Key Points
Transcriptomes cluster most AEL apart from other myeloid malignancies.
Alterations of AEL erythroid master regulators impair GATA1 activity and induce the disease in mice.

Abstract
Acute erythroleukemia (AEL or acute myeloid leukemia [AML]-M6) is a rare but aggressive hematologic malignancy. Previous studies showed that AEL leukemic cells often carry complex karyotypes and mutations in known AML-associated oncogenes. To better define the underlying molecular mechanisms driving the erythroid phenotype, we studied a series of 33 AEL samples representing 3 genetic AEL subgroups including TP53-mutated, epigenetic regulator-mutated (eg, DNMT3A, TET2, or IDH2), and undefined cases with low mutational burden. We established an erythroid vs myeloid transcriptome-based space in which, independently of the molecular subgroup, the majority of the AEL samples exhibited a unique mapping different from both non-M6 AML and myelodysplastic syndrome samples. Notably, >25% of AEL patients, including in the genetically undefined subgroup, showed aberrant expression of key transcriptional regulators, including SKI, ERG, and ETO2. Ectopic expression of these factors in murine erythroid progenitors blocked in vitro erythroid differentiation and led to immortalization associated with decreased chromatin accessibility at GATA1-binding sites and functional interference with GATA1 activity. In vivo models showed development of lethal erythroid, mixed erythroid/myeloid, or other malignancies depending on the cell population in which AEL-associated alterations were expressed. Collectively, our data indicate that AEL is a molecularly heterogeneous disease with an erythroid identity that results in part from the aberrant activity of key erythroid transcription factors in hematopoietic stem or progenitor cells.
Introduction
Acute myeloid leukemia (AML) of the erythroid lineage (acute erythroleukemia [AEL] or AML-M6) accounts for 3% to 5% of AML patients and is inherently associated with poor outcome.1-3 Although AEL can occur at any age, the majority of patients are >65 years, and the disease often occurs secondary to other neoplasms, including myeloproliferative neoplasms (MPNs) or myelodysplastic syndrome (MDS), or after cytotoxic cancer treatment. Two major morphological subtypes have been proposed: pure erythroleukemia (PEL; AML-M6b, also known as Di Guglielmo disease) with >80% of blasts committed to the erythroid lineage and AML-M6a characterized by the presence of both erythroid precursors and myeloid blasts.1-3 The 2016 World Health Organization (WHO) classification integrated AML-M6a into MDSs or not otherwise specified AML (AML-NOS), but this classification remains a matter of debate.4-6
Functional studies have suggested that 2 to 5 genetic driver lesions on a background of preexisting alterations in hematopoietic stem or progenitor cells (HSPCs) might be sufficient to induce AML.7,8 For AEL, earlier work showed that leukemic cells often have complex karyotypes, and targeted DNA sequencing revealed the presence of several known AML-associated mutations,9-12 but AEL-driving molecular mechanisms remain incompletely understood and erythroleukemia-specific mutations have seldom been functionally validated. Strikingly, single or multiple TP53 mutations have been shown to be a molecular hallmark of PEL.13
Normal erythroid differentiation is controlled by the activity of both extrinsic signaling factors, including erythropoietin (EPO) mediating its effects through the EPO receptor (EPOR) signaling pathways, and intrinsic multimeric transcription complexes.14-16 The latter includes hematopoietic master regulators like GATA-binding protein 1 (GATA1), T-cell acute lymphocytic leukemia protein 1 (TAL1), LIM domain-only 2 (LMO2), CBFA2/RUNX1 partner transcriptional corepressor 3 (CBFA2T3, also known as ETO2), and LIM-domain-binding protein 1 (LDB1), thereafter broadly named GATA1 complexes, which can activate or repress transcription of target genes. These GATA1 complexes contribute to terminal erythroid differentiation through binding to gene loci and transcription of essential erythroid genes (eg, hemoglobin). This process is also regulated by Krüppel-like factor 1 (KLF1), which binds DNA next to the GATA1 complexes to coregulate erythroid genes.17,18 To establish the erythroid differentiation program, functional synergism between these transcriptional complexes and the EPO/EPOR signaling is mediated by the presence of phosphorylated STAT5 binding in the neighborhood of GATA1 and KLF1.19,20 Accordingly, mutations in these factors have been associated with altered erythropoiesis.21,22 For example, GATA1 mutations are associated with congenital erythroid hypoplasia (Diamond-Blackfan anemia [DBA]) or X-linked dyserythropoietic anemia.23 Moreover, the identification of a NFIA-ETO2 fusion in pediatric PEL24 suggests that an altered activity of these complexes may contribute to human erythroid leukemogenesis.
To better understand the molecular mechanisms that control the erythroid feature, we characterized the genetic and transcriptional landscape in leukemic cells from 33 AEL patients. We identified distinct molecular subgroups composed of patients carrying (1) TP53 mutations, (2) various combinations of mutations previously found in AML and MDS such as DNMT3A, TET2 or IDH2, and (3) those with none of these recurrent alterations. Comparative transcriptomics established an erythro/myeloid differentiation expression signature space that distinguished the majority of AEL cases from MDS or other AML forms. Notably, leukemic cells from >25% of AEL patients showed aberrant expression of key transcriptional regulators including SKI, ERG, and ETO2, which interfere with the activity of the erythroid master regulator GATA1. Combinatorial experimental expression in HSPC fractions induced lethal erythroid or mixed erythroid/myeloid diseases in mice phenocopying several aspects of the human disease, underlining their importance in the molecular pathogenesis of AEL.
Materials and methods
Patient samples
Fifty-eight human patient samples were obtained with the informed consent of the patient and approved by the local ethics committees in accordance with national ethics rules. AEL patient diagnostics were established according to the WHO 2008 classification and criteria described recently.16 Cytogenetic risk groups were defined according to the revised International Prognostic Scoring System (IPSS-R).25 Mononuclear cell fractions were obtained from patient blood or bone marrow (BM) samples by Ficoll gradient, and frozen in fetal bovine serum (FBS; Gibco) supplemented with 10% dimethyl sulfoxide (DMSO). DNA and RNA extraction were done on fresh or frozen samples. DNA was extracted using bulk or sorted cells from patient samples (n = 7, CD36+ for blast cell population and CD3+ or CD19+ for nonneoplastic cell populations) or from xenograft-amplified samples (n = 4). RNA was extracted from patient samples (n = 22) and xenograft-amplified samples (n = 7) from bulk or sorted cells (CD36+ or CD45+ cells), respectively. We obtained appropriate sequencing material for 33 patients (11 paired patient samples for exome sequencing and 29 patient samples for RNA sequencing).
Murine models
C57BL/6JOlaHsd mice (named C57BL/6J) were purchased from Envigo and NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice from The Jackson Laboratory (005557). TP53R248Q/+ knock-in mice were described previously.26 To generate double transgenic TET2−/−/GATA1s mice, we intercrossed Tet2−/− and Gata1Δe2 (here named Gata1s) mice.27,28 Mice were maintained at the Gustave Roussy preclinical facility and all experiments were approved by the French National Animal Care and Use Committee (CEEA 26: projects 2017-082-12726 and 2017-084-12799).
Flow cytometry and cell sorting
Antibodies used for flow cytometry are listed in supplemental Table 1 (available on the Blood Web site). Cells were stained in 1× phosphate-buffered saline (PBS) supplemented with 2% FBS at 4°C for 30 minutes and washed prior to analysis. Whole BM or spleen cells were analyzed without red blood cell lysis. For cell sorting, total BM cells underwent red blood cell lysis. To obtain HSPCs, total BM was depleted of all major hematopoietic cell lineage (Lin−) using the Mouse Hematopoietic Progenitor (Stem) Cell Enrichment Set (Becton Dickinson [BD]]. Progenitor populations were further purified by fluorescence-activated cell sorting (FACS) according to the following phenotypes: Hematopoietic stem cells (HSC) were defined as Lin-/Sca1+/KIT+/CD34−/CD48−, megakaryocytic-erythroid progenitors (MEPs) were defined as Lin−Sca1−/KIT+/CD34−/CD16/32− and granulocyte-macrophage progenitors (GMP) were defined as Lin−/Sca1−/KIT+/CD34+/CD16/32+. To obtain mouse erythroid progenitors, BM cells were first depleted using biotin-conjugated antibodies against CD3, B220, Gr-1, and CD11b (BD) followed by FACS according to the population described as CD71+/Ter119+/KIT+. Flow cytometric analysis was performed using ARIAII, CANTO-II, or CANTO-X instruments (BD), and data were analyzed using the FlowJo software (Flowjo 9.3.2).
Cell culture
Mouse erythroid progenitor cells were expanded in StemSpan serum-free expansion medium (SFEM; Stem Cell Technologies) supplemented with penicillin (100 U/mL)-streptomycin (100 μg/mL), murine stem cell factor (mSCF) (10 ng/mL), murine interleukin 3 (mIL3) (10 ng/mL), mIL6 (10 ng/mL), human EPO (hEPO) (2 U/mL), 0.4% cholesterol, and dexamethasone (10−6 M). Mouse erythroleukemia (MEL) cells were maintained in RPMI 1640 (Gibco) supplemented with 10% FBS, penicillin (100 U/mL)-streptomycin (100 μg/mL) and 2 mM l-Glutamine (Gibco). Murine G1E cells, a generous gift from M. Weiss,29 were maintained in Iscove modified Dulbecco medium (Gibco) supplemented with 15% FBS, penicillin (100 U/mL)-streptomycin (100 μg/mL), mSCF (10 ng/mL), hEPO (2 U/mL), monothioglycerol (4.5 × 10−5 M), and 2 mM l-Glutamine (Gibco). Murine growth factor-dependent Ba/F3 cells was maintained in RPMI 1640 (Gibco) supplemented with 10% FBS, penicillin (100 U/mL)-streptomycin (100 μg/mL), mIL3 (10 ng/mL), and 2 mM L-Glutamine (Gibco). Human embryonic kidney (HEK-293T) cells were grown in Dulbecco modified Eagle medium (Gibco) supplemented with 10% FBS, penicillin (100 U/mL)-streptomycin (100 μg/mL), and 2 mM L-Glutamine (Gibco).
