Key Points
First comprehensive genomic and transcriptomic profiling of CNL, aCML, and MDS/MPN-U.
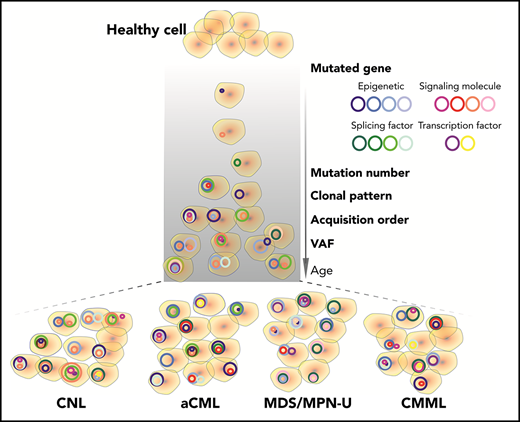
Diagnoses represent a continuum of related diseases rather than discrete diagnostic entities.

Abstract
Chronic neutrophilic leukemia (CNL), atypical chronic myeloid leukemia (aCML), and myelodysplastic/myeloproliferative neoplasms, unclassifiable (MDS/MPN-U) are a group of rare and heterogeneous myeloid disorders. There is strong morphologic resemblance among these distinct diagnostic entities as well as a lack of specific molecular markers and limited understanding of disease pathogenesis, which has made diagnosis challenging in certain cases. The treatment has remained empirical, resulting in dismal outcomes. We, therefore, performed whole-exome and RNA sequencing of these rare hematologic malignancies and present the most complete survey of the genomic landscape of these diseases to date. We observed a diversity of combinatorial mutational patterns that generally do not cluster within any one diagnosis. Gene expression analysis reveals enrichment, but not cosegregation, of clinical and genetic disease features with transcriptional clusters. In conclusion, these groups of diseases represent a continuum of related diseases rather than discrete diagnostic entities.
Introduction
Chronic neutrophilic leukemia (CNL), atypical chronic myeloid leukemia (aCML), and myelodysplastic/myeloproliferative neoplasms, unclassifiable (MDS/MPN-U) are a group of heterogeneous MPN or MDS/MPN overlap syndromes.1-5 Traditionally, these diseases have been diagnosed based on morphologic and exclusion criteria. However, some patients with MDS/MPN overlap syndromes fulfill the criteria for either multiple or none of the MDS/MPN entities. Furthermore, due to the lack of specific molecular markers and a limited understanding of the underlying pathogenic mechanisms, treatment of these diseases has largely remained empiric and ineffective. The outcomes of patients have remained dismal, with a median survival of 11 to 40 months.6
In recent years, next-generation sequencing techniques have identified recurrent mutations in ASXL1, TET2, SRSF2, SETBP1, and signaling pathway genes (eg, CSF3R in CNL) in patients with these diseases.2,7-16 However, many of these genetic mutations overlap with mutations seen in other myeloid malignancies, including MDS,17 acute myeloid leukemia (AML),18,19 MPN,20 chronic myelomonocytic leukemia (CMML),16,21 and/or juvenile myelomonocytic leukemia.22,23 In addition, the limited number of somatic mutations present in the patients does not easily explain the morphologic and clinical heterogeneity of CNL/aCML/MDS/MPN-U, and their rarity has limited deep sequencing. We, therefore, collected and analyzed a cohort of over 100 patients with these rare disorders using whole-exome sequencing (WES) and RNA sequencing (RNA-seq) to characterize the full genomic landscape of CNL/aCML/MDS/MPN-U. Importantly, we provide a comprehensive analysis of the co-occurrence and order of acquisition of gene mutations, as well as the association of different mutations with disease subtypes and clinical outcomes, which could facilitate improvements in diagnosis and treatment of patients with these diseases.
Materials and methods
Patients and samples
The study was approved by the institutional review boards of all participating institutions. Samples were obtained with written, informed consents obtained from all patients according to the Declaration of Helsinki.
Cell lines and reagents
HEK 293T/17 cells (provided by Richard Van Etten) were maintained in Dulbecco’s modified Eagle medium (Invitrogen) supplemented with 10% fetal bovine serum (Atlanta Biologicals), l-glutamine, penicillin/streptomycin (Invitrogen), and fungizone (Fisher). Ba/F3 cells (ATCC) were maintained in RPMI 1640 (Invitrogen) supplemented with 10% fetal bovine serum, 15% WEHI-conditioned media, l-glutamine, penicillin/streptomycin, and fungizone. Mycoplasma contamination was routinely tested (once per month). Only mycoplasma-free cells were used in the experiments.
WES and RNA-seq
Genomic DNA was extracted from cryopreserved bone marrow (BM) and/or peripheral blood mononuclear cells using Qiagen DNeasy columns. Library preparation and sequencing (using an Illumina HiSeq 2500) was performed by the Oregon Health & Science University Massively Parallel Sequencing Shared Resource. Detailed information is provided in the supplemental Materials and methods (available at the Blood Web site) information. For RNA-seq, libraries were constructed using the SureSelect stranded RNA-seq protocol (Agilent) on the Bravo robot (Agilent) and sequenced on the HiSeq 2500 using a 100-cycle paired-end protocol. All sequence was performed at 2 time points (November 2015 and April 2016). Detailed WES and gene expression analysis is provided in the supplemental Materials and methods.
Retroviral vector production and transduction
CSF3R and NRAS mutations were generated using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) on the respective pENTR vectors (GeneCopoeia GC-Z2134 and Invitrogen clone IOH11942) and cloned into a gateway compatible MSCV-IRES-puromycin and MSCV-IRES-GFP retroviral vector via Gateway Cloning System (Invitrogen). Retrovirus was produced by transfecting HEK 293T/17 cells together with an EcoPac helper plasmid. After 2 days, the virus-containing supernatants were filtered and infected to Ba/F3 cells followed by flow cytometry (fluorescence-activated cell sorter) selection and puromycin double selection.
Inhibitor assay
Transformed Ba/F3 cells were seeded in 384-well plates (1250 cells per well) and exposed to increasing concentrations of ruxolitinib (Selleck, #S1378), trametinib (Selleck, #S2673), or the combination of both drugs for 72 hours. Cell viability was measured using a methanethiosulfonate-based assay (CellTiter96 Aqueous One Solution; Promega) and read at 490 nm after 1 to 3 hours using a BioTek Synergy 2 plate reader (BioTek). Cell viability was determined by comparing the absorbance of drug-treated cells to that of untreated controls (4 replicates for each condition) set at 100%. Half-maximal inhibitory concentration (IC50) values were calculated by regression curve fit analysis using GraphPad Prism software.
