic leukemia51 and LMO2 in T-cell acute lymphoblastic leukemia.52 Driver mutations perturb several molecular and cellular pathways53 and their identification has improved classification of hematological disease.54,55 However, little is known about when driver mutations actually occur over the lifetime of an individual. Do driver mutations occur early in life but confer no clonal advantage until other cell-intrinsic or environmental changes result in realization of their oncogenic potential or an enhanced cellular fitness compared with the normal stem cell pool? Alternatively, is chance acquisition of the first driver mutation the rate-limiting step in driving the development of clonal expansions and subsequent malignancy?
The size of the cell population that is the target for acquisition of driver mutations, as well as the mutation rate in this pool of cells, are both central to estimating the rate at which driver mutations may be acquired. Lee-Six et al provide us with some estimates relating to HSC dynamics in steady-state human hematopoiesis.44 In this study, individual bone marrow–derived colonies from a healthy 59-year-old man underwent whole-genome sequencing, and the pattern of shared and unique somatic mutations in individual colonies was then used to derive a phylogenetic tree depicting the ancestral relationship between all of the individual colonies. Using methods adopted from population genetics, the size of the HSC pool was shown to expand during childhood and adolescence and then remain fairly stable over subsequent adult life. The total number of adult HSCs was estimated to be much larger than previously thought (between 44 000 and 215 000), with each HSC dividing every 2 to 20 months and acquiring 1 to 2 somatic mutations per cell division.44
Using these figures, one can begin to make rough estimates of the chance of randomly acquiring a driver mutation by 60 years of age. As shown in Figure 2, we estimate that there is a 0.5% to 2.4% chance of acquiring 1 driver mutation (single-nucleotide substitution) within the entire HSC pool by 60 years of age. This is of course oversimplified because it restricts the tumor cell of origin to only the HSC pool, and it accounts for neither variability in mutation rates across different genomic regions nor tissue-specific mutational processes that have been demonstrated to play a role in acquiring driver mutations.56 Nevertheless, this rough estimate of HSC driver mutation acquisition can be compared with the reported prevalence of driver mutations in blood from healthy individuals. For example, there may be some driver mutations that confer a strong enough selective advantage that their reported frequency in the general population might largely reflect their rate of acquisition in HSCs. Such drivers might include JAK2V617F or DNMT3AR882H, the frequency of which appears to increase linearly with age, both reaching an incidence of 2% to 3% in 80- to 89-year olds.29 In another study of ∼50 000 normal adults (median age, 56 years), the frequency of JAK2V617F was 0.1% with the majority of patients subsequently developing evidence of a myeloproliferative neoplasm (MPN).57 These figures are consistent with estimates of the number and mutation rate of human HSCs (Figure 2). However, age-related clonal hematopoiesis can result from several other driver mutations, including loss-of-function mutations occurring across the entire length of the genes such as TET2, ASXL1, and DNMT3A.7-9,29 Considering this, the reported prevalence of only ∼5% for age-related clonal hematopoiesis (defined by a variant allele fraction >2% in blood) by 60 years of age across these studies appears somewhat lower than expected. Notwithstanding the observation that studies utilizing greater depth or error correction of sequencing reveal higher rates of clonal hematopoiesis, additional factors may also influence clonal expansion.

Estimating the chance of acquiring a specific driver mutation within any single HSC by 60 years of age. Calculations use estimates provided by Lee-Six et al44 for the total HSC population size (between 44 000 and 215 000 cells) and the rate of mutation acquisition in individual HSCs (estimated to be 1 mutation every 3 weeks, or 17 mutations per year). The total number of base pairs in the human genome is approximated at 3 billion.
Estimating the chance of acquiring a specific driver mutation within any single HSC by 60 years of age. Calculations use estimates provided by Lee-Six et al44 for the total HSC population size (between 44 000 and 215 000 cells) and the rate of mutation acquisition in individual HSCs (estimated to be 1 mutation every 3 weeks, or 17 mutations per year). The total number of base pairs in the human genome is approximated at 3 billion.