Retroviral constructs, particle production, and cell transduction
The SKI complementary DNA (cDNA) was a kind gift from Suzana Atansoski (Basel, Switzerland). The other cDNAs were synthesized. All cDNAs were cloned into retroviral pMSCV-IRES-EGFP or -mCherry backbones. GATA1 cDNA was cloned into lentiviral pLT3-GEPIR-IRES-EGFP expression vector. For retroviral or lentiviral particles production, HEK-293T cells were plated 1 day before cotransfection with the expression constructs coexpressing EGFP or mCherry and cDNA using the X-tremeGENE-9 DNA Transfection Reagent (Roche) or jetPRIME reagent (Polyplus transfection), respectively, according to the manufacturer’s recommendations. Culture media were changed 24 hours posttransfection and supernatants containing viral particles were harvested 48 hours and 72 hours posttransfection. Murine cells were transduced by spinoculation (90 minutes at 2500 rpm, 33°C) with supernatants containing viral particles supplemented with 5 μg/mL polybrene in 7.5 mM HEPES buffer.
BM transplantation
Total BM (0.4 × 106 cells) and/or transduced progenitor cells were transplanted through IV injection in lethally (9.5 Gy) or sublethally (5 Gy) irradiated 8- to 10-week-old C57BL/6J recipient mice.
RNA extraction and RT-qPCR
RNA was extracted using a RNeasy Mini kit (Qiagen) or AllPrep DNA/RNA Mini kit (Qiagen), according to the manufacturer’s recommendations and quantified using NanoDrop (ThermoScientific). Reverse transcription (RT) was performed using SuperScript II (Invitrogen). Quantitative polymerase chain reaction (qPCR) was performed using SYBR Select Master mix or TaqMan Gene Expression Master mix (Applied Biosystems) on a 7500HT Fast Real-Time PCR System (Applied Biosystems) following the manufacturer;’s recommendations. Primer sequences are listed in supplemental Table 2.
Whole-exome sequencing
Whole-exome sequencing was conducted as described previously30 on paired-samples from 11 patients. DNA from sorted CD3+ or CD19+ nonneoplastic cells was used for exome capture using SureSelect All Exon V4 or V5 kits (Agilent Technologies). We performed paired-end sequencing (100 bp) using HiSeq2000 sequencing instruments at Gustave Roussy genomic platform. Reads were mapped to the reference genome hg19 using the Burrows-Wheeler Aligner (BWA) alignment tool version 0.7.10. PCR duplicates were removed using Picard tools–Mark Duplicates (version 1.119). Local realignment around indels and base quality score recalibration was performed using GATK 3.3 (Genome Analysis Tool Kit). Reads with a mapping quality score < 30 < 20 were removed. Somatic single-nucleotide variations (SNVs) and indels were called in the leukemic sample using Varscan (v2.3.7) by comparison with the paired nonneoplastic samples for exomes, and by comparison with the reference genome for RNA-seq. For candidate somatic mutations, the variants were adopted as candidate mutations when P value was <.001 and allele frequency was <.1 in the reference sample. Variants were annotated with Annovar (v141112). We excluded synonymous SNVs, variants located in intergenic, intronic, untranslated regions and noncoding RNA regions. The mean coverage in the targeted regions was, respectively, 85,4× and 91,2× for leukemic and nonneoplastic samples. The functional variants were predicted using the open platform Cancer Genome Interpreter31 (CGI) and only known-variants or predicted driver variants were confirmed through visualization with IGV (2.3.88) and finally kept in this study.
RNA sequencing
RNA sequencing (RNA-seq) was performed as described.27 Sequences were aligned to the reference genome with TopHat2 version 2.0.9 using the following parameters: –bowtie1–fusion-search–library-type fr-firststrand–read-realign-edit-dist 0 -p 8 -r 50 (or 2.0.14 for mice data sets) and Bowtie1 version 1.0.0. The number of reads per genes (RefSeq database) was counted with HTSeq-count version 0.5.4p5 using the “union” mode. The counts were then normalized with the DESeq2 method, which takes into account the library size of each sample.
ATAC sequencing
ATAC-sequencing (ATAC-seq) analysis has been previously described32 Briefly, after lysis of 50 000 cells, transposition, and purification steps, the transposed DNA fragments were amplified by PCR (12 cycles) using adapters from the Nextera Index Kit (Illumina). PCR purification was performed using Agencourt AMPure XP magnetic beads (Beckman Coulter) to remove large fragments and remaining primers. Library quality was assessed using an Agilent 2100 Bioanalyzer using a High Sensitivity DNA Chip (Agilent Technologies). Libraries were sequenced using NovaSeq 6000 sequencer (Illumina; 50 bp paired-end reads). Quality control of reads was performed using FastQC 0.11.7 and multiQC 1.5. The reads were aligned to the reference genome mm10 with bwa (aln 0.7.17). After alignment, we removed reads mapping to the mitochondrial genome, PCR duplicate reads, and reads with a mapping quality lower than 20 using samtools (v 1.9). Final read counts for all mouse data sets ranged from 42 to 128 million reads. Mapped reads were normalized to bins per million and were converted to bigwig format using deeptools (v3.3.0). Peak calling, differential analysis, annotation, and motif analysis were performed using macs2 (v 2.1.1) and homer (v4.10.4, annotatePeak.pl and findMotifsGenome.pl).
PCA of data from differentiation map
To define the hematopoietic space described (Figure 3A), we built a principal component analysis (PCA) of cell types from Differentiation Map [DMAP]33 (DMAP_PCA), excluding NK cells, B cells, T cells, and dendritic cells. As features we included ranks of differentially expressed genes (DMAP_DE) (FDR < 0.05, logFC > 2) determined with limma.34 A Loess regression line was fitted in PCA space to erythroid cells (all erythrocytes and MEP), and Myelocytes (HSC, CMP, GMP, GRAN, MONO, BASO, and EOS). New data points (NP) was projected into the DMAP_PCA space as a dot product between scaled NP vector and DMAP_PCA rotation. By these calculation, we applied the same transformation to NP and added them to DMAP_PCA without recalculation of principal components.
Transcription factor activity inference
For gene regulatory network inference, the ARACNe-AP software was used to infer a Gene Regulatory Network using scRNA-seq data from healthy human progenitors to predict a list of target genes for each transcription factor (TF).28,29 ARACNe was run over the log2 normalized counts in bootstrap mode (100 iterations), with a P value threshold of 1e-8 and a custom curated list of 2171 TFs. Therefore, the activity of each TF in a normal context was computed in a network. For each AEL sample, TF activities were inferred by interrogating this network with AEL transcriptome data and expressed as Normalized Enrichment Score (NES) using the R library viper, as described in the bioconductor package manual.30 NES were used to test differential activity by Student t test and P value correction by Benjamini-Hochberg (FDR cutoff at 0.05). Differentially activated gene lists were established by PCA analysis using predicted activated gene matrix (previously computed using ARACNE and VIPER algorithm), then genes driving PCA dimensions were identified and ranked by contribution (using FactoMineR v1.41 and factoextra v1.0.5 R packages). Finally, heatmap of activated genes was obtained by plotting the top 50 most contributed genes from the first PCA dimension (using pheatmap v1.0.12 R package).
Data sets
Sequencing data were deposited into EBI - Array-Express under the accession E-MTAB-9012 (ATAC-seq) and European Genome-Phenome Archive (EGA) under the accession EGAS00001004203 (Exome/RNA-seq). Available GATA1 ChIPseq on mouse erythroblasts were obtained from ENCODE (GSE36029; SRA accession: SRR492437) and available ATAC-seq data sets from mouse MEP, CFU-E, and proerythroblasts were previously published.35
Statistical analysis
Statistical significance was calculated using Prism (version 6.0a) and is indicated as P values (Student t test except when otherwise specified). *P < .05, **P < .01, ***P < .001.
Results
Molecular alterations in AEL patients
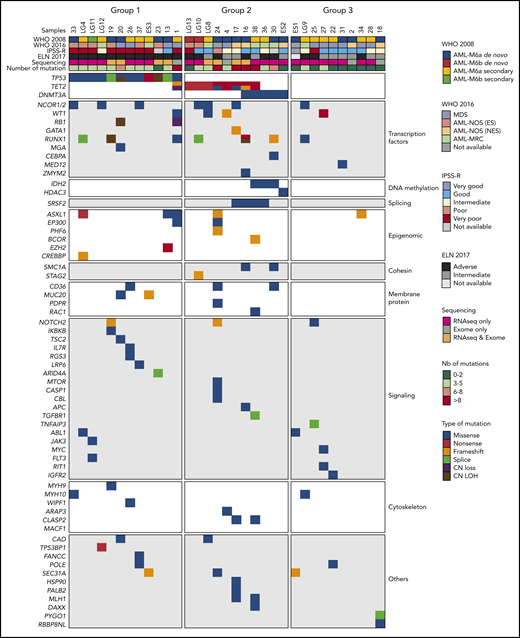
We collected samples from 58 AEL patients, including 34 adults >60 years, 14 between 40 and 59 years, 8 young adults (21-39 years), and 2 pediatric patients. According to the 2008 WHO classification, 33 patients were diagnosed with de novo AEL, including 29 AML-M6a and 4 AML-M6b; 20 patients were diagnosed with AML-M6a secondary to MDS/CML/ALL; 1 with AML-M6b secondary to plexus choroid carcinoma; and a more precise diagnosis was lacking for 4 patients (supplemental Figure 1A; supplemental Table 3). Thereafter, the term “AEL” was used for all patients. Several AEL samples lacking sufficient number of viable cells, were expanded by xenografting them in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice. This approach provided additional leukemic material to isolate RNA (7 patients) and DNA (4 patients). Together, we obtained appropriate sequencing material for 33 patients and performed exome sequencing on 11 paired leukemic and nonneoplastic (either CD3+ or CD19+ cells from the same patient) samples and RNA sequencing of 29 leukemic samples. Combining exome and RNA-seq data, we identified sequence variants with predicted functional consequences in 62 genes (Figure 1).