Data availability
All sequence data are being deposited to dbGaP and Genomic Data Commons (phs001799). In addition, all data can be accessed and queried through our online, interactive user interface, Vizome, at www.vizome.org (this platform was first described by Tyner et al24 ).
Statistical analysis
Statistical analyses were performed on GraphPad Prism 8. The data were expressed as the mean ± standard error of the mean (SEM). Statistical significance was determined using 2-tailed nonparametric Student t tests (Mann-Whitney U test), 1-way analyses of variance (ANOVAs), χ2 tests, Fisher’s exact tests, or log-rank tests for survival curves as indicated. Clustering analyses and related figures were generated using R.
Results
The landscape of somatic mutations in CNL/aCML/unclassifiable (MPN-U or MDS/MPN-U)/CMML
We performed WES on 158 specimens from patients with a diagnosis of CNL (n = 39), aCML (n = 27), unclassifiable-MPN (n = 13), or -MDS/MPN (n = 12), CMML (n = 29), and unavailable or ambiguous diagnosis (n = 38).
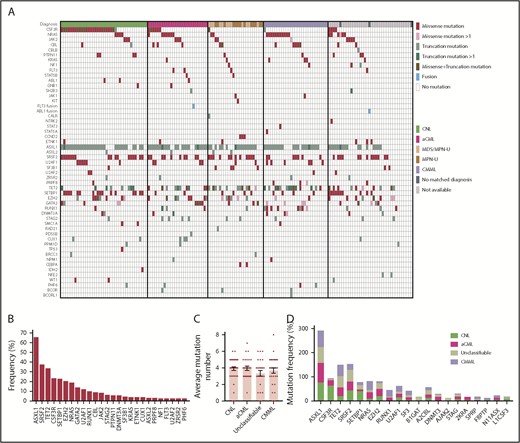
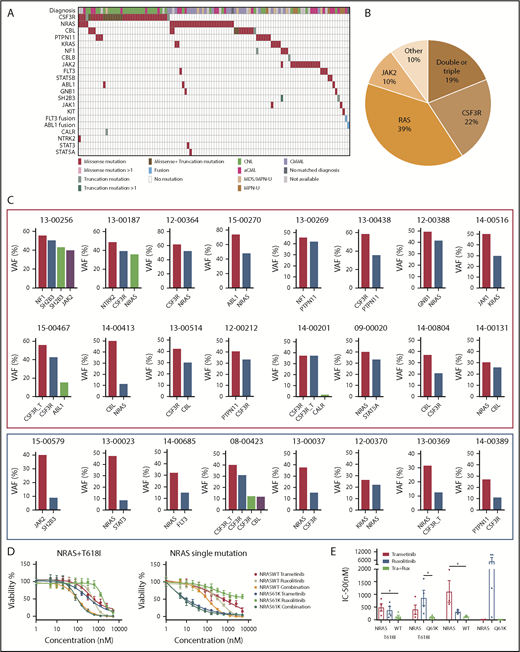
We observed recurrent mutations in known oncogenes and tumor suppressors that broadly fall into categories of genes involved in epigenetic regulation, signal transduction, the spliceosome complex, and transcription factors (Figure 1A-B; supplemental Table 1; supplemental Data 1). The most prevalently mutated gene pathway was chromatin modification (ASXL1, 65.8%; EZH2, 19%; and ASXL2, 3.2%) (Figure 1A-B; supplemental Figure 1). We also observed that 69.6% (110 out of 158) of cases harbored ≥1 signaling pathway driver mutations largely involving JAK/STAT and RAS signaling pathways (CSF3R, 23.4%; NRAS, 19%; and JAK2, 8.2%).7 Interestingly, MPL mutations (n = 0) and CALR indels (n = 1) were rare in our cohort, supportive of the distinct phenotype of these diseases from classical MPN.25 High frequencies of mutations in genes that modify DNA were also observed in our cohorts (TET2 , 33.5%; and DNMT3A, 5.7%), whereas mutations in other common epigenetic regulators, such as IDH1, IDH2, and WT1, were infrequent. Members of the spliceosome complex, including SRSF2, U2AF1, SF3B1, ZRSR2, and RPRF8, were mutated at high frequencies, with a total incidence of 55.7% (Figure 1A; supplemental Figure 2A-B). In addition to the most common hotspot mutations at codon P95 of SRSF2, we identified recurrent microdeletions of SRSF2 (10 out of 60) (supplemental Figure 2C), which were detected by a previous study.26 Interestingly, nearly all mutations in U2AF1 were observed at or near the less commonly mutated codon 157 hotspot and not at codon S34, which is the more commonly mutated U2AF1 hotspot in AML and MDS27 (supplemental Figure 2D). Mutations in SF3B1 were infrequent (4.4%) (supplemental Figure 2E). Frequent mutations were also observed in the transcription factors GATA2 (13.9%) and RUNX1 (10.1%), but not in CEBPA (1.3%). Consistent with previous studies,14,15,28 a high frequency of SETBP1 mutations (22.8%) were detected, whereas mutations in the cohesion factor family (STAG2, RAD21, SMC1A, SMC3, and PDS5B), tumor suppressors (TP53 and PHF6), and NPM1 were infrequent. Notably, high frequencies of ≥2 variants were observed in GATA2, TET2, EZH2, and CSF3R (supplemental Data 1).
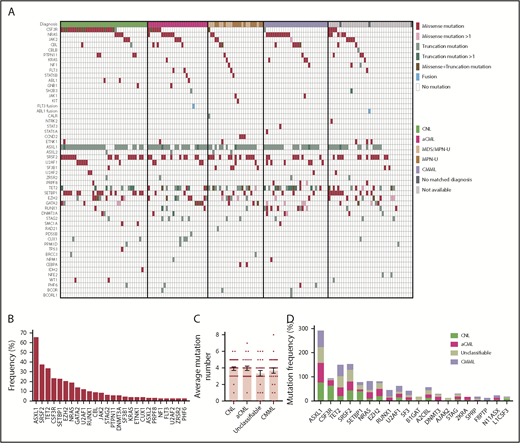
Genomic landscape of CNL/aCML/unclassifiable/CMML. (A) The mosaic plot depicts distributions of recurrent gene mutations in 158 patients. Each column displays each patient, and each row denotes a specific gene. Variant types are color coded as indicated. (B) The graph depicts frequencies of recurrent gene mutations in this cohort of CNL/aCML/unclassifiable/CMML patients. (C) The graph depicts the mean ± SEM of the number of mutations in different diagnostic groups. Statistical analysis was performed using a 1-way ANOVA. (D) The graph depicts the frequencies of recurrently mutated genes in different diagnostic groups. Statistical analysis was performed using a contingency table χ2 test followed by the Bonferroni correction and expressed as P < .0001.