Clonal selection and expansion
Assuming a single somatic driver mutation has been acquired, it then becomes important to understand whether it is sufficient to result in clonal expansion and overt hematological disease and, if not, what additional permissive factors may be required.
Whole-exome and targeted sequencing studies show that a significant proportion of patients with hematological neoplasms harbor only a single driver mutation. For example, MPNs are often found only to harbor mutated JAK2, CALR, or MPL.58 Similarly, the BCR-ABL translocation or MLL rearrangement are commonly found as isolated genetic lesions in patients with chronic myeloid leukemia59,60 and infant acute lymphoblastic leukemia,61,62 respectively. This suggests that single mutations can drive disease phenotype in some hematological neoplasms.
The MPNs, in particular, provide a powerful model for understanding how a single mutation results in a clinical phenotype, due to their disease chronicity and the ready growth of hematopoietic colonies which allow intrapatient comparisons with unmutated control colonies. Such approaches in MPNs have shown, inter alia, that: (i) JAK2V617F drives the growth of erythropoietin-independent burst forming unit–erythroid colonies from MPN patients,63,64 (ii) JAK2V617F has distinct signaling and transcriptional consequences in patients with essential thrombocythemia compared with those with polycythemia vera,65 (iii) clones harboring homozygosity for JAK2V617F are a pathognomic feature of polycythemia vera highlighting the importance of gene dosage in determining phenotype,66,67 and (iv) the order of acquisition of driver mutations also impacts their functional consequences as discussed in “Cooperating somatic mutations.”68,69
In vivo murine transplantation assays have been used to address whether JAK2V617F is sufficient to drive clonal outgrowth. Using limiting-dilution transplants of JAK2V617F- positive murine cells, Lundberg et al70 have shown that an MPN phenotype (either erythrocytosis or thrombocytosis) was only infrequently initiated by a single JAK2-mutant HSC.70 Consistent with this, some other murine models of JAK2V617F have shown that mutant HSCs do not have an enhanced ability to outgrow when compared with normal HSCs (reviewed in Li et al71 ), suggesting that additional factors may be required to promote clonal expansion at the level of mutant stem cells. However, it is worth bearing in mind that in these studies, HSC advantage is measured by the ability of mutant cells to outcompete normal competitors following transplantation into irradiated mice. Such a context may not reflect the in vivo clonal competition occurring in humans that acquire driver mutations. An additional complication comes from observations of heterogeneity in cell-fate potential within the HSC/progenitor compartment in mice,72,73 raising the possibility that differences in the HSC cell of origin may affect the ability of a driver mutation to confer an advantageous cellular phenotype. Future studies using somatic mutations to track clonal dynamics in vivo74,75 will be better able to address whether driver mutations are sufficient, or require additional factors, for clonal outgrowth.