Genetic alterations of a human AEL cohort. Heatmap representing 3 molecular AEL subgroups: TP53-mutated (group 1), epigenetic modifier-mutated (group 2), and others (with no recurrent mutation; group 3), according to WHO 2008 and 2016 diagnosis classifications, cytogenetic risk group (IPSS-R), ELN 2017 risk stratification, methods of sequencing, number of predicted driver mutations, and type of predicted driver mutations.
Genetic alterations of a human AEL cohort. Heatmap representing 3 molecular AEL subgroups: TP53-mutated (group 1), epigenetic modifier-mutated (group 2), and others (with no recurrent mutation; group 3), according to WHO 2008 and 2016 diagnosis classifications, cytogenetic risk group (IPSS-R), ELN 2017 risk stratification, methods of sequencing, number of predicted driver mutations, and type of predicted driver mutations.
These data, including high variant allele frequency (supplemental Figure 1B), support classification of patients into 3 molecular subgroups (Figure 1). Subgroup 1, presenting with TP53 mutations (n = 12, 36.3% of patients), had in average 4.41 mutations per sample and was associated with both a higher cytogenetic risk and a poorer outcome (Figure 1; supplemental Figure 1C-F). Subgroup 2 (n = 11, 33.3%) mostly presented with TET2 nonsense mutations (n = 8) and DNMT3A mutations (n = 5), including 2 patients with both TET2 and DNMT3A mutations, and had in average 5.72 mutations per sample. Several patients with TET2 and/or DNMT3A mutations also carried SRSF2P95H/R or IDH2R140Q mutations. Of note, in the only sample presenting both a TET2 and an IDH2 mutation, the variant allele frequencies were 60% and 13%, respectively (data not shown), possibly reflecting 2 independent clones. Interestingly, 1 case (#17) of subgroup 2 harbored a TET2 loss-of-function mutation and a GATA1 mutation, predicted to encode the short isoform GATA1s. Additional mutations affected transcription factors (eg, WT1, RUNX1), epigenetic regulators (eg, ASXL1, EP300, BCOR), signaling mediators (eg, NOTCH2, IL7R), and other genes in this group of patients. Finally, subgroup 3 (n = 10, 30.4%) contained samples without TP53 or epigenetic variants. On average, these AEL showed 1.60 mutations per sample, a significantly lower value than for subgroups 1 and 2 (Figure 1; supplemental Figure 1G-H). Overall, our data confirmed that AEL is a molecularly heterogeneous disease characterized by a high prevalence of genetic variants in TP53 and epigenetic regulators comparable to other published cohorts.11,36
AEL gene-expression signatures correlate with erythroid differentiation
As the heterogeneous genetic alterations did not provide any strong rationale for the erythroid phenotype of these leukemia, we investigated the erythroid feature by comparing gene-expression signatures (GES). PCA did not reveal any significant correlation between the GES and the 3 previously identified molecular subgroups (Figure 2A). Similarly, the percentage of erythroblasts in the patient BM at diagnosis was poorly reflected by GES (Figure 2B).

AEL transcriptomes reflect erythroid differentiation rather than genetic mutations. (A) PCA of AEL patient samples based on gene expression. Each point represents 1 sample, colored according to molecular subgroup. (B) PCA of AEL patient samples based on gene expression. Samples are colored according to the amount (percentage) of erythroid cells in the patient’s BM at diagnosis. (C) Heatmap showing correlations between AEL patient samples and the Merryweather-Clarke et al erythroid expression signatures.38 Merryweather-Clarke et al described the expression profiles of enriched human colony-formation unit-erythroid (CFU-E; CD71+CD235−), proerythroblasts (Pro-E; CD71+CD235low), intermediate (Int-E; CD71+CD235high), and late (Late-E; CD71lowCD235high) erythroblast, based on surface cell marker expression. AEL patient-derived expression signatures clustered in an unsupervised manner into the 4 “Merryweather-Clark groups” according to the differentiation stage. (D) PCA of AEL patient samples, colored according to differentiation stage groups defined by Merryweather-Clarke et al. (E) Heatmap of correlations between AEL samples from Iacobucci et al and the Merryweather-Clarke et al data set, clustered in an unsupervised manner into 4 groups according to differentiation stage: CFU-E (CD71+CD235−), proerythroblasts (Pro-E; CD71+CD235low), intermediate (Int-E; CD71+CD235high), and late (Late-E; CD71lowCD235high) erythroblast. (F) PCA of AEL samples from Iacobucci et al36 colored according to differentiation stage groups found when compared with Merryweather-Clarke et al data set.
AEL transcriptomes reflect erythroid differentiation rather than genetic mutations. (A) PCA of AEL patient samples based on gene expression. Each point represents 1 sample, colored according to molecular subgroup. (B) PCA of AEL patient samples based on gene expression. Samples are colored according to the amount (percentage) of erythroid cells in the patient’s BM at diagnosis. (C) Heatmap showing correlations between AEL patient samples and the Merryweather-Clarke et al erythroid expression signatures.38 Merryweather-Clarke et al described the expression profiles of enriched human colony-formation unit-erythroid (CFU-E; CD71+CD235−), proerythroblasts (Pro-E; CD71+CD235low), intermediate (Int-E; CD71+CD235high), and late (Late-E; CD71lowCD235high) erythroblast, based on surface cell marker expression. AEL patient-derived expression signatures clustered in an unsupervised manner into the 4 “Merryweather-Clark groups” according to the differentiation stage. (D) PCA of AEL patient samples, colored according to differentiation stage groups defined by Merryweather-Clarke et al. (E) Heatmap of correlations between AEL samples from Iacobucci et al and the Merryweather-Clarke et al data set, clustered in an unsupervised manner into 4 groups according to differentiation stage: CFU-E (CD71+CD235−), proerythroblasts (Pro-E; CD71+CD235low), intermediate (Int-E; CD71+CD235high), and late (Late-E; CD71lowCD235high) erythroblast. (F) PCA of AEL samples from Iacobucci et al36 colored according to differentiation stage groups found when compared with Merryweather-Clarke et al data set.
Because the AEL WHO classification is based on the number of erythroid and myeloid blasts present in the BM, we used a digital cellular deconvolution method (xCell) to compute a GES-based enrichment in erythroid, myeloid, and other hematopoietic cell types (supplemental Figure 2A).37 The majority of samples had a prominent “erythrocyte” signature (n = 20), whereas some AEL samples presented a higher signal for the immature (MPP, CMP, GMP) or mature myeloid (monocyte, neutrophil) signatures (n = 9). To further explore the link between AEL transcriptomes and different stages of human erythroid maturation, we compared GES from the patients with those obtained experimentally after in vitro differentiation of human peripheral blood mononuclear cells into colony-forming unit erythroid (CFU-E; CD71+CD235-), proerythroblasts (Pro-E; CD71+CD235low), intermediate (Int-E; CD71+CD235high), and late erythroblasts (Late-E; CD71lowCD235high)38 and observed clustering according to these maturation stages (Figure 2C-D). Importantly, transcriptomes from an independent larger cohort of AEL patients36 clustered similarly (Figure 2E-F).
Together, AEL gene expression programs are influenced by the erythroid differentiation stages rather than by the presence of particular genetic lesions, suggesting that the erythroid identity in human AEL relates to the cellular origin and the activity of transcriptional regulators driving cellular differentiation.
A transcriptome-based space maps AEL, MDS, and other AML to erythroid- and myeloid-lineage trajectories
As the 2016 WHO classification assigns most cases previously diagnosed as AEL to MDS or other AML,7 we aimed at designing a transcriptome-based space that is able to distinguish AEL from MDS and other non-AEL AML subtypes. To this end, we retrieved cellular signatures from the DMAP database32 and computed erythroid and myeloid differentiation expression trajectories (Figure 3A). As expected, relative differential gene expression clustered our AEL samples between the erythroid and myeloid trajectories. The majority of cases (n = 25) mapped closer to the erythroid axis whereas the rest mapped closer to the myeloid trajectory (n = 7) and closer to MDS transcriptomes (Figure 3B-C). Likewise, the samples from a recently published large AEL cohort36 mostly clustered apart from MDS samples39,40 (Figure 3C) and apart from non-M6 AML samples41 (Figure 3D). Notably, AEL samples mostly projected between HSC and mature erythroid cells supporting that only part of the maturation-associated erythroid program is expressed in these samples. Interestingly, among our AEL samples that mapped closer to the myeloid axis and other AML samples, sample 24 showed a high expression of SPI1 (Figure 3D; supplemental Figure 2B) also seen in other AML subtypes and was actually independently reclassified as AML-M5 by clinicians during the course of this study. These data support the idea that the transcriptional programs of the majority of AEL cases differs from those of MDS and other AML subtypes. They also support the existence of an overlapping continuum between these entities and the WHO-2016 reclassification of some AEL cases as AML-NOS.