Genomic landscape of CNL/aCML/unclassifiable/CMML. (A) The mosaic plot depicts distributions of recurrent gene mutations in 158 patients. Each column displays each patient, and each row denotes a specific gene. Variant types are color coded as indicated. (B) The graph depicts frequencies of recurrent gene mutations in this cohort of CNL/aCML/unclassifiable/CMML patients. (C) The graph depicts the mean ± SEM of the number of mutations in different diagnostic groups. Statistical analysis was performed using a 1-way ANOVA. (D) The graph depicts the frequencies of recurrently mutated genes in different diagnostic groups. Statistical analysis was performed using a contingency table χ2 test followed by the Bonferroni correction and expressed as P < .0001.
The median number of mutations per patient is 3.6 (range, 0-8), underscoring complex genetic pathogenesis. We did not observe differences in the average number of mutations per case among different diagnosis groups (Figure 1C). Consistent with previous studies,3,10 CSF3R and SETBP1 mutations are more frequent in CNL (Figure 1D; supplemental Table 2).
Co-occurrence and exclusivity analysis
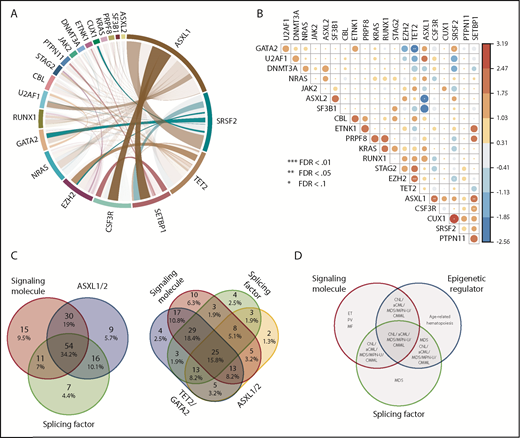
The majority of gene variants coexisted with ≥1 other gene mutation (Figures 1A and 2A). In addition to the previously observed co-occurrence of CSF3R and SETBP1 mutations,3 we found significant co-occurrence of ASXL1 with CSF3R, SETBP1 with U2AF1, CUT1 with SRSF2, and EZH2 with TET2, which have not previously been observed in other hematologic malignancies (Figure 2B). Conversely, mutual exclusivity of mutations in TET2 and GATA2 was observed, indicating mutations of these 2 genes may act redundantly or form a synthetic lethal combination.
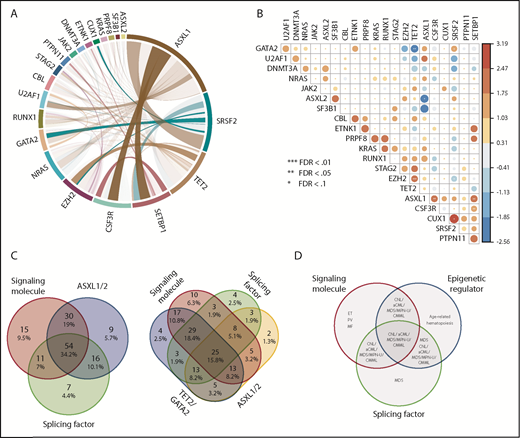
Mutations in signaling molecules, epigenetic regulators, and splicing factors are frequently co-occurring in CNL/aCML/unclassifiable/CMML. (A) The circos plot depicts the relative frequency and pairwise co-occurrence of molecular mutations. Outer segments indicate a particular subcohort being positive for the given gene mutations. Relative frequencies of pairwise co-occurrences are indicated by ribbon widths. The transparency of the lines is tied to their significance in panel B (no transparency indicates the co-occurrence is significant). (B) Exclusivity and co-occurrence between different gene mutations. The circles were sized and colored according to their log(odds) from a Fisher’s exact test (red color series represent co-occurrence and blue series represent exclusivity). (C) Venn diagrams depict distribution and co-occurrence frequencies of mutations of ASXL1/2, signaling molecules, and splicing factors (left panel) or mutations of ASXL1, signaling molecules, splicing factors, and TET2/GATA2 mutations (right panel). (D) Venn diagram showing potential association and distribution of different pathway mutations with different disease diagnosis. ET, essential thrombocythemia; MF, myelofibrosis; PV, polycythemia vera.
Mutations in signaling molecules, epigenetic regulators, and splicing factors are frequently co-occurring in CNL/aCML/unclassifiable/CMML. (A) The circos plot depicts the relative frequency and pairwise co-occurrence of molecular mutations. Outer segments indicate a particular subcohort being positive for the given gene mutations. Relative frequencies of pairwise co-occurrences are indicated by ribbon widths. The transparency of the lines is tied to their significance in panel B (no transparency indicates the co-occurrence is significant). (B) Exclusivity and co-occurrence between different gene mutations. The circles were sized and colored according to their log(odds) from a Fisher’s exact test (red color series represent co-occurrence and blue series represent exclusivity). (C) Venn diagrams depict distribution and co-occurrence frequencies of mutations of ASXL1/2, signaling molecules, and splicing factors (left panel) or mutations of ASXL1, signaling molecules, splicing factors, and TET2/GATA2 mutations (right panel). (D) Venn diagram showing potential association and distribution of different pathway mutations with different disease diagnosis. ET, essential thrombocythemia; MF, myelofibrosis; PV, polycythemia vera.
We further observed that the majority of patients exhibited mutations of ≥3 major pathways/processes; 34.2% of patients demonstrated mutation of 3 pathways and 15.8% of 4 pathways, which collectively involved ASXL1/2, TET2/GATA2, a signaling gene, and/or a splicing factor mutation (Figure 2C-D). There were ≥15 different combination patterns of these genes/pathways, indicating functional cooperation and heterogeneity of these pathway alterations in driving disease. The unique combination of pathway mutation patterns that we observed in this CNL/aCML/unclassifiable cohort is similar to CMML, whereas distinct from the patterns of combinatorial pathway mutations observed in age-related hematopoiesis or other cohorts of chronic myeloid malignancies as summarized in Figure 2D.