Several lines of evidence provide clues as to the nature of additional factors that may aid positive selection of cells carrying a driver mutation (Figure 3). Clonal hematopoiesis increases exponentially with advancing age in keeping with a role for aging in promoting clonal expansion.7-9,29 The effect of age may operate at a cell-intrinsic level, and it is recognized that HSCs undergo a range of age-related biochemical and functional changes thought to result in reduced stem cell fitness.76 Such alterations could favor HSCs with mutations that enhance aspects of stem cell fitness such as self-renewal, as shown for mutations in DNMT3A77 and TET2.78,79 Aging may also operate at a cell-extrinsic level, with substantial changes known to occur in the bone marrow microenvironment, including increased inflammatory signaling and reactive oxygen species.80
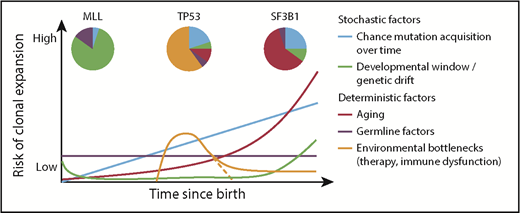
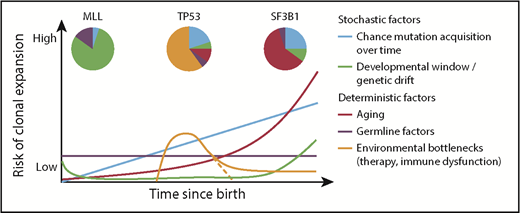
Factors influencing hematopoietic clonal expansion. A model of various stochastic and deterministic factors that may affect the likelihood of clonal expansion of a cell that has acquired a driver mutation. Combinations of risks at a given time point would additively affect the total risk. The impact of each factor, and thus the shape and angle of the depicted lines, would vary depending on the specific driver mutation. Pie charts show potential differences in the contribution of the various factors to clonal expansion in the context of different driver mutations.29,61,62,82
Factors influencing hematopoietic clonal expansion. A model of various stochastic and deterministic factors that may affect the likelihood of clonal expansion of a cell that has acquired a driver mutation. Combinations of risks at a given time point would additively affect the total risk. The impact of each factor, and thus the shape and angle of the depicted lines, would vary depending on the specific driver mutation. Pie charts show potential differences in the contribution of the various factors to clonal expansion in the context of different driver mutations.29,61,62,82
Other environmental factors are recognized to play a significant role in promoting clonal selection. Mutations in TP53 and PPM1D, thought to confer resistance to DNA damage, are prevalent in clonal expansions arising after chemotherapy.81 In patients with therapy-related myelodysplastic syndrome or therapy-related AML, Wong et al have shown that TP53-mutated clones were present at low levels prior to any exposure to genotoxic agents,82 and tracking of transplanted TP53 mutant cells in recipient mice demonstrated that the mutant HSCs only had a clonal advantage in the context of chemotherapy. Environmental selection also operates in aplastic anemia and paroxysmal nocturnal hemoglobinuria which are believed to result from immunological or complement-mediated destruction of normal hematopoietic cells.81,83 This situation is thought to select for genetic alterations that allow immune escape, including the expansion of clones with loss of heterozygosity for chromosome 6p (involving the HLA locus) and/or mutations in PIG-A.84 Complex cross talk between mutant clones and an inflammatory environment is increasingly being recognized.85 Proinflammatory cytokine secretion by TET-mutant clones has been shown to modify the environment to enhance clonal expansion.86,87 In addition, cytokine secretion by circulating mutant leukocytes has been shown to result in nonhematopoietic effects, such as increased atherosclerosis and cardiovascular disease.88,89
The genetic background of an individual is also likely to influence the response to a given driver mutation. For example, multiple single-nucleotide polymorphisms predispose to the development of an MPN.90 Moreover, in a recent study of clonal hematopoiesis driven by chromosomal alterations, several germline loci were found to be associated not only with the acquisition of specific chromosomal alterations, but also with the extent of subsequent clonal expansion.91 Lastly, it is important to remember that clonal expansion may also be the end product of stochastically acting neutral genetic drift.30,92
Cooperating somatic mutations and tumor evolution
Despite the observation that certain hematological malignancies appear to be driven by single-gene mutations, the vast majority of patients with hematological cancers have additional mutations. Furthermore, the absolute risk of developing overt malignancy in the context of age-related clonal hematopoiesis remains relatively low (0.5% to 1% per year),8,9,93 demonstrating that these drivers are usually insufficient for disease progression. Armitage and Doll hypothesized that cancer requires the successive and stepwise accumulation of multiple mutations94 to account for the age incidence of cancer (Figure 4), and subsequent investigators have suggested that varying numbers of rate-limiting events are required in different tissues, from the Knudson 2-hit model for the development of retinoblastoma,95 to at least 4 to 5 stages in colorectal cancers.96 In AML, multiple lines of evidence suggest that at least 2 driver mutations are required for disease development.97 The dynamics of hematological disease development remain unclear and various models can be envisaged that might differ between them in the number of events, mutations or otherwise, required for disease development, and the rate of clonal expansion (Figure 4). In addition to the hypothesized number of lesions required to drive disease onset, sequencing of established hematological cancers has identified substantial intratumor subclonal genomic heterogeneity confirming ongoing acquisition of somatic mutations during tumor evolution. Clonal approaches allow assessment of how different mutations cooperate within cells to promote disease phenotypes, and more precise analyses of tumor evolutionary patterns.