Transcriptome-based mapping and aberrant erythroid regulators in AEL. (A) PCA of data from Differentiation Map (DMAP)33 with regression to cell types in an erythroid and myeloid compartment. The regression line fits to erythroid (green) and myeloid (gray) cells in the PCA space of genes significantly (FDR > 0.05, LogFC > 2) segregating each hematopoietic population: basophiles (BASO), common myeloid progenitor cells (CMP), eosinophil (EOS), erythrocytes (ERY), granulocyte-monocyte progenitor cells (GMP), granulocytes (GRAN), hematopoietic stem cells (HSC), megakaryocytes (MEGA), megakaryocytes-erythroid progenitor cells (MEP), and monocytes (MONO). (B) PCA with regression lines from plot (A) with projection of AEL patient samples. (C) PCA with regression lines from plot (A) with projection of AEL patient samples from our cohort (AEL cohort 1), from Iacobucci et al36 (AEL cohort 2) and MDS samples.39,40 (D) PCA with regression lines from plot (A) with projection of our AEL patient samples, AML and APL samples from the Blueprint consortium database. (E) PCA with regression lines from plot (A) with projection of AEL patient samples colored with KLF1, GATA1, and CEBPA expression and predicted activity. (F) Histogram representation of ERG, GFI1, RUNX1T1, ETO2, GATA3, and SKI gene expression in AEL patients. Positive patient samples (red bars) were defined as presenting an expression above the threshold set as fourfold the average of AEL samples. Dotted bars represent the average expression of AEL samples. (G) Table indicate patient samples presenting with genetic alteration (orange) or transcriptional alteration (blue) of GATA1-associated genes67-70 in 3 molecular subgroups of AEL: TP53-mutated, epigenetics, and others. (H) Kaplan-Meier survival plot of AEL patients grouped according to the presence (or absence) of genetic or transcriptional alterations defined in panel G. P value using log-rank Mantel-Cox test is indicated. (I) Kaplan-Meier survival plot of AEL patients from Iacobucci et al36 and grouped according to the presence (or not) of genetic or transcriptional alterations defined in panel G. P value using log-rank Mantel-Cox test is indicated.
Transcriptome-based mapping and aberrant erythroid regulators in AEL. (A) PCA of data from Differentiation Map (DMAP)33 with regression to cell types in an erythroid and myeloid compartment. The regression line fits to erythroid (green) and myeloid (gray) cells in the PCA space of genes significantly (FDR > 0.05, LogFC > 2) segregating each hematopoietic population: basophiles (BASO), common myeloid progenitor cells (CMP), eosinophil (EOS), erythrocytes (ERY), granulocyte-monocyte progenitor cells (GMP), granulocytes (GRAN), hematopoietic stem cells (HSC), megakaryocytes (MEGA), megakaryocytes-erythroid progenitor cells (MEP), and monocytes (MONO). (B) PCA with regression lines from plot (A) with projection of AEL patient samples. (C) PCA with regression lines from plot (A) with projection of AEL patient samples from our cohort (AEL cohort 1), from Iacobucci et al36 (AEL cohort 2) and MDS samples.39,40 (D) PCA with regression lines from plot (A) with projection of our AEL patient samples, AML and APL samples from the Blueprint consortium database. (E) PCA with regression lines from plot (A) with projection of AEL patient samples colored with KLF1, GATA1, and CEBPA expression and predicted activity. (F) Histogram representation of ERG, GFI1, RUNX1T1, ETO2, GATA3, and SKI gene expression in AEL patients. Positive patient samples (red bars) were defined as presenting an expression above the threshold set as fourfold the average of AEL samples. Dotted bars represent the average expression of AEL samples. (G) Table indicate patient samples presenting with genetic alteration (orange) or transcriptional alteration (blue) of GATA1-associated genes67-70 in 3 molecular subgroups of AEL: TP53-mutated, epigenetics, and others. (H) Kaplan-Meier survival plot of AEL patients grouped according to the presence (or absence) of genetic or transcriptional alterations defined in panel G. P value using log-rank Mantel-Cox test is indicated. (I) Kaplan-Meier survival plot of AEL patients from Iacobucci et al36 and grouped according to the presence (or not) of genetic or transcriptional alterations defined in panel G. P value using log-rank Mantel-Cox test is indicated.
Expression and activities of erythroid regulators in AEL
Myelo/erythroid differentiation is controlled by expression and activity of a relatively small group of transcription factors. Using the ARACNe and VIPER packages42-44 and a large data set from human healthy progenitor cell transcriptomes,45 we computed the activity of transcription factors and inferred lists of putative target genes (supplemental Figure 3A-B). Interestingly, we observed a gradual decrease in expression of erythroid transcription factors (eg, KLF1, GATA1, NFE2, TAL1, NFIA) and their predicted activity when going from the erythroid to the myeloid trajectories and an inverse correlation with myeloid factors (eg, CEPBA and SPI1) (Figure 3E; supplemental Figure 3C). This finding indicates that AEL is characterized by the transcriptional proximity to the normal erythroid lineage trajectory and by the relative activity of master transcription factors that control erythroid differentiation.
These data led us to hypothesize that some AEL cases might be driven by aberrant expression and activity of erythroid transcription factors. We focused on factors known to be predominantly expressed during erythroid differentiation and/or to control the activity of the GATA1 erythroid master regulator.14 Using a threshold of fourfold higher expression level than the average, we observed that some AEL patients indeed expressed abnormally high levels of ERG (n = 2), GFI1 (n = 1), RUNX1T1 (= 1), and ETO2 (n = 1) (Figure 3F). GATA3, which enforced expression previously resulted in erythroid bias,46 was also highly expressed in 3 samples. Notably, we also found high expression of the transcriptional corepressor SKI (v-Ski avian sarcoma viral oncogene homolog) in 2 patients from molecular subgroup 3 (Figure 3F). Interestingly, v-Ski was previously reported to transform chicken erythroid cells, and to directly interact with GATA1 to repress erythroid differentiation.47-50 Our findings suggest that SKI not only influences experimental erythroid differentiation but could also contribute to human AEL pathogenesis.
The search for fusion transcripts using RNA-seq data revealed additional alterations, including 1 in-frame BCR-ABL1 fusion gene (expected in this secondary-to-CML sample) and 3 novel out-of-frame fusion transcripts, notably 2 of them in a TP53-mutated context (supplemental Figure 4A-B). Sample 37 harbored an out-of-frame fusion of YWHAE (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein ε) with EPO and sample ES3 showed an out-of-frame fusion of HSD17B11 (hydroxysteroid 17-β dehydrogenase 11) with B4GALNT3 (β-1,4-N-acetyl-galactosaminyltransferase 3). These fusions were associated with ectopic expression of EPO and B4GALNT3, respectively. Interestingly, the YWHAE-EPO+ sample also showed high EPOR, suggesting an autocrine mechanism of proliferation/survival in this case (supplemental Figure 4C). Although the role of B4GALNT3 in human erythropoiesis remains unclear, an out-of-frame fusion leading to overexpression of B4GALNT3 has been previously reported in thyroid carcinoma.51 Sample ES1 from subgroup 3 presented with an out-of-frame fusion targeting the middle of the DNMT3B locus and associated with lower DNMT3B expression as compared with other samples, supporting DNMT3B inactivation. Notably, Dnmt3a and Dnmt3b expression were previously reported to be tightly controlled during erythroid maturation in mice.52
Overall, genetic and transcriptional alterations in erythroid regulators, including physical or functional interactors of the GATA1 transcriptional complexes were found in 9 of 33 patients (27%) (Figure 3G). Notably, these patients showed a trend toward poorer overall survival (Figure 3H), which became significant upon analysis of a larger AEL data set (Figure 3I).36 Together, transcriptome analysis revealed that the majority of AEL samples are significantly different from MDS and other AML subtypes and that AEL frequently presents with epigenomic alterations that converge on factors interfering with GATA1 activity.
Overexpression of AEL-associated GATA1-interfering factors transforms mouse erythroid progenitors
To functionally test whether the aberrant expression of GATA1-interfering factors identified in AEL samples may contribute to the transformation of the erythroid lineage, we explored the consequences of ectopic expression of SKI, ERG, ETO2, GATA1s, EPO, SPI1, and B4GALNT3 on murine erythroid progenitors. FACS-purified KIT+CD71+Ter119+ cells were transduced with retroviruses encoding these genes and grown in vitro (Figure 4A). In contrast to vector-transduced controls that proliferated for only ∼7 days, ectopic expression of ERG, SPI1, ETO2, SKI, and B4GALNT3 significantly maintained proliferation of erythroid cells presenting with an immature CD71+KIT+Ter119- phenotype and a proerythroblast morphology for >30 days (Figure 4B; supplemental Figure 5A). Although the precise comparison between the overexpression level observed in human AEL samples and those achieved in murine models is technically challenging in this setting, a similar range of overexpression was observed for ERG, ETO2, SKI, and B4GALNT3 (supplemental Figure 5B). Notably, ectopic expression of EPO or GATA1s alone was not sufficient to expand erythroblasts longer than 10 days (Figure 4B).