Clonal architecture of different pathway mutations
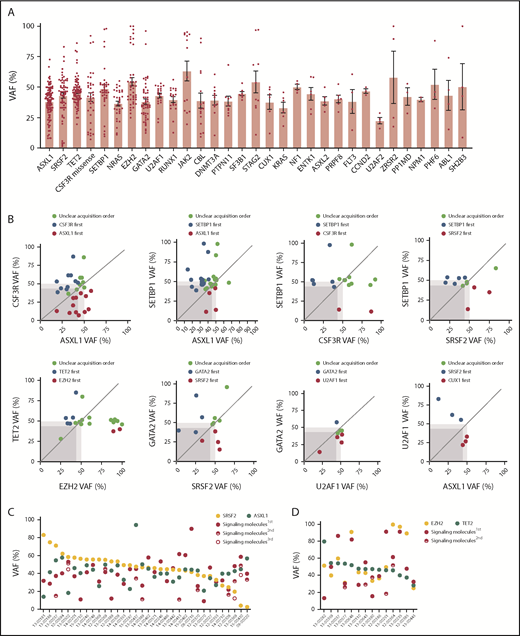
Next, we used the variant allele frequency (VAF) to study the acquisition order and clonal architecture of the cases with multiple pathway mutations. We observed that a majority of variants of EZH2, SETBP1, TET2, U2AF1, and SF3B1 had high VAFs slightly <50% (Figure 3A; supplemental Figure 3A). This is consistent with the presence of heterozygous mutations present in the majority of tumor cells and suggests that these mutations were acquired by an early founder clone. In contrast, mutations in ASXL1, SRSF2, CSF3R, CBL, and NRAS showed a wide range of VAFs, indicating that these gene mutations may occur in the founder clone in some cases and are acquired in later subclones in other cases. Of note, loss of heterozygosity was frequently observed with EZH2, JAK2, GATA2, and CSF3R missense mutations (supplemental Figure 3A). We further compared VAFs of recurrent gene mutations of different myeloid malignancies. We observed that EZH2 and SRSF2 mutations had higher VAFs in CNL/aCML/unclassifiable/CMML compared with AML (supplemental Figure 3B). In addition, a trend of increased VAFs of the signaling gene (NRAS, PTPN11, and NF1) mutations was observed in our cohort compared with the VAFs of signaling gene mutations in AML (supplemental Figure 3B),17,29 indicating that these signaling mutations may contribute to the unique phenotype of CNL/aCML/unclassifiable/CMML and may represent important pharmaceutical targets.30
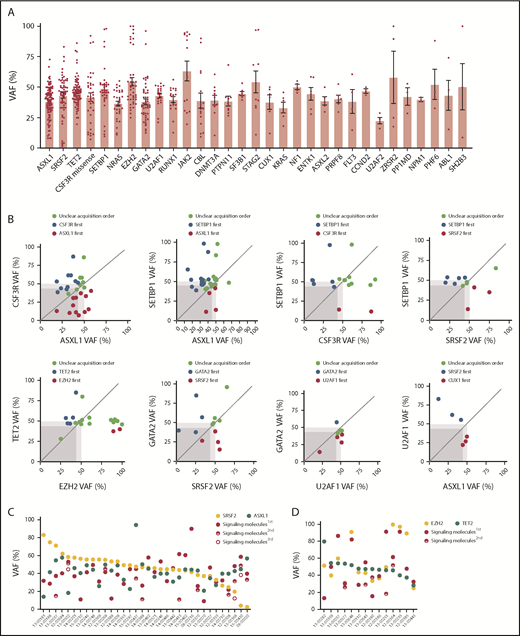
Clonal architecture of different pathway mutations. (A) Mean ± SEM of VAFs of common driver mutations in the cohort. (B) Dot plots depict VAFs of pairwise co-occurring mutations. Gene mutations with higher VAFs are considered to occur earlier then variants with lower VAFs. Dark gray square highlights 45% VAF. Dots appearing in the dark gray square are possibly acquired as subclones. Dot plot shows different mutation acquisition orders of SRSF2, ASXL1, and signaling molecules (C) and TET2, EZH2, and signaling molecules (D).
Clonal architecture of different pathway mutations. (A) Mean ± SEM of VAFs of common driver mutations in the cohort. (B) Dot plots depict VAFs of pairwise co-occurring mutations. Gene mutations with higher VAFs are considered to occur earlier then variants with lower VAFs. Dark gray square highlights 45% VAF. Dots appearing in the dark gray square are possibly acquired as subclones. Dot plot shows different mutation acquisition orders of SRSF2, ASXL1, and signaling molecules (C) and TET2, EZH2, and signaling molecules (D).
Since we observed multiple pairs of coexisting mutations (Figure 2B), we next determined the order of acquisition of these events and whether they were predicted to exist within the same tumor clones. We observed that U2AF1 mutations were mostly acquired earlier than GATA2 mutations and TET2 acquired earlier than EZH2 (Figure 3B). The other pairs (ASXL1/CSF3R, CSF3R/SETBP1, ASXL1/SETBP1, SETBP1/SRSF2, and CUT1/SRSF2) were observed in variable orders of acquisition, with 1 event appearing first in some cases and the other appearing first in others (Figure 3B). In addition, while we found evidence that CSF3R and ASXL1 may be present in distinct subclones in some cases, the other pairs were predicted to occur within the same clone, since ≥1 of the mutations was observed with a VAF close to 50%, representing the presence of this mutation in essentially all of the tumor cells. We next analyzed the clonal architecture pattern of patients harboring SRSF2, ASXL1, and signaling gene mutations. We observed that the majority of SRSF2 mutations were present in the predominant clone, whereas signaling genes and ASXL1 mutations were observed in the dominant clone in some cases and in a subclonal pattern in other cases (Figure 3C). Similar variability for the order of acquisition was also observed for cases with EZH2, TET2, and signaling gene mutations (Figure 3D). In addition, we observed that for the majority of cases, VAFs of 2 of the 3 pathway mutations was >50%, suggesting the presence of both of these mutations in the dominant clone. The third mutation was at a lower VAF, suggestive of subsequent acquisition of this mutation in a subclone harboring both original mutations. This finding suggests a linear evolution of a single, dominant clone rather than a branching pattern of mutational acquisition. However, a more sensitive and accurate sequencing method (eg, single-cell sequence, allele-specific polymerase chain reaction, or digital polymerase chain reaction) is needed to confirm the clonal architectural data.
Diverse signaling gene mutations are identified in CNL/aCML/unclassifiable/CMML
We next analyzed the mutation profile of patients harboring CSF3R, RAS pathway, and JAK2 mutations (supplemental Table 3). In line with previous studies, CSF3R mutations are more prevalent in CNL and RAS mutations are more frequently diagnosed with CMML. Furthermore, CSF3R mutation demonstrated a higher frequency of SETBP1 mutation co-occurrence, consistent with the previous studies,10,31,32 whereas RAS pathway mutations were more often seen concomitantly with RUNX1, GATA2, and STAG2 mutation. JAK2 mutations showed higher co-occurrence with TET2 and SF3B1 mutations. ASXL1 co-occurred with CSF3R and RAS pathway mutations more commonly than with JAK2 mutations.