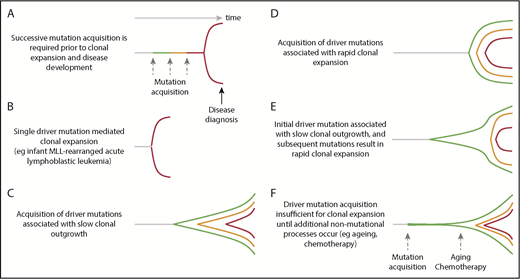
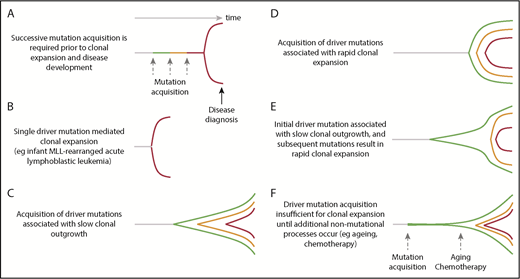
Dynamics of clonal evolution during the development of hematological malignancies. Little is known about the dynamics of clonal evolution following successive mutation acquisition. Six different models are presented (A-F). The horizontal axis is time and the vertical space represents clonal expansion. Green, orange, and red lines represent sequential driver mutation acquisition, with red representing the final malignant clone. The Armitage and Doll multistep model of cancer development,94 in which there is no premalignant clonal expansion, is shown in panel A. Rare, single-driver–mediated malignant transformation is shown in panel B. In models (panels C-F), each mutation results in sequential clonal expansion along a path to malignancy. However, the dynamics of clonal expansion show different patterns. MLL, mixed lineage leukemia.
Dynamics of clonal evolution during the development of hematological malignancies. Little is known about the dynamics of clonal evolution following successive mutation acquisition. Six different models are presented (A-F). The horizontal axis is time and the vertical space represents clonal expansion. Green, orange, and red lines represent sequential driver mutation acquisition, with red representing the final malignant clone. The Armitage and Doll multistep model of cancer development,94 in which there is no premalignant clonal expansion, is shown in panel A. Rare, single-driver–mediated malignant transformation is shown in panel B. In models (panels C-F), each mutation results in sequential clonal expansion along a path to malignancy. However, the dynamics of clonal expansion show different patterns. MLL, mixed lineage leukemia.
Cooperating somatic mutations
Using limiting-dilution xenograft transplantation of peripheral blood–derived HSCs from patients with DNMT3A and NPM1c-mutated AML, Schlush et al identified that the majority of transplanted mice that showed normal multilineage engraftment were positive only for the DNMT3A mutation.98 In contrast, mice that regenerated hematopoiesis with a dominant myeloid blast population harbored the additional NPM1c mutation. This study provided 2 key insights. First, that AML could result from the sequential acquisition of 2 mutations, with the first mutation conferring a clonal advantage and the second mutation triggering a malignant phenotype. Second, that cells carrying only a DNMT3A mutation retained multilineage potential, providing evidence for the existence of a “preleukemic” multipotent stem cell. Similarly, in MPNs, Triviai et al have shown that mutations in the chromatin modifiers ASXL1 and EZH2 have a synergistic effect on levels of HSC engraftment in mice.99
Studies of colonies have shown that the order in which driver mutations are acquired can affect the behavior of stem/progenitor cells and thus influence clonal evolution, clinical presentation, and response to targeted therapy.68,69 Using MPN samples harboring mutations in both JAK2 and TET2, Ortmann et al analyzed colonies to determine which mutation had been acquired first. JAK2-first and TET2-first HSCs and progenitors differed in their functional properties with, for example, single JAK2-first HSCs, giving rise to more proliferative progeny compared with TET2-first HSCs. JAK2-first patients were shown to present with disease at a younger age, most commonly with polycythemia vera, and had an HSC/progenitor compartment composed mostly of double-mutant JAK2 and TET2 cells. In contrast, TET2-first patients presented at an older age, more commonly with essential thrombocythemia, and had a hematopoietic stem and progenitor cell compartment dominated by single-mutant TET2-mutated cells. Outcomes and response to targeted therapy also differed significantly between the 2 cohorts.68 These data revealed the importance of mutation order for the first time in any cancer and suggested that mutation of TET2 alters the epigenetic landscape and thus modifies the transcriptional response to a subsequent JAK2 mutation.