AEL-associated transcription factors transform erythroid progenitors and impair GATA1 activity. (A) Experimental design: mouse erythroid progenitors (CD71+Ter119+KIT+/low) were sorted from lineage marker–depleted BM, transduced with retrovirus encoding SKI, ERG, ETO2, GATA1s, EPO, SPI1, or B4GALNT3 combined with IRES-GFP (GFP expression is a surrogate marker for transgene expression), or an empty vector (Ctrl), and maintained in StemSpan SFEM with cytokines (mSCF, mIL3, mIL6, hEPO, cholesterol, and dexamethasone). (B) A total of 5 × 104 transduced mouse erythroid progenitors were cultured for 15 days and viable cells were enumerated by trypan-blue exclusion. Mean plus or minus SD number of cells is represented. SKI (n = 5), ERG (n = 4), ETO2 (n = 3), GATA1s (n = 3), EPO (n = 3), SPI1 (n = 6), B4GALNT3 (n = 3), or empty vector (Ctrl) (n = 5). (C) Experimental design: erythroid progenitors (CD71+ Ter119+ KIT+/low) from WT, Tet2−/−, Gata1s (G1s), or Tet2−/−+ Gata1s (Tet2−/−+G1s) mice were sorted from lineage marker–depleted BM and maintained for 15 days in StemSpan SFEM with cytokines (mSCF, mIL3, mIL6, hEPO, cholesterol, and dexamethasone). (D) A total of 3.5 × 104 sorted erythroblasts from WT, Tet2−/−, Gata1s (G1s), or Tet2−/−+ Gata1s (Tet2−/−+G1s) mice were grown in liquid cultures over 15 days and viable cells were counted by trypan-blue exclusion. Mean plus or minus SD (n = 3) is shown. (E) Dot-plot showing the log(fold changes) of the percentage of sequence for a given motif found under ATAC-seq peaks, between normal and transformed erythroblasts (expressing ERG, ETO2, SKI, and SPI1). (F) Histogram representation of the percentage of sequence with GATA1 (top) or ERG (bottom) motif found under ATAC-seq peaks of normal (Ctrl) and transformed erythroblasts. Statistical differences were calculated using the χ2 test. For GATA1 motif, Ctrl vs ERG: P = .0013; Ctrl vs ETO2: P = .0001; Ctrl vs SKI: P < .00001; Ctrl vs SPI1: P = .0032. For ERG motif, Ctrl vs ERG: P = .00001; Ctrl vs ETO2: P < .00001; Ctrl vs SKI: P = .0498; Ctrl vs SPI1: P = .0415. (G) Heatmap representing the hierarchical clustering of ATAC-seq signals, performed using normal (Ctrl) and transformed erythroblast by either ERG, ETO2, SKI, or SPI1, focused on GATA1-binding sites in normal Ter119+ erythroblast (ENCODE). Heatmaps were focused on peak centers with ±5 kb. (H) Profile plot representing ATAC-seq signals performed using normal (Ctrl) and transformed erythroblast expressing either ERG, ETO2, SKI, or SPI1 on ATAC-seq specific peaks previously identified in mouse MEP, CFU-E, or proerythroblasts (proE).35 Profile-plot were focused on peaks centers with ±5 kb. (I) Visualization of GATA1 ChIP-seq peaks performed in normal erythroblast (ENCODE, first lane, gray) and ATAC-seq peaks of normal MEP, proerythroblast, orthochromatic erythroblast, and transformed erythroblasts expressing ERG, ETO2, SKI, or SPI1, focused on Nfe2, Hba-a1, and Hbq-a1 genes, using IGV software (v 2.3.88). (J) Quantitative Nfe2 mRNA expression measured by RT-qPCR in WT erythroblast (Ctrl) or erythroblast transformed with either SKI, ERG, ETO2, or SPI1 overexpression. Expression levels were also compared with the erythroleukemia MEL or the Ba/F3 myelolymphoid cell line. (K) Experimental design: G1E cell lines were cotransduced with a GATA1 doxycycline-inducible vector and with MSCV-vector expressing either ERG, ETO2, SKI, SPI1, or empty. (L) Flow cytometry histogram analysis of Ter119 expression in G1E cells expressing ERG (green), ETO2 (orange), SKI (blue), SPI1 (red), or empty control (gray), without (left) or with (right) induction of GATA1 expression. (M) Histogram representation of Ter119 expression detected by flow cytometry analysis of G1E cells expressing ERG, ETO2, SKI, SPI1, or empty control, without (gray) or with (red) induction of GATA1 expression. Statistical significance (in panels B, D, J, and M) is indicated as P values (Student t test except when otherwise specified). *P < .05; **P < .01; ***P < .001.
AEL-associated transcription factors transform erythroid progenitors and impair GATA1 activity. (A) Experimental design: mouse erythroid progenitors (CD71+Ter119+KIT+/low) were sorted from lineage marker–depleted BM, transduced with retrovirus encoding SKI, ERG, ETO2, GATA1s, EPO, SPI1, or B4GALNT3 combined with IRES-GFP (GFP expression is a surrogate marker for transgene expression), or an empty vector (Ctrl), and maintained in StemSpan SFEM with cytokines (mSCF, mIL3, mIL6, hEPO, cholesterol, and dexamethasone). (B) A total of 5 × 104 transduced mouse erythroid progenitors were cultured for 15 days and viable cells were enumerated by trypan-blue exclusion. Mean plus or minus SD number of cells is represented. SKI (n = 5), ERG (n = 4), ETO2 (n = 3), GATA1s (n = 3), EPO (n = 3), SPI1 (n = 6), B4GALNT3 (n = 3), or empty vector (Ctrl) (n = 5). (C) Experimental design: erythroid progenitors (CD71+ Ter119+ KIT+/low) from WT, Tet2−/−, Gata1s (G1s), or Tet2−/−+ Gata1s (Tet2−/−+G1s) mice were sorted from lineage marker–depleted BM and maintained for 15 days in StemSpan SFEM with cytokines (mSCF, mIL3, mIL6, hEPO, cholesterol, and dexamethasone). (D) A total of 3.5 × 104 sorted erythroblasts from WT, Tet2−/−, Gata1s (G1s), or Tet2−/−+ Gata1s (Tet2−/−+G1s) mice were grown in liquid cultures over 15 days and viable cells were counted by trypan-blue exclusion. Mean plus or minus SD (n = 3) is shown. (E) Dot-plot showing the log(fold changes) of the percentage of sequence for a given motif found under ATAC-seq peaks, between normal and transformed erythroblasts (expressing ERG, ETO2, SKI, and SPI1). (F) Histogram representation of the percentage of sequence with GATA1 (top) or ERG (bottom) motif found under ATAC-seq peaks of normal (Ctrl) and transformed erythroblasts. Statistical differences were calculated using the χ2 test. For GATA1 motif, Ctrl vs ERG: P = .0013; Ctrl vs ETO2: P = .0001; Ctrl vs SKI: P < .00001; Ctrl vs SPI1: P = .0032. For ERG motif, Ctrl vs ERG: P = .00001; Ctrl vs ETO2: P < .00001; Ctrl vs SKI: P = .0498; Ctrl vs SPI1: P = .0415. (G) Heatmap representing the hierarchical clustering of ATAC-seq signals, performed using normal (Ctrl) and transformed erythroblast by either ERG, ETO2, SKI, or SPI1, focused on GATA1-binding sites in normal Ter119+ erythroblast (ENCODE). Heatmaps were focused on peak centers with ±5 kb. (H) Profile plot representing ATAC-seq signals performed using normal (Ctrl) and transformed erythroblast expressing either ERG, ETO2, SKI, or SPI1 on ATAC-seq specific peaks previously identified in mouse MEP, CFU-E, or proerythroblasts (proE).35 Profile-plot were focused on peaks centers with ±5 kb. (I) Visualization of GATA1 ChIP-seq peaks performed in normal erythroblast (ENCODE, first lane, gray) and ATAC-seq peaks of normal MEP, proerythroblast, orthochromatic erythroblast, and transformed erythroblasts expressing ERG, ETO2, SKI, or SPI1, focused on Nfe2, Hba-a1, and Hbq-a1 genes, using IGV software (v 2.3.88). (J) Quantitative Nfe2 mRNA expression measured by RT-qPCR in WT erythroblast (Ctrl) or erythroblast transformed with either SKI, ERG, ETO2, or SPI1 overexpression. Expression levels were also compared with the erythroleukemia MEL or the Ba/F3 myelolymphoid cell line. (K) Experimental design: G1E cell lines were cotransduced with a GATA1 doxycycline-inducible vector and with MSCV-vector expressing either ERG, ETO2, SKI, SPI1, or empty. (L) Flow cytometry histogram analysis of Ter119 expression in G1E cells expressing ERG (green), ETO2 (orange), SKI (blue), SPI1 (red), or empty control (gray), without (left) or with (right) induction of GATA1 expression. (M) Histogram representation of Ter119 expression detected by flow cytometry analysis of G1E cells expressing ERG, ETO2, SKI, SPI1, or empty control, without (gray) or with (red) induction of GATA1 expression. Statistical significance (in panels B, D, J, and M) is indicated as P values (Student t test except when otherwise specified). *P < .05; **P < .01; ***P < .001.
To address whether a cooperation between Tet2-inactivating and Gata1s mutations could transform erythroblasts in vitro, we purified erythroid progenitors from wild-type, Tet2-deficient27 (thereafter named Tet2−/−), Gata1Δe2 knock-in28 (thereafter named Gata1s), and double Tet2−/−+Gata1s transgenic mice and compared their proliferation (Figure 4C). Although Gata1s- or Tet2−/−-only erythroblasts did not expand for >10 to 15 days, Tet2−/−+Gata1s erythroblasts proliferated >2 months and exhibited an erythroid morphology (Figure 4D; supplemental Figure 5C).
Collectively, these data demonstrate that ectopic expression of ERG, ETO2, SKI, or the combination of Tet2 loss-of-function and Gata1s mutations can efficiently immortalize murine erythroblasts in vitro.