Notably, we observed that 19% of patients harbor ≥2 different signaling pathway mutations (Figure 4A-B; supplemental Table 4). CSF3R was shown to co-occur with NRAS, CBL, PTPN11, SH2B3, NTRK2, and ABL1 mutations; NRAS mutations were present in co-occurrence with CSF3R, KRAS, JAK2, CBL, STAT3, NTRK2, FLT3, and GNB1 mutations. VAF analysis demonstrated that the majority of the multiple different signaling pathway mutations were present in the same clone (Figure 4C, red rectangle).
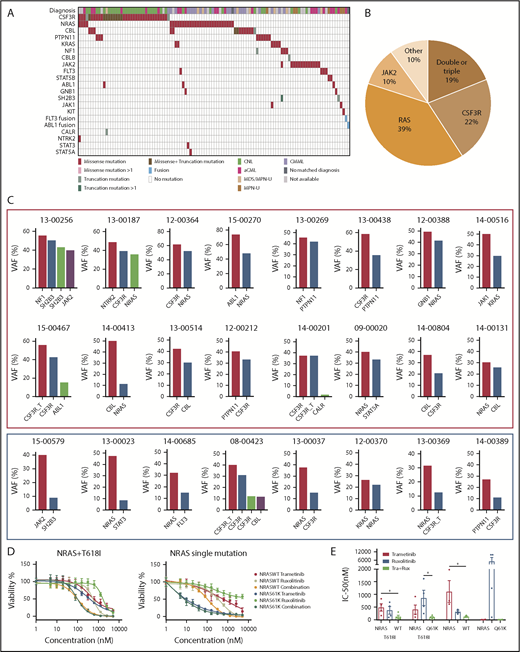
Diverse signaling molecule mutations are identified in CNL/aCML/unclassifiable/CMML. (A) The mosaic plot depicts the spectrum of different signaling molecule mutations in the cohort. (B) The pie chart depicts the frequencies of different signaling pathway mutations. (C) The graph depicts the VAFs of coexisting signaling pathway mutations showing co-occurrence pattern (red rectangle) and potential subclone pattern (blue rectangle). (D) Mean ± SEM of cell viability of Ba/F3 cells expressing CSF3R T618I with NRAS wild-type or mutant treated with gradient concentrations of indicated drugs for 72 hours (left). Mean ± SEM of cell viability of Ba/F3 cells expressing NRAS wild-type or mutant treated with a gradient concentration of indicated drugs for 72 hours (right). (E) Mean ± SEM of drug IC50 of Ba/F3 cells expressing single or compound mutation treated with indicated drugs. Statistical significance was assessed using 1-way ANOVA and Kruskal-Wallis tests (*P < .05; **P < .01).
Diverse signaling molecule mutations are identified in CNL/aCML/unclassifiable/CMML. (A) The mosaic plot depicts the spectrum of different signaling molecule mutations in the cohort. (B) The pie chart depicts the frequencies of different signaling pathway mutations. (C) The graph depicts the VAFs of coexisting signaling pathway mutations showing co-occurrence pattern (red rectangle) and potential subclone pattern (blue rectangle). (D) Mean ± SEM of cell viability of Ba/F3 cells expressing CSF3R T618I with NRAS wild-type or mutant treated with gradient concentrations of indicated drugs for 72 hours (left). Mean ± SEM of cell viability of Ba/F3 cells expressing NRAS wild-type or mutant treated with a gradient concentration of indicated drugs for 72 hours (right). (E) Mean ± SEM of drug IC50 of Ba/F3 cells expressing single or compound mutation treated with indicated drugs. Statistical significance was assessed using 1-way ANOVA and Kruskal-Wallis tests (*P < .05; **P < .01).
Since CSF3R membrane proximal mutation has been shown to be sensitive to treatment with the JAK inhibitor ruxolitinib from ex vivo drug assay results, in vivo mouse modeling, and clinical case reports.10,33-35 We further performed drug sensitivity assays of cells transformed by coexisting CSF3R and NRAS mutations. We observed reduced drug sensitivity (increased IC50) to either ruxolitinib or a MEK inhibitor, trametinib, which was rescued with the drug combination targeting both mutated pathways (Figure 4D-E).
Somatic alterations implicate diagnosis and predict the outcome
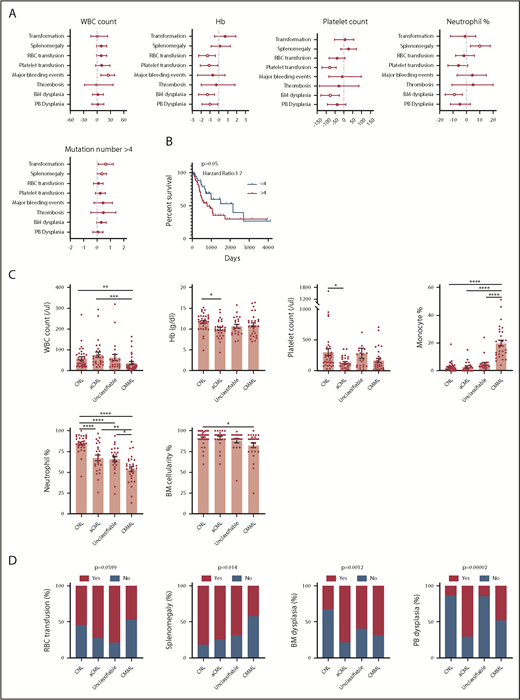
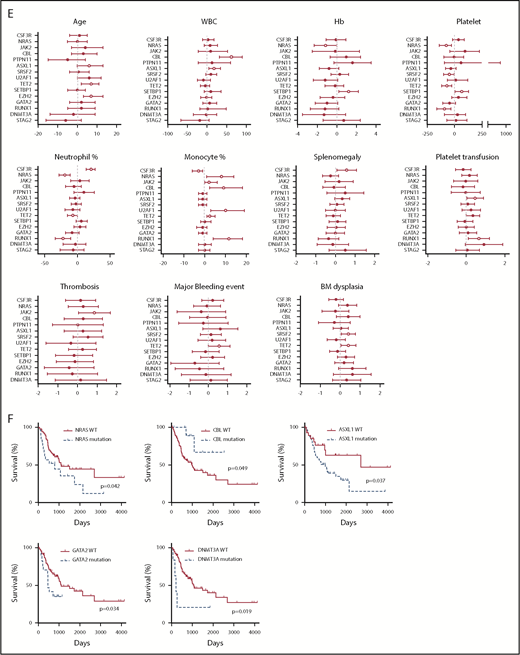
We next analyzed the correlation between clinical parameters and clinical outcomes. Interestingly, a high white blood cell (WBC) count is associated with more major bleeding events. Consistent with previous studies, low hemoglobin (Hb) is associated with more red blood cell transfusion, whereas low Hb level and low platelet count are associated with more platelet transfusion (Figure 5A). A high neutrophil percentage is associated with splenomegaly (Figure 5A). A higher mutation number (≥4) was associated with leukemia transformation and shorter survival (Figure 5A-B). No association was found for age and gender (supplemental Figure 4A-B). The median age at diagnosis was 68 (27-90) years. There was no age difference between different diagnostic groups (supplemental Figure 4C). Consistent with previous studies, there was a male predominance (1.6-fold of the female for the whole cohort), especially for aCML patients (3.7-fold of the female) (supplemental Figure 4D). We also observed that CNL demonstrated high WBC, high neutrophil percentage, high BM cellularity, and more splenomegaly cases, whereas CMML was associated with a low WBC count, high monocyte count, low BM cellularity, and a lower rate of splenomegaly (Figure 