Tumor evolutionary dynamics
From bulk tumor samples, differences in the variant allele frequencies of mutations can be used to infer the genetic subclonal composition and phylogenetic relationships between tumor subclones.100 However, such studies have only limited resolution, as one is restricted to analysis of only a subset of samples wherein the spectrum of variant allele fractions is both sufficiently large and broadly distributed. Genomic characterization of either single cells or colonies provides a “gold standard” approach to construction of accurate tumor phylogenies, and has revealed previously hidden layers of clonal complexity in a range of hematological malignancies.101,102 For example, in acute lymphoblastic leukemia, targeted genomic sequencing of single cells showed that structural rearrangements are acquired prior to other driver mutations, and that KRAS mutations are late events in tumor evolution.36 Furthermore, previously hidden patterns of tumor evolution have been identified. Characterization of the breakpoints of uniparental disomy in JAK2V617F-mutated colonies has shown that most patients with MPN harbor multiple independent acquisitions of JAK2V617F homozygosity,66 and that independently acquired mutant clones, which may also arise from distinct HSCs, have also been shown to coexist in some individuals with MPN.69,103,104 Similarly, single-cell polymerase chain reaction analysis of the immunoglobulin heavy chain locus in chronic lymphocytic leukemia has identified the coexistence of multiple independent clones in lymphoid malignancies.105 Such parallel evolution suggests strong selection for specific genomic events during tumor evolution.
Future directions
Studies of normal and malignant hematopoiesis have pioneered the use of a range of clonal approaches. Through in vitro analysis of single-cell–derived colonies, direct single-cell characterization, and a variety of transplantation strategies, significant progress has been made in understanding the consequences of somatic mutations. Lineage-tracing studies using genetic markers combined with phylogenetic assessment has allowed us to estimate the number of human HSCs in the bone marrow. Such studies should be extended to studying clonal hematopoiesis and hematological malignancies to understand questions such as the timing of clonal expansion in individuals, the dynamics of clonal outgrowth, and the way in which different driver mutations exert a selective advantage within the stem cell pool. Technological advances in the fields of single-cell “omics” and gene editing will further enhance our ability to explore such areas. The mechanisms that control clonal expansion and evolution, and whether these can be prevented, remain exciting questions for the future.
Acknowledgments
The authors thank Peter Campbell for his valuable comments on the manuscript.
E.M. was supported by the Wellcome Trust UK. J.N. was supported by a fellowship from Cancer Research UK and funding from the European Hematology Association. Work in the Green laboratory was supported by the Wellcome–MRC Stem Cell Institute, the Cancer Research UK–Cambridge Cancer Centre, the NIHR Cambridge Biomedical Research Centre, Cancer Research UK, the Wellcome Trust, Bloodwise, and the WBH Foundation.
Authorship
Contribution: J.N., E.M., and A.R.G. designed and cowrote the review.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Anthony R. Green, Cambridge Institute for Medical Research, Hills Rd, Cambridge CB2 0QQ, United Kingdom; e-mail: arg1000@cam.ac.uk.