Aberrantly expressed AEL-associated transcriptional regulators interfere with GATA1 chromatin accessibility and function
To better understand how aberrantly expressed transcription factors (ERG, ETO2, SKI, SPI1) immortalize erythroblasts, we studied chromatin accessibility by ATAC-seq. Motif analysis revealed a lower representation of GATA1 and KLF1 (also known as EKLF) motifs and a global increase of ETS-associated motifs (including ERG and SPI1 motifs) in all transformed cells compared with Ctrl (Figure 4E-F; supplemental Figure 5D).
To investigate chromatin accessibility at erythroid GATA1-binding sites and at sites that are regulated at specific stages of healthy erythroid differentiation, we interrogated previously published data sets.35 Interestingly, ERG-, ETO2-, SKI-, and SPI1-overexpressing erythroblasts showed a decreased chromatin accessibility at erythroid GATA1-binding sites compared with vector-transduced control cells (Figure 4G). ERG-, ETO2-, SKI-, and SPI1-expressing erythroblasts also showed a decreased chromatin accessibility at sites open in healthy CFU-E and proerythroblasts, whereas there was no difference in chromatin accessibility at sites open in less differentiated MEP cells (Figure 4H; supplemental Figure 5E). Notably, these observations correlated with a decreased chromatin accessibility and mRNA expression at GATA1-controlled erythroid genes such as nuclear factor erythroid 2 (Nfe2) or hemoglobin A1 (Hba-a1) (Figure 4I; supplemental Figure 5F).
Finally, we investigated the consequence of aberrant expression of these transcription factors on GATA1 activity in the GATA1-deficient G1E erythroid cell line in which terminal erythroid maturation can be induced by expression of exogenous Gata129 (Figure 4K). As expected doxycycline-induced Gata1 expression restored G1E erythroid differentiation with upregulation of Ter119 expression (supplemental Figure 5G). In contrast, ectopic expression of ETO2, ERG, SKI, or SPI1 significantly inhibited GATA1-induced differentiation (Figure 4L-M).
Collectively, these data indicate that aberrant expression of ETO2, ERG, SKI, and SPI1 functionally interferes with GATA1 activity and restrains GATA1-dependent erythroid differentiation consistent with impaired differentiation observed in primary human AEL cells.
In vivo modeling of AEL from immortalized erythroblasts
We used complementary strategies to model in vivo the leukemogenic potential of AEL-associated alterations of the different molecular subgroups. First, to ascertain that in vitro–transformed erythroblasts can induce disease in vivo, we injected them into irradiated syngeneic recipients. ERG-, ETO2-, SKI-, or Tet2−/−+Gata1s-transformed, but not SPI1- or B4GALNT3- transformed, cells rapidly induced a fully penetrant fatal disease characterized mostly by the accumulation of CD71+Ter119− and few CD71+Ter119+ blasts lacking expression of myeloid markers (supplemental Figure 6A-C). Histopathological analysis of symptomatic mice showed infiltration of BM, spleen, and livers by erythroblasts expressing nuclear GATA1 (supplemental Figure 6D-E). These results show that some epigenomic alterations found in human AEL have the potential to immortalize murine erythroblasts, which can then induce an AEL-like disease.
In vivo modeling of functional cooperation between AEL-associated alterations
Next, to define in vivo transforming capacities starting from healthy hematopoietic progenitors, we obtained oncogene-expressing Lin− HSPCs (either by retroviral transduction or by breeding transgenic models) and assessed disease development upon engraftment into lethally irradiated recipients. Based on our observations that AEL subgroups 1 and 2 showed frequent cooccurrence of mutations (Figure 1B) and that GATA1 activity is targeted either directly (TET2+GATA1s and IDH2+GATA1 mutations in another cohort36 ) or through associated factors (TP53+ERGhigh), we investigated these 2 representative potential functional cooperation schemes.
Previous work has shown that both Tet2 loss-of-function and Gata1s alter erythroid differentiation but do not induce bona fide leukemia in vivo alone27,28,53-57 (supplemental Figure 7A). To address functional cooperation, we transplanted Tet2−/−+Gata1s Lin− HSPCs into lethally irradiated recipients (Figure 5A). As opposed to recipients of Tet2−/−-only cells, recipients of Tet2−/−+Gata1s cells developed a rapid and fully penetrant lethal disease associated with high WBC, anemia, thrombocytopenia, and splenomegaly (Figure 5B-C; supplemental Figure 7B). Flow cytometry analysis indicated that leukemic blasts were primarily CD11b+Gr1+ myeloid cells (Figure 5D) and histopathological analysis confirmed that BM and spleen were highly infiltrated by blasts with myeloid features (supplemental Figure 7C). Notably, we also observed emperipolesis that was previously described in murine GATA1s models. Together, these data demonstrate that Tet2 loss of function cooperates with Gata1s mutation to promote an AML-like phenotype in vivo.

In vivo modeling of functional cooperation between AEL-associated alterations. (A) Experimental design: mouse CD45.2+ HSPCs from WT, G1s, Tet2−/−, or Tet2−/−+ G1s mice were sorted and injected into sublethally irradiated CD45.1+ recipients followed for disease development. (B) Kaplan-Meier plot of diseased recipients of HSPC cells from Tet2−/− (n = 5) or Tet2−/−+G1s (n = 5) mice. (C) Peripheral blood counts (WBC, RBC, and PLT) of immunodeficient mice engrafted with HSPC cells from Tet2−/− (n = 5) or Tet2−/−+G1s (n = 5) mice. (D) Flow cytometry analysis of myeloid cells (Gr1+CD11b+), erythroid progenitors (CD71+ Ter119+), B cells (B220+), and T cells (CD4+CD8+) gated for CD45.2+ donor cells from Tet2−/− or Tet−/−+G1s mice. (E) Experimental design: HSPCs from WT or TP53R248Q mice were retrovirally transduced with ERG or an empty vector (Ctrl), containing both an IRES-GFP expression cassette, and transplanted into WT primary recipients. Top, Primary recipients of ERG-transduced cells developed T-ALL (supplemental Figure 6). Bottom, to investigate cooperation specifically in erythroid progenitors, ERG-expressing erythroid progenitors (GFP+CD71+Ter119+KITlow/+) were sorted from primary recipient mice and transplanted into secondary recipients to follow disease development. (F) Kaplan-Meier plot of diseased secondary recipient mice (described in panel A). WT (Ctrl or ERG): n = 4, TP53R248Q (Ctrl or ERG): n = 6. (G) Peripheral white blood cell (WBC), red blood cell (RBC), and platelet (PLT) counts of diseased mice transplanted with WT or TP53R248Q HSPC retrovirally transduced with ERG or empty vector (Ctrl). WT (Ctrl or ERG): n = 4, TP53R248Q (Ctrl or ERG): n = 4. (H) Flow cytometry analysis of myeloid cells (CD11b+Gr-1+), erythroid progenitors (CD71+Ter119+), B cells (B220+), and T cells (CD4+CD8+) from diseased secondary recipients of TP53R248Q HSPCs retrovirally transduced with ERG gated for GFP+ or GFP− cells. Statistical significance (in panels B, C, F, and G) is indicated as P values (Student t test except when otherwise specified). *P < .05; **P < .01; ***P < .001.
In vivo modeling of functional cooperation between AEL-associated alterations. (A) Experimental design: mouse CD45.2+ HSPCs from WT, G1s, Tet2−/−, or Tet2−/−+ G1s mice were sorted and injected into sublethally irradiated CD45.1+ recipients followed for disease development. (B) Kaplan-Meier plot of diseased recipients of HSPC cells from Tet2−/− (n = 5) or Tet2−/−+G1s (n = 5) mice. (C) Peripheral blood counts (WBC, RBC, and PLT) of immunodeficient mice engrafted with HSPC cells from Tet2−/− (n = 5) or Tet2−/−+G1s (n = 5) mice. (D) Flow cytometry analysis of myeloid cells (Gr1+CD11b+), erythroid progenitors (CD71+ Ter119+), B cells (B220+), and T cells (CD4+CD8+) gated for CD45.2+ donor cells from Tet2−/− or Tet−/−+G1s mice. (E) Experimental design: HSPCs from WT or TP53R248Q mice were retrovirally transduced with ERG or an empty vector (Ctrl), containing both an IRES-GFP expression cassette, and transplanted into WT primary recipients. Top, Primary recipients of ERG-transduced cells developed T-ALL (supplemental Figure 6). Bottom, to investigate cooperation specifically in erythroid progenitors, ERG-expressing erythroid progenitors (GFP+CD71+Ter119+KITlow/+) were sorted from primary recipient mice and transplanted into secondary recipients to follow disease development. (F) Kaplan-Meier plot of diseased secondary recipient mice (described in panel A). WT (Ctrl or ERG): n = 4, TP53R248Q (Ctrl or ERG): n = 6. (G) Peripheral white blood cell (WBC), red blood cell (RBC), and platelet (PLT) counts of diseased mice transplanted with WT or TP53R248Q HSPC retrovirally transduced with ERG or empty vector (Ctrl). WT (Ctrl or ERG): n = 4, TP53R248Q (Ctrl or ERG): n = 4. (H) Flow cytometry analysis of myeloid cells (CD11b+Gr-1+), erythroid progenitors (CD71+Ter119+), B cells (B220+), and T cells (CD4+CD8+) from diseased secondary recipients of TP53R248Q HSPCs retrovirally transduced with ERG gated for GFP+ or GFP− cells. Statistical significance (in panels B, C, F, and G) is indicated as P values (Student t test except when otherwise specified). *P < .05; **P < .01; ***P < .001.