5C-D). aCML and unclassifiable were associated with more red blood cell transfusion dependence (Figure 5D). Moreover, aCML showed a higher WBC count, lower platelet count, lower Hb, and more cases of BM and peripheral blood dysplasia (Figure 5D). No difference of survival was observed among different diagnostic groups (supplemental Figure 4E). We further analyzed associations between somatic mutations and clinical parameters/outcome (Figure 5E-F; supplementary Figure 4F-G). We observed that the mutation of ASXL1, TET2, or EZH2 was correlated with older age. The presence of CSF3R mutations was associated with a high neutrophil percentage, low monocyte count, and the presence of splenomegaly. The presence of NRAS mutations was associated with low Hb, low platelet count, low neutrophil percentage, and high monocyte percentage. ASXL1 mutations were associated with a high WBC count and more platelet transfusion. Moreover, we observed that the presence of TET2 mutations was associated with a low platelet count, consistently high platelet transfusion requirement, low neutrophil percentage, high monocyte percentage, more major bleeding events, and BM dysplasia cases. Interestingly, the presence of SETBP1 mutations was associated with a high Hb level and high platelet count. Moreover, the presence of NRAS, ASXL1, GATA2, and DNMT3A mutations demonstrated a trend of shorter overall survival, whereas CBL mutations predicted favorable overall survival.
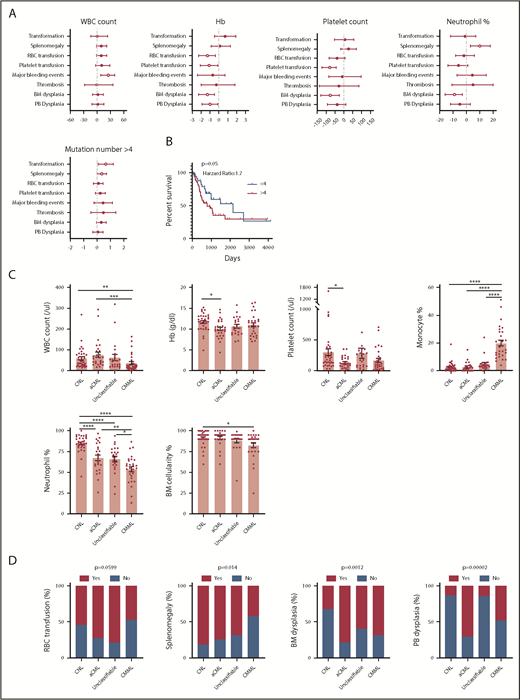
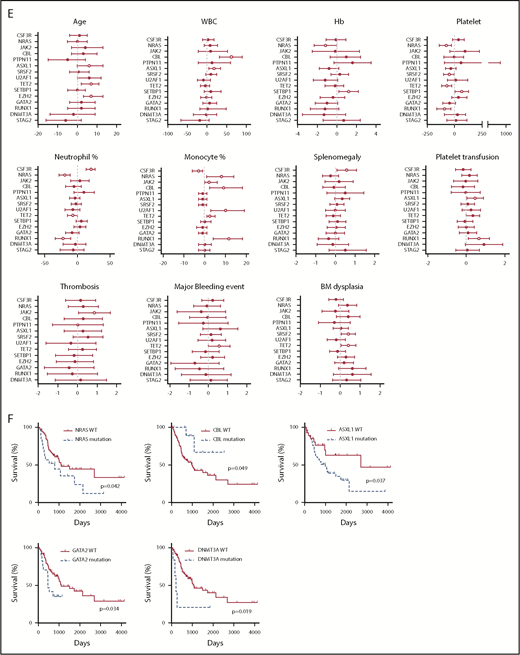
Disease diagnosis and the presence of certain mutations predict clinical outcomes. (A) The graph depicts 95% confidence interval and Hodges-Lehmann median differences of odds ratios of different clinical parameters for indicated clinic outcomes. (B) The graph depict the Kaplan-Meier survival curve of patients with ≥4 mutations or patients with <4 mutations. Statistical significance of the Kaplan-Meier survival curve was analyzed by the log-rank test (C) Graphs depict the mean ± SEM of indicated clinic parameters in different disease subgroups. Statistical significance was assessed using 1-way ANOVA and Kruskal-Wallis tests. (D) Graphs depict the comparison of frequencies of indicated clinic outcomes in different disease groups. Statistical significance was analyzed using contingency table χ2 tests. (E) The graph depicts 95% confidence interval and Hodges-Lehmann median differences for different clinical parameters in the presence or absence of mutations in a given gene. Statistical significance of continual variables was calculated using Mann-Whitney U tests, whereas for bilinear variables, significance was calculated by Fisher's exact test, and the log10-transformed odds ratios are shown. (F) Graphs depict Kaplan-Meier survival curves of patients in the presence or absence of given gene mutations. Statistical significance of the Kaplan-Meier survival curve was analyzed by the log-rank test. WT, wild-type.
Disease diagnosis and the presence of certain mutations predict clinical outcomes. (A) The graph depicts 95% confidence interval and Hodges-Lehmann median differences of odds ratios of different clinical parameters for indicated clinic outcomes. (B) The graph depict the Kaplan-Meier survival curve of patients with ≥4 mutations or patients with <4 mutations. Statistical significance of the Kaplan-Meier survival curve was analyzed by the log-rank test (C) Graphs depict the mean ± SEM of indicated clinic parameters in different disease subgroups. Statistical significance was assessed using 1-way ANOVA and Kruskal-Wallis tests. (D) Graphs depict the comparison of frequencies of indicated clinic outcomes in different disease groups. Statistical significance was analyzed using contingency table χ2 tests. (E) The graph depicts 95% confidence interval and Hodges-Lehmann median differences for different clinical parameters in the presence or absence of mutations in a given gene. Statistical significance of continual variables was calculated using Mann-Whitney U tests, whereas for bilinear variables, significance was calculated by Fisher's exact test, and the log10-transformed odds ratios are shown. (F) Graphs depict Kaplan-Meier survival curves of patients in the presence or absence of given gene mutations. Statistical significance of the Kaplan-Meier survival curve was analyzed by the log-rank test. WT, wild-type.