TP53-mutated AEL samples are associated with other alterations, including aberrant expression of the transcription factor ERG (Figure 3F). Most AEL-associated TP53 alterations are DNA-binding missense mutations13 including TP53R248Q.26 To address functional cooperation, we transplanted TP53R248Q Lin− HSPCs transduced with an ERG-expressing retrovirus. Because ectopic ERG expression in adult murine hematopoiesis was shown to primarily induce T-cell leukemia58,59 (supplemental Figure 7D-E), we assessed the long-term consequences of high ERG expression specifically in erythroid progenitors by transplanting purified ERG-transduced (GFP+) wild-type or TP53R248Q erythroblasts obtained from primary recipients, into secondary recipients (Figure 5E). All recipients of TP53R248Q erythroblasts overexpressing ERG developed a fatal leukemia with a median survival of 60 days, whereas recipients of ERG-expressing wild-type erythroblasts developed disease after 4 months (Figure 5F). The TP53R248Q+ERG-induced disease was characterized by anemia, thrombocytopenia (Figure 5G), and the accumulation of CD71+Ter119+ erythroid and to a lesser extent CD11b+Gr1+ myeloid progenitors in the BM (Figure 5H), with infiltration in spleen and liver (supplemental Figure 7F). These data indicate that an AEL-associated TP53 DNA-binding mutation cooperates with aberrantly high ERG expression to enhance the proliferative capacity of erythroid progenitors leading to leukemia with several features of the human disease.
Taken together, these results demonstrate that mutation associations in human AEL functionally cooperate to induce murine AML-like leukemia in vivo. Notably, although both combinations could readily induce an AEL-like phenotype when expressed in erythroid-restricted progenitors, their expression in HSPCs led to other mostly mixed leukemia phenotypes. Therefore, these data also suggest that the target cell in which these mutations are active impacts the disease phenotype.
In vivo modeling of erythroid transformation by aberrant SKI expression
To further explore the relevance of the cell context for consequences of the transcriptional alterations, we investigated in vivo disease development upon high expression of SKI, which was observed in 2 AEL samples of subgroup 3 (Figure 3G). First, we confirmed previous work49 showing that transplantation of Lin− HSPCs retrovirally overexpressing SKI induced a lethal disease (supplemental Figure 8A) characterized by anemia, thrombocytopenia, and increased myeloid cells in the periphery (supplemental Figure 8B) associated with hypercellular BM and spleens showing high percentage of mostly myeloid or erythroid transgene-expressing cells (supplemental Figure 8C-E). Notably, GFP detection in all 3 myeloid, erythroid, and platelet lineages (supplemental Figure 8F) suggested that SKI overexpression may affect early multipotent stem or progenitor cells.
To investigate whether the transforming activity of SKI depends on the hematopoietic target cell, we purified, transduced and transplanted long-term multipotent HSC, erythroid-enriched (MEP), or myeloid-committed granulocyte-macrophage (GMP) progenitors (Figure 6A). Three weeks posttransplant, transduced cells were detectable in the blood for all groups but engraftment in the BM was only observed in recipients from SKI-transduced HSCs and MEPs that later developed symptomatic diseases characterized by anemia, thrombocytopenia, (supplemental Figure 8H) and presenting with both CD11b+Gr1+ myeloid and CD71+Ter119+ erythroid features (Figure 6B-E). SKI-transduced GMP recipients did not develop disease. Symptomatic recipients of HSCs or MEPs showed an increase in basophilic, polychromatophilic, and orthochromatic erythroblasts and in reticulocytes associated with a relative decrease in mature red cells (Figure 6F), suggesting that SKI delays but does not fully block erythroid differentiation in vivo. Histopathological analyses confirmed BM hypercellularity and revealed infiltration of erythroid cells in the spleen and liver (Figure 6G; supplemental 8I).

In vivo modeling of erythroid transformation by aberrant SKI expression. (A) Experimental design: hematopoietic stem cells (HSC), granulocyte-monocyte progenitors (GMP), and megakaryocyte-erythroid progenitors (MEP) were sorted from WT mice, retrovirally transduced with SKI or an empty vector (Ctrl) carrying an IRES-GFP expression cassette, and injected into lethally irradiated recipients. (B) Left, GFP+ cells (percentage) in the BM of transplanted mice. Mean plus or minus SD is shown (n = 5 per group). Right, Kaplan-Meier plot of disease symptoms in transplanted mice. Only mice transplanted with SKI-transduced HSCs and MEP developed disease. (C) Flow cytometry analysis of myeloid cells (CD11b+Gr-1+), erythroid progenitors (CD71+Ter119+), B cells (B220+), and T cells (CD4+CD8+) gated for viable GFP+ cells, in spleens of mice transplanted with HSC (Ctrl vs SKI) or MEP (SKI) cells. (D) Percentage of myeloid cells (CD11b+Gr1+), erythroid progenitors (CD71+Ter119+), B cells (B220+), and T cells (CD4+CD8+) within viable GFP+ cells, in HSC-Control (Ctrl), HSC-SKI, and MEP-SKI in primary mice BM and spleen. Mean plus or minus SD is shown (n = 5 per group). (E) Number of GFP+ and GFP− total BM cells, myeloid cells (CD11b+Gr1+), and erythroid progenitors (CD71+Ter119+) in the BM of diseased primary recipients. Mean plus or minus SD is shown (n = 5 per group). (F) Flow cytometry analysis of terminal erythroid differentiation in the BM of diseased HSC (Ctrl and SKI) and MEP-SKI recipients, determined by forward scatter (FSC-A) and CD44 expression gated for viable Ter119+ cells. Bottom panel, Mean plus or minus SD (n = 5 per group) of the percentage of each population. (G) Histopathology analysis of BM spleen and liver of mice transplanted with HSC-Ctrl (left row) or HSC-SKI (right row) stained with hematoxylin-eosin (top 6 photomicrographs; original magnification ×20) or with a GATA1 antibody (bottom 4 photomicrographs; original magnification ×20).
In vivo modeling of erythroid transformation by aberrant SKI expression. (A) Experimental design: hematopoietic stem cells (HSC), granulocyte-monocyte progenitors (GMP), and megakaryocyte-erythroid progenitors (MEP) were sorted from WT mice, retrovirally transduced with SKI or an empty vector (Ctrl) carrying an IRES-GFP expression cassette, and injected into lethally irradiated recipients. (B) Left, GFP+ cells (percentage) in the BM of transplanted mice. Mean plus or minus SD is shown (n = 5 per group). Right, Kaplan-Meier plot of disease symptoms in transplanted mice. Only mice transplanted with SKI-transduced HSCs and MEP developed disease. (C) Flow cytometry analysis of myeloid cells (CD11b+Gr-1+), erythroid progenitors (CD71+Ter119+), B cells (B220+), and T cells (CD4+CD8+) gated for viable GFP+ cells, in spleens of mice transplanted with HSC (Ctrl vs SKI) or MEP (SKI) cells. (D) Percentage of myeloid cells (CD11b+Gr1+), erythroid progenitors (CD71+Ter119+), B cells (B220+), and T cells (CD4+CD8+) within viable GFP+ cells, in HSC-Control (Ctrl), HSC-SKI, and MEP-SKI in primary mice BM and spleen. Mean plus or minus SD is shown (n = 5 per group). (E) Number of GFP+ and GFP− total BM cells, myeloid cells (CD11b+Gr1+), and erythroid progenitors (CD71+Ter119+) in the BM of diseased primary recipients. Mean plus or minus SD is shown (n = 5 per group). (F) Flow cytometry analysis of terminal erythroid differentiation in the BM of diseased HSC (Ctrl and SKI) and MEP-SKI recipients, determined by forward scatter (FSC-A) and CD44 expression gated for viable Ter119+ cells. Bottom panel, Mean plus or minus SD (n = 5 per group) of the percentage of each population. (G) Histopathology analysis of BM spleen and liver of mice transplanted with HSC-Ctrl (left row) or HSC-SKI (right row) stained with hematoxylin-eosin (top 6 photomicrographs; original magnification ×20) or with a GATA1 antibody (bottom 4 photomicrographs; original magnification ×20).
Taken together, these data indicate that high SKI expression transforms HSCs and MEPs, but not myeloid-restricted progenitors like GMPs. Although aberrant SKI expression in erythroid-restricted progenitors leads to pure erythroid proliferation, expression in more immature HSPCs resulted in increased self-renewal capacity with aberrant differentiation toward both myeloid and erythroid lineages indicative of an AEL/MDS-like disease.
Discussion
AEL is an aggressive human cancer, often difficult to diagnose due to its close resemblance to other forms of hematopoietic malignancies presenting with variable compositions of cells with erythroid features, like MDS or certain AML subtypes. Here, we describe novel features of AEL that shed light on the pathophysiology of this disease. First, our data indicate that the majority of human AEL exhibit a unique erythroid transcriptional signature that differs from those found in patients with non-M6 AML or MDS without prominent erythropoiesis. Second, aberrant expression of various transcriptional regulators known to modulate GATA1 activity was frequently found in AEL and may represent a common molecular module that controls erythroid differentiation. Third, in vivo models demonstrate that the relative composition of the erythroid and myeloid features is strongly dependent on the hematopoietic target cell in which a driving oncogene is expressed, providing a basis for a better understanding of the highly heterogeneous clinical appearance of AEL.