Aberrant karyotypes were detected in 34 samples with available information, with trisomy 8 (n = 5, 6%) and trisomy 14 (n = 3, 4%) being the most common abnormalities (supplemental Data 2). High relevant fusions or microdeletions were detected in 10 patients, with chromosome 13 and 17 being the most commonly affected chromosomes (supplemental Data 3). Known pathogenic fusions include MYO18A-FLT336 and ETV6-ABL1. We also performed a copy-number variation (CNV) analysis. We observed frequent CNVs on chromosomes 3, 8, 9, 14, 19, and 21 (supplemental Figure 5). CNVs on the 170 hematological-malignancy–related genes were also detected (supplemental Data 4).
Gene expression signatures identify 3 main sample clusters
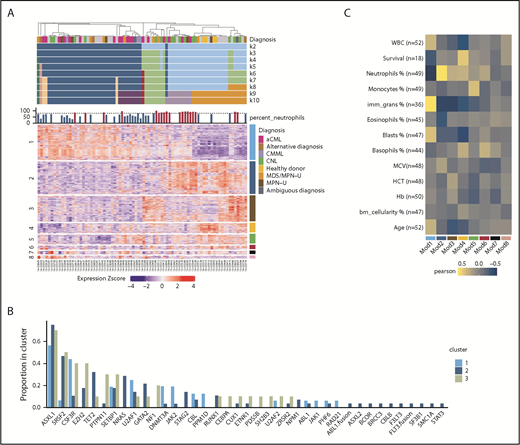
We performed RNA-seq on 76 individual patient samples and 4 normal neutrophil controls to identify gene expression signatures associated with disease classification and/or prognostic markers. We used the consensus clustering approach37 to cluster the 80 samples using the top 2000 most variable genes. We examined a range of expected cluster numbers (k) from 2 to 10, with 7 being selected as the final k, after examination of the cumulative distribution function38 distributions and comparison with simulated null datasets. At the chosen k = 7 clustering, the majority of samples (74/80) were assigned to 3 main sample clusters (Figure 6A).
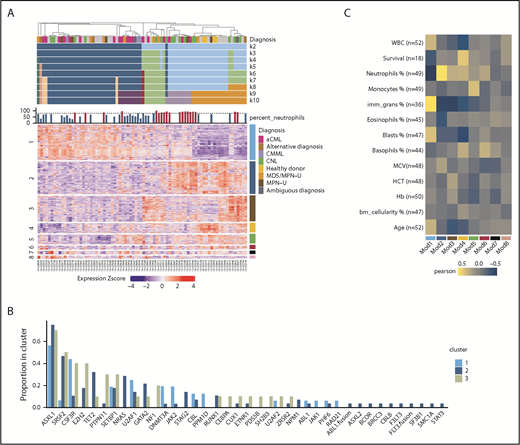
Differential gene expression may guide clinical diagnosis. (A) Consensus clustering heatmap of expression values (Z score; mean centered and divided by standard deviation) (k = 2 through k = 10). Rows are first split by their WGCNA module membership and then clustered by expression values. The module number is on the left, and the corresponding color is on the right. Heatmap columns were clustered by consensus clustering (k = 7). (B) The barplot demonstrates the gene mutation frequencies colored by the consensus clustering result. (C) Heatmap summary of Pearson correlations of the WGCNA module eigengenes and the numeric clinical variables. The y-axis indicates the clinical variables with the number of samples used in the correlation shown as n. The x-axis indicates the module name/color. Note that the survival data are only limited to samples with noncensored data. HCT, hematocrit; MCV, mean corpuscular volume.
Differential gene expression may guide clinical diagnosis. (A) Consensus clustering heatmap of expression values (Z score; mean centered and divided by standard deviation) (k = 2 through k = 10). Rows are first split by their WGCNA module membership and then clustered by expression values. The module number is on the left, and the corresponding color is on the right. Heatmap columns were clustered by consensus clustering (k = 7). (B) The barplot demonstrates the gene mutation frequencies colored by the consensus clustering result. (C) Heatmap summary of Pearson correlations of the WGCNA module eigengenes and the numeric clinical variables. The y-axis indicates the clinical variables with the number of samples used in the correlation shown as n. The x-axis indicates the module name/color. Note that the survival data are only limited to samples with noncensored data. HCT, hematocrit; MCV, mean corpuscular volume.
We then examined the relation between consensus cluster and WES or clinical parameters. We observed that consensus cluster 2 (dark blue) showed a higher average number of mutations per patient and more samples with multiple mutations per gene (eg, >1 in-frame, missense, or truncation), a higher percentage of immature granulocytes, and a higher incidence of leukemia transformation and platelet transfusion (supplemental Figure 6A-D). In terms of mutations in specific genes, cluster 2 showed higher frequencies of NRAS and TET2 mutations and lower frequencies of CSF3R, PTPN11, NF1, and JAK2 mutations (Figure 6B). Moreover, cluster 2 has more cases diagnosed with aCML and CMML, whereas clusters 1 and 3 have more CNL cases (supplemental Figure 6E).
To further explore the gene expression differences underlying the clustering of the patient samples, we applied the weighted gene coexpression network analysis (WGCNA) methodology.39 This approach produced 9 sets of genes termed modules or subnetworks (Figure 6A). We first tested whether the expression pattern of a given module differed with respect to the patient sample clustering. Of the 9 modules, 6 had patterns that noticeably differed among the 3 patient sample clusters (supplemental Figure 7A). Among these modules, we observed that module 1 (turquoise), is enriched in genes related to cell cycle and DNA repair and is positively correlated with the percentage of immature granulocyte and negatively correlated with neutrophil percentage and healthy donor diagnosis, whereas module 2 (blue) is negatively correlated with immature granulocytes and positively correlated with neutrophil percentage (Figure 6C; supplemental Figure 7B). In addition, module 4 (yellow) is positively correlated with patient survival and included the healthy donor controls that were included in the study (supplemental Figure 7B).
Discussion
CNL/aCML/unclassifiable/CMML are a group of rare diseases with diverse clinical and morphological features resembling both MDS and MPN.1 The diversity of these diseases is appreciated at all levels ranging from disease history, patient symptomatology, clinical manifestations, blood and BM cell morphologies, a variable risk of transformation to AML, and differential response to therapy. This can create challenges in diagnosis, prediction of outcomes, and identification of effective therapies. To further understand the molecular pathogenesis of these diseases, we performed WES and RNA-seq on the largest ever cohort of patients with the clinically diagnoses of CNL, aCML, CMML, MPN-U, or MDS/MPN-U.