The genomic lesions described here are in line with previous reports, including the largest genetic landscape study of human erythroleukemia to date,9-12,36 and support classification of AEL patients into molecular subgroups. In our study, 3 subgroups were identified, including patients with TP53 mutations (36.3% of cases), patients with mutations in epigenetic regulators previously associated with clonal hematopoiesis of indeterminate potential (CHIP) and MDS (eg, DNMT3A, TET2, and IDH1/2 mutations) (33.3% of cases) and another group of patients presenting with none of these recurrent alterations (30.4% of cases). Although TP53 mutations and epigenetic mutations are not mutually exclusive, their frequencies within AEL samples are similar.36 In our sequenced AEL cases, we did not detect the other recently described subgroups, including those with NUP98, KMT2A, and other in-frame fusions,36 which could reflect the limited number of pediatric patients in our cohort. Also, consistent with the frequent association between FLT3 mutations and NPM1 or KMT2A alterations,33 our patient cohort lacked samples with FLT3 or NPM1 mutations. For samples sequenced with RNA-seq only, we cannot exclude the possibility that some structural variants or low expressed mutated transcripts remained undetected.
In contrast to previous studies, we also found out-of-frame fusion transcripts associated with altered expression of 1 of the partner genes. For example, the fusion between YWHAE and EPO in a TP53-mutated patient was associated with ectopic expression of EPO, and the concomitant high expression of EPOR suggested an autocrine EPO/EPOR-signaling mechanism.15 Interestingly, alterations of multiple signaling intermediates, including downstream of EPO/EPOR, were recently found in up to 48% of human AEL samples36 and acquired activating KIT mutations were also essential to induce a bona fide erythroleukemia in a transgenic murine model,60 indicating the importance of signaling alterations for efficient oncogenic transformation of the erythroid lineage.
As the vast majority of the AEL-associated mutations are also found in a wide spectrum of human myeloid malignancies, it is essential to gain insights into their functional role in the erythroid phenotype that leads to a diagnosis of AEL. Together with the molecular alterations targeting EPO (YWHAE-EPO fusion) and the erythroid transcription factor GATA1 found here, the recently reported APLP2-EPOR and MYB1-GATA1 fusion genes36 further support the relevance of alterations in erythroid master regulators as underlying the erythroid phenotype in some AEL cases. However, most AEL do not present with erythroid-specific genetic alterations. Our patient-based transcriptional data, together with chromatin accessibility and functional analyses in cellular and in vivo models revealed that at least 25% of AEL cases present with transcriptional alterations ultimately interfering with GATA1 activity through direct or functional interaction within the GATA1 transcriptional complexes (eg, aberrantly expressed ETO2, ERG, SKI, SPI1). Although some of these transcriptional alterations (eg, ERGhigh expression) were recently reported to have a genetic bases,61,62 the origin of some others remains to be determined (eg, SKIhigh). As reported previously,60,63 we noted that ectopic SPI1 expression was sufficient to immortalize erythroblasts in vitro but not to induce the disease in vivo, supporting the idea that cooperating alterations that have yet to be identified are required in SPI1high human leukemia.
Although the basis for the erythroid phenotype remains to be demonstrated in many cases, several epigenomic AEL alterations may also functionally converge on aberrant activity of erythroid master regulators. Indeed, a novel signaling pathway based on JAK2-mediated phosphorylation of TET2 leading to interaction with KLF1 was recently reported.64 Combined TET2 and DNMT3A inactivation was also reported to upregulate expression of KLF1 and EPOR in HSCs.55 Therefore, the concomitant TET2 and DNMT3A mutations observed in 2 AEL patients and the presence of TET2 and GATA1s mutations in another AEL sample support a functional synergism between alterations of KLF1 and GATA1 transcriptional programs leading to differentiation blockage. Based on these observations, we hypothesize that the erythroid phenotype in AEL results from a cooperation between genetic and transcriptional alterations. As proposed for other subtypes of leukemia, interference with the activity of altered erythroid master regulators, for example, through targeting of critical protein-protein interactions may therefore represent promising therapeutic strategies for AEL.65,66
Our observations also have implications for the classification of AEL patients into molecular and/or prognosis subgroups. Comparative analysis of AEL expression signatures with normal erythroid and myeloid differentiation indicated that AEL is heterogeneously spread along a differentiation-associated trajectory with some patient samples clustering next to progenitors retaining myeloid features and other patient samples clustering closer to the erythroid trajectory. Also, although several oncogenes (eg, SKI) can transform restricted erythroid lineages, they led to mixed erythroid/myeloid hematopoietic malignancies upon expression in multipotent murine progenitors. These data indicate that the relative composition of myeloid vs erythroid elements at time of diagnosis is not solely based on the type of mutations but likely also reflects the type of progenitor targeted by these mutations. Notably, the relationship between gene-expression signatures and normal differentiation trajectories was not clearly visible when comparing the reported immunophenotypes of the blasts, and no correlation was found with the different molecular subgroups. These data strongly suggest that, in some AEL patients, the erythroid phenotype maybe initiated either by strong mutations that interfere with erythroid differentiation, or by mutations that provide advantages to erythroid-restricted progenitors. Alternatively, in others, the erythroid phenotype may originate from mutations in multipotent progenitors with a subsequent epigenetic drift toward the erythroid lineage.
Taken together, our work provided insights into the molecular mechanisms of the erythroid identity in AEL. Future studies need to resolve, likely at the single-cell level, the clonal genetic and epigenomic heterogeneous architecture in prospectively collected fresh samples as a further step toward the development of specific therapies.
Contact the corresponding authors for original data.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are grateful to Olivier Hermine, Françoise Moreau-Gachelin, Michaela Fontenay, and Françoise Pflumio for expert advice and useful discussions. The authors also thank Suzana Atanasoski (Basel, Switzerland) for providing the human SKI cDNA, and Stéphanie Ranga and Michael Finnegan for handling human AEL samples and helping with cDNA cloning. One patient’s samples were handled, conditioned, and stored by the FILOtheque (no. BB-0033-00073), Tumor Bank of the French Innovative Leukemia Organization (FILO) Group (Cochin Hospital, Paris, France).
This work was supported by Institut National du Cancer (PLBIO-2014-176 and PLBIO-2018-169 [T.M.]), Ligue Contre le Cancer (PhD grant [A.F.], Équipe Labelisée [T.M.]), Institut National du Cancer (INCa)-PlanCancer ‘‘Soutien pour la Formation’’ 2009-2013 (C.I.), Cancéropôle Ile-de-France (2014-2017 [C.K.L.]), Sites de Recherche Intégrée sur le Cancer (SIRIC)-SOCRATE (INCa-DGOS-INSERM 6043 [T.M.] and INCa-DGOS-INSERM_12551 [O.A.B.]), Fondation pour la Recherche Médicale (C.I., Z.A. [FRM-ING20150532273], and C.K.L.), Fondation de France (FdF-00057925 [C.T. and T.M.]), and the Gustave Roussy Genomic Core Facility (Taxe d’Apprentissage TA2018-ALFA [A.F.]). J.S. was supported by grants from the Swiss Cancer League (KFS-3487-08-2014), the Gertrude von Meissner Foundation (Basel, Switzerland), the San Salvatore Foundation (201525; Lugano, Switzerland), the Wilhelm Sander Foundation (2017.035.1; Munich, Germany) and the Swiss National Science Foundation (SNF, 31003A_173224/1). U.M. was supported by the National Institutes of Health, National Cancer Institute (2RO1CA176647) and the Stony Brook Foundation (Carol Baldwin Foundation). P. Valent was supported by the Austrian Science Fund (FWF) grant F4704-B20 and a stem cell grant of the Medical University of Vienna. P. Vyas was supported by the Bloodwise and Children with Cancer Specialist Programme (grant 13001), the National Institute for Health Research (NIHR) Oxford Biomedical Research Fund, and the Medical Research Council Molecular Haematology Unit (MRC MHU; MC_UU_12009/11). E.A. was supported by the Fundación Hay Esperanza. C.M. was supported by the Leukemia & Lymphoma Society Translational Research Program, National Institutes of Health and National Cancer Institute Outstanding Investigator Award R35 CA197695, and the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital. E.S. was supported by Fondation pour la Recherche Médicale (Equipe FRM DEQ20180339221), the ATIP-Avenir Program (Plan Cancer), and Labex EpiGenMed (Investissements d’Avenir Program, reference ANR-10-LABX-12-01).
Authorship
Contribution: A.F., M.-R.P.-B., C.I., A.C., C.K.L., B.U., Z.A., C.T., S.T., B.L., V.D., S.M., L.G., E.S., J.S., and T.M. performed and analyzed experiments; A.F., M.-R.P.-B., F.O.B., and E.R. performed bioinformatics analyses; V.G.-B., A.K.-K., J.M., C.D., O.S., S.S., C.S., V.D.M., T.P., K.S., H.L., S.d.B., J.-B.M., I.I., C.G.M., B.K., C.L.C., M.C., P. Valent, E.D., P. Vyas, D.B., and E.A. provided patient samples and clinical information; A.F., U.M., Z.K., S.M., O.A.B., D.B., E.A., L.G., E.S., J.S., and T.M. provided major intellectual inputs and/or reagents; T.M. and J.S. conceived and supervised the project and drafted the manuscript; and all authors revised and approved the final version of the manuscript.
Conflict-of-interest disclosure: C.G.M. received research funding from Abbvie, Loxo Oncology, and Pfizer; and speaking and travel fees from Illumina and Amgen. The remaining authors declare no competing financial interests.
Correspondence: Thomas Mercher, Institut Gustave Roussy, INSERM U1170, 39 rue Camille Desmoulins, 94800 Villejuif, France; e-mail: thomas.mercher@inserm.fr; or Juerg Schwaller, University Children’s Hospital Basel (UKBB), Department of Biomedicine (DBM), University of Basel, ZLF-Laboratory 202, Hebelstrasse 20, CH-4031 Basel, Switzerland; e-mail: j.schwaller@unibas.ch.