We observed unique patterns of combinatorial pathway mutations in CNL/aCML/unclassifiable, which are not observed in other hematological malignancies with the exception of CMML. More than half of the CNL/aCML/unclassifiable/CMML patients demonstrate ≥3 different pathway co-occurring mutations involving chromatin modifiers, epigenetic regulators, splicing complex, and signaling genes (Figure 2B-C). In contrast, high frequencies of signaling genes, but not epigenetic or splicing complex mutations, are seen in classical MPN, a high incidence of mutations in the splicing complex but low frequencies of signaling gene mutations are seen in MDS, and only RAS pathway mutations with rare coexisting mutations in splicing complex or epigenetic regulators are seen in juvenile myelomonocytic leukemia.22,40-43 Although CNL is classified as an MPN, the multiple pathway mutation co-occurring pattern closely resembles aCML/MDS/MPN-U and CMML, indicating that CNL, aCML, MDS/MPN-U, and CMML are a group of similar diseases. Moreover, in the majority of cases, the VAF clonal structure analysis demonstrated ≥2 common mutations involving epigenetic regulators and splicing factors in the dominant clone with the acquisition of other mutations in linear, nonbranching fashion, which is distinct from AML, where one can observe the presence of multiple independent subclones.19 Notably, it is possible that these mutations are acquired separately and the co-occurring mutations are selected during evolution before clinical presentation.
CNL/aCML/unclassifiable/CMML represent diseases with a wide range of phenotypic states, which must be driven, at least in part, by the genetic makeup of the tumor cells. Our detailed co-occurrence analyses led us to observe ≥15 groups of different combination patterns involving chromatin modifiers, DNA methylation regulators, splicing factors, and signaling pathway genes in our cohort. If each different gene within these pathways is considered distinctly, the different combinatorial patterns are even more numerous. This is especially true for the signaling pathway mutations, where mutations from 19 different signaling genes were identified, with different combinations of double or triple mutations in signaling pathway genes. Moreover, high frequencies of double variants were also observed for GATA2, TET2, CSF3R, and EZH2. Moreover, if we take clonal hierarchy into account, the variation further increases with a wide range of mutations demonstrating variable orders of acquisition. In sum, although CNL/aCML/unclassifiable/CMML demonstrate a limited overall number of recurrent mutations, the high frequencies of double mutations within the same genes, the diverse combinatorial patterns within the same pathway and among different pathways, and the variable order of acquisition may confer complex combinatorial and temporal effects that lead to the diverse clinical manifestations of these diseases.
The RNA-seq analysis demonstrated 3 major groups, with different proportions of all 5 diagnoses in each group, indicating that expression alone could not clearly distinguish these different diagnostic groups into 5 distinct categories. Overall, cluster 2 demonstrated a worse prognosis and harbored high frequencies of NRAS and TET2 mutations. We also identified a set of genes that predicting overall survival, neutrophil percentage, and healthy donor signature.
Therefore, we propose that CNL/aCML/unclassifiable/CMML are a group of diseases with a similar combination of genetic and epigenetic alterations. The dose and temporal effect of single and combinational gene mutations contribute to the heterogeneity and diversity of the gene expression and clinical manifestations of this group of diseases, partially explaining the difficulty in making clear diagnostic classifications based solely on histology for some cases. However, key pathway mutations implicate distinct clinical, co-occurring mutation, gene expression, and outcome predictions. Therefore, pharmacological targeting of these pathways may be considered in the context of the clinical management of these diseases.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Dorian LaTocha and Brianna Garcia for help with fluorescence-activated cell sorting, Armon Azizi for computing coding assistance, and Kara Johnson for general advice and support.
This work was supported by the National Cancer Institute, National Institutes of Health (grants 1U01CA217862-01, 1U54CA224019-01, and 3P30CA069533-18S5). J.W.T. was supported by the V Foundation for Cancer Research, the Gabrielle’s Angel Foundation for Cancer Research, and the National Cancer Institute, National Institutes of Health (5R00CA151457-04 and 1R01CA183947-01). H.Z. received the Medical Research Foundation grant, the Collins Trust Award, and K99 Career Transition Award (1K99CA237630-01 from the National Institutes of Health, National Cancer Institute). J.E.M. is supported by the National Cancer Institute, National Institutes of Health (R00-CA190605) and an ASH Scholar Award.
Authorship
Contribution: A.R., S.S., and A.R.S. extracted genomic DNA and RNA from patient samples; A.C., R.H., C.L., and R.S. performed the WES and RNA-seq; D.B., B.W., and S.K.M. carried out bioinformatics analyses of genomic data; L.W. and B.W. developed the CNL instance of the Vizome platform, in close consultation with H.Z. N.L., J.E.M., E.S., and V.K. organized patient sample information; H.Z. performed the experiments; C.A.E. participated in analyzing and graphing the data; K.-H.T.D., R.H.C., D.J.D., M.W.D., T.D., T.H., M.R.L., B.C.M., S.T.O., D.A.P., D.P.S., R.M.S., and J.R.G. collected the patient samples, organized patient sample information, and participated in data analysis and discussion; B.J.D. provided important research materials and oversight; H.Z. and J.W.T. analyzed and interpreted the data and wrote the manuscript; and all authors contributed to the final manuscript.
Conflict-of-interest disclosure: J.W.T. has received research support from Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Seattle Genetics, Syros, and Takeda and is co-founder of Vivid Biosciences. B.J.D. is on the scientific advisory board of Aileron Therapeutics, ALLCRON, Cepheid, Gilead Sciences, Vivid Biosciences, Celgene, and Baxalta (inactive); is on the scientific advisory board of and owns stock in Aptose Biosciences, Blueprint Medicines, Beta Cat, GRAIL, Third Coast Therapeutics, and CTI BioPharma (inactive); is a scientific founder of and owns stock in MolecularMD; is on the board of directors and owns stock in Amgen; is on the board of directors of Burroughs Wellcome Fund and CureOne; is on the Joint Steering Committee of Beat AML LLS; and receives clinical trial funding from Novartis, Bristol-Myers Squibb, and Pfizer and royalties from patent 6958335 (Novartis exclusive license), Oregon Health & Science University, and Dana-Farber Cancer Institute (1 Merck exclusive license). K.-H.T.D. is a member of the advisory board of Incyte and received funding from Incyte for research study. The remaining authors declare no competing financial interests.
Correspondence: Jeffrey W. Tyner, Knight Cancer Institute, Oregon Health & Science University 3181 SW Sam Jackson Park Rd, KCRB 2122, Mailcode KR-HEM, Portland, OR 97239; e-mail: tynerj@ohsu.